Defects and Calcium Diffusion in Wollastonite


 ,
, 
Abstract

1. Introduction
2. Computational Methods
3. Results
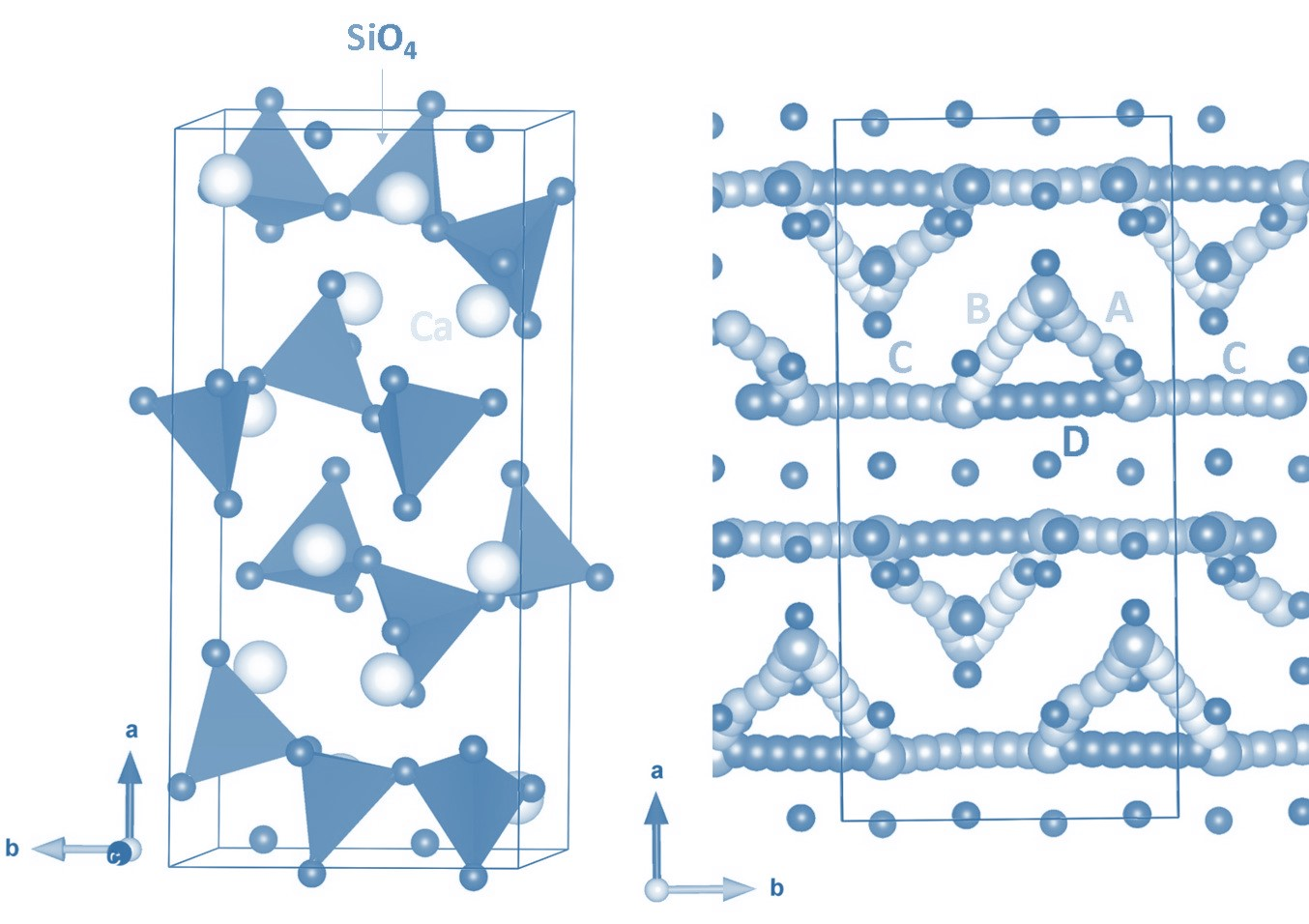
3.1. Crystal Structure of CaSiO3 and Validation of Potentials
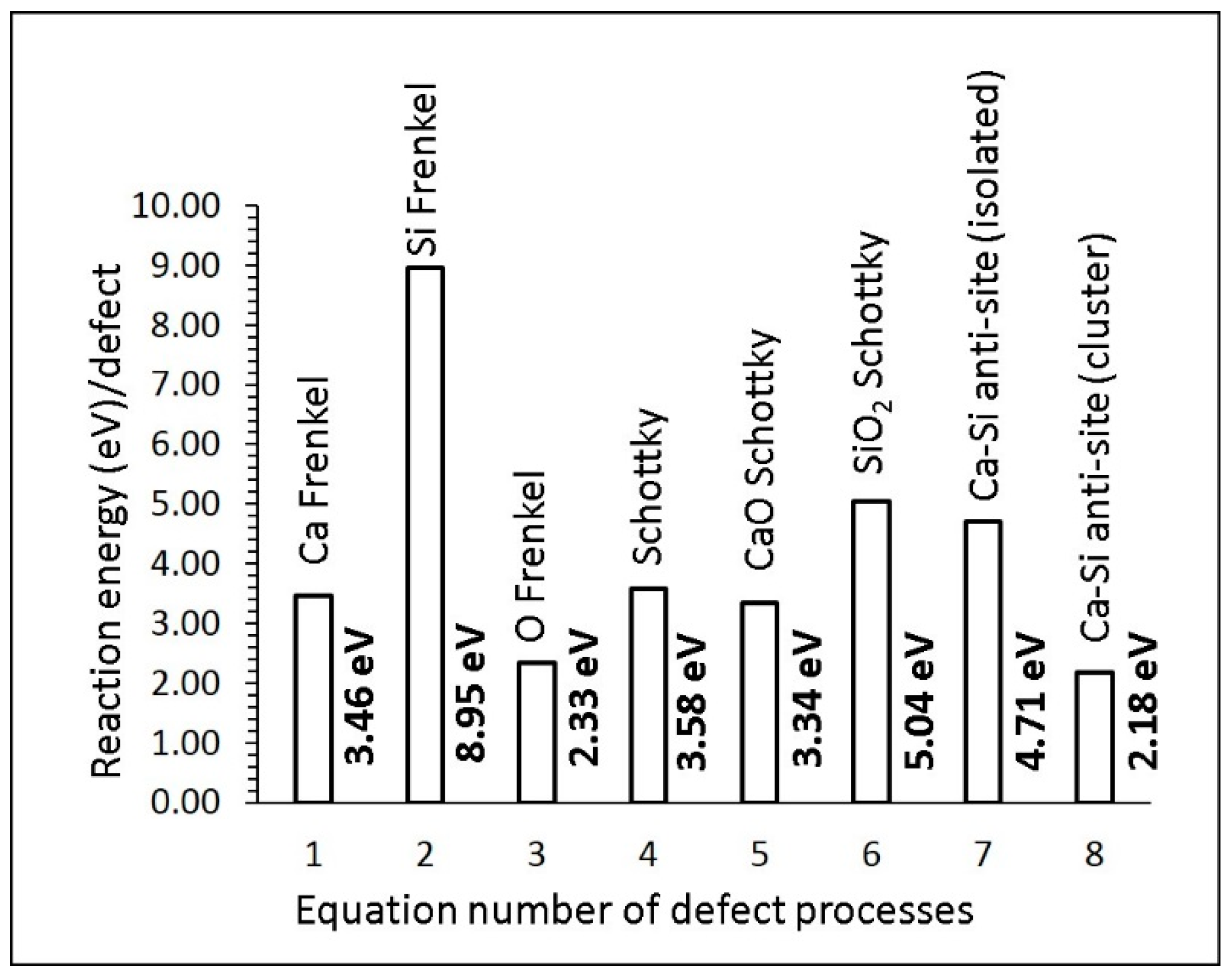
3.2. Defect Energetics
3.3. Calcium Ion Diffusion
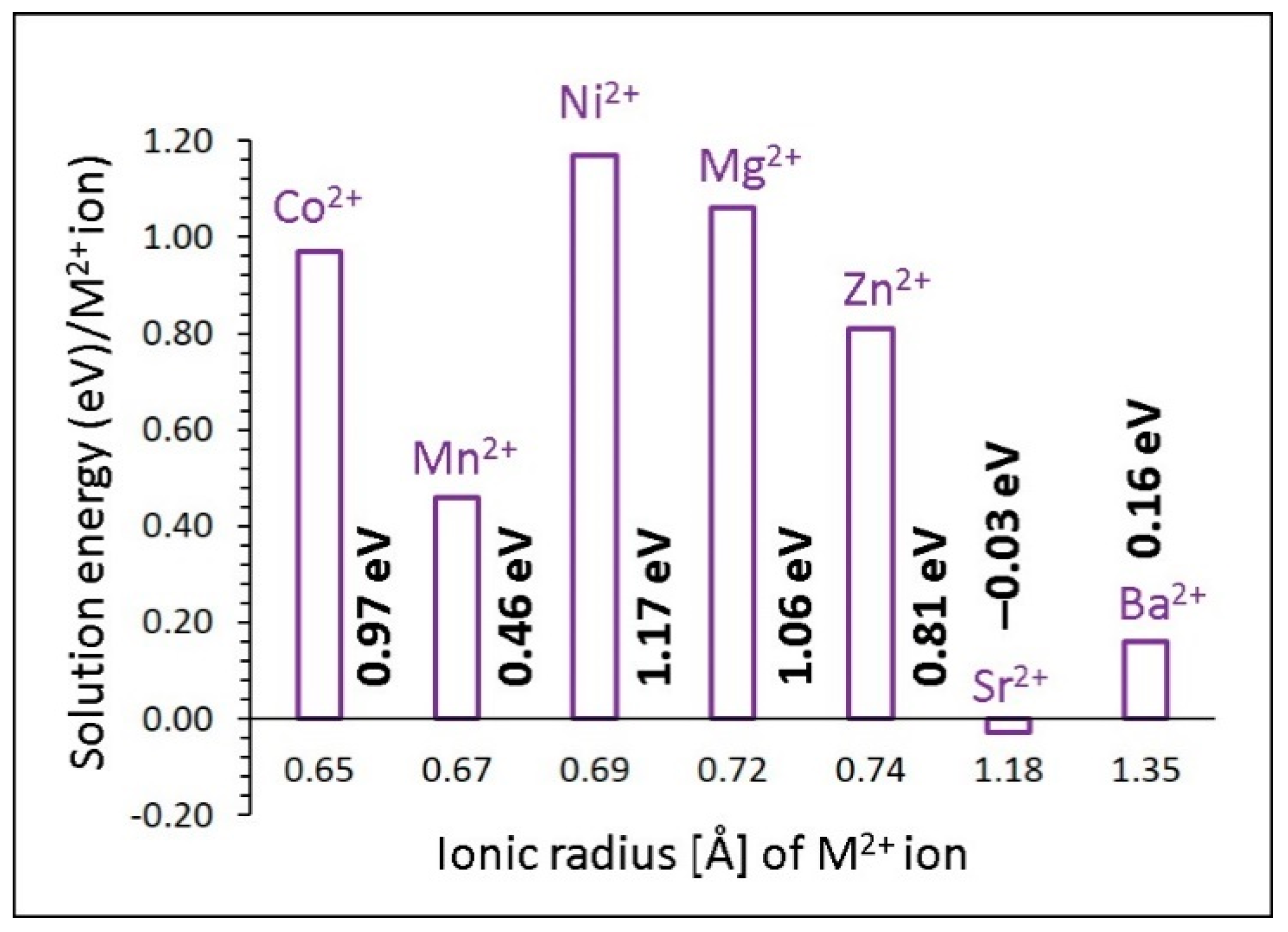
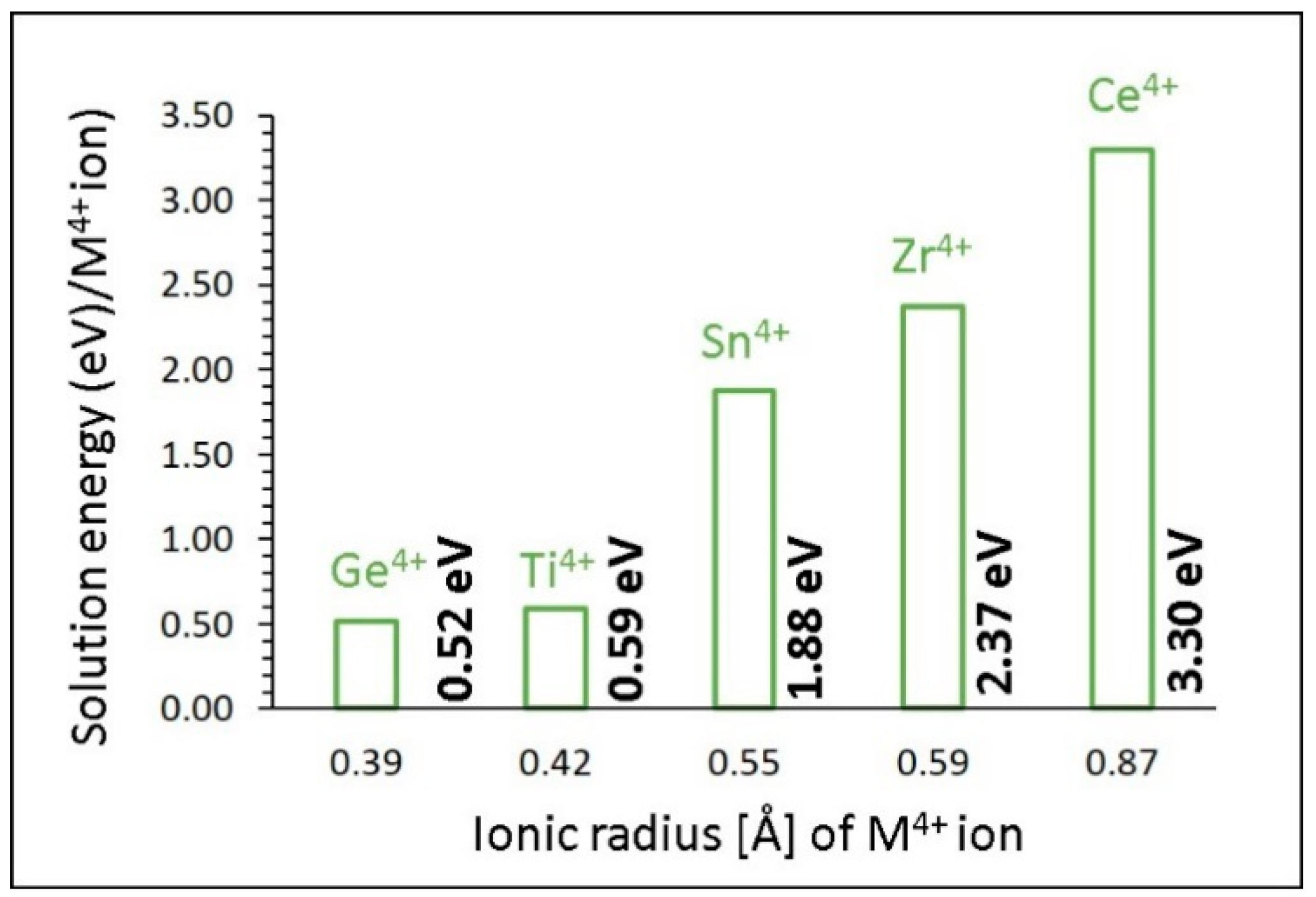
3.4. Solution of Dopants
3.4.1. Divalent Dopants
3.4.2. Trivalent Dopants
3.4.3. Tetravalent Dopants
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Abd Rashid, R.; Shamsudin, R.; Abdul Hamid, M.A.; Jalar, A. Low temperature production of wollastonite from limestone and silica sand through solid-state reaction. J. Asian Ceram. Soc. 2014, 2, 77–81. [Google Scholar] [CrossRef]
- Demidenko, N.I.; Podzorova, L.I.; Rozanova, V.S.; Skorokhodov, V.A.; Shevchenko, V.Y. Wollastonite as a New Kind of Natural Material (A Review). Glass Ceram. 2001, 58, 308–311. [Google Scholar] [CrossRef]
- Kangal, M.O.; Bulut, G.; Guven, O. Physicochemical Characterization of Natural Wollastonite and Calcite. Minerals 2020, 10, 228. [Google Scholar] [CrossRef]
- Serghiou, G.C.; Hammack, W.S. Pressure-induced amorphization of wollastonite (CaSiO3) at room temperature. J. Chem. Phys. 1993, 98, 9830–9834. [Google Scholar] [CrossRef]
- Kotsis, I.; Balogh, A. Synthesis of wollastonite. Ceram. Int. 1989, 15, 79–85. [Google Scholar] [CrossRef]
- Svensson, K.; Neumann, A.; Menezes, F.F.; Lempp, C.; Pöllmann, H. The Conversion of Wollastonite to CaCO3 Considering Its Use for CCS Application as Cementitious Material. Appl. Sci. 2018, 8, 304. [Google Scholar] [CrossRef]
- Harabi, A.; Chehlatt, S. Preparation process of a highly resistant wollastonite bioCeramics using local raw materials: Effect of B2O3 additions on sintering and mechanical properties. J. Therm. Anal. Calorim. 2013, 111, 203–211. [Google Scholar] [CrossRef]
- Ismail, H.; Shamsudin, R.; Abdul Hamid, M.A.; Jalar, A. Synthesis and characterization of nano-wollastonite from rice husk ash and limestone. Mater. Sci. Forum 2013, 756, 43–47. [Google Scholar] [CrossRef]
- Lin, K.; Chang, J.; Lu, J. Synthesis of wollastonite nanowires via hydrothermal microemulsion methods. Mater. Lett. 2006, 60, 3007–3010. [Google Scholar] [CrossRef]
- Yun, Y.H.; Yun, S.D.; Park, H.R.; Lee, Y.K.; Youn, Y.N. Preparation of β-wollastonite glass-Ceramics. J. Mater. Synth. Process. 2002, 10, 205–209. [Google Scholar] [CrossRef]
- Ming, H.; Ren, Z.S.; Hua, Z.X. Characterization and analysis of CaO-SiO2-B2O3 ternary system Ceramics. J. Mater. Sci. Mater. Electron. 2011, 22, 389–393. [Google Scholar] [CrossRef]
- Zilles, J.U. Wollastonites. In Encyclopedia of Polymers and Composites; Palsule, S., Ed.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 1–21. [Google Scholar]
- Gineika, A.; Siauciunas, R.; Baltakys, K. Synthesis of wollastonite from AlF3-rich silica gel and its hardening in the CO2 atmosphere. Sci. Rep. 2019, 9, 18063. [Google Scholar] [CrossRef] [PubMed]
- Zulumyan, N.; Mirgorodski, A.; Isahakyan, A.; Beglaryan, H.; Gabrielyan, A.; Terzyan, A. A low-temperature method of the β-wollastonite synthesis. J. Therm. Anal. Calorim. 2015, 122, 97–104. [Google Scholar] [CrossRef]
- Wan, X.; Chang, C.; Mao, D.; Jiang, L.; Li, M. Preparation and in vitro bioactivities of calcium silicate nanophase materials. Mater. Sci. Eng. C 2005, 25, 455–461. [Google Scholar] [CrossRef]
- Long, L.H.; Chen, L.D.; Bai, S.Q.; Chang, J.; Lin, K.L. Preparation of dense β-CaSiO3 Ceramics with high mechanical strength and HAp formation ability in simulated body fluid. J. Eur. Ceram. Soc. 2006, 26, 1701–1706. [Google Scholar] [CrossRef]
- Hazar, A.B.Y. Preparation and in vitro bioactivity of CaSiO3 powders. Ceram. Int. 2007, 33, 687–692. [Google Scholar] [CrossRef]
- Zhao, S.; Zhou, Z.; Wu, J.; Wang, S.; Guo, X.; Zhang, Q. Preparation and characterization of a novel hydroxyapatite-wollastonite/silk fibroin composite. J. Compos. Mater. 2011, 46, 1571–1581. [Google Scholar] [CrossRef]
- De La Casa-Lillo, M.A.; Velásquez, P.; De Aza, P.N. Influence of thermal treatment on the “in vitro” bioactivity of wollastonite materials. J. Mater. Sci. Mater. Med. 2011, 22, 907–915. [Google Scholar] [CrossRef]
- Edrees, S.J.; Shukur, M.M.; Obeid, M.M. First-principle analysis of the structural, mechanical, optical and electronic properties of wollastonite monoclinic polymorph. Comput. Condens. Matter. 2018, 14, 20–26. [Google Scholar] [CrossRef]
- Kundu, T.K.; Hanumantha Rao, K.; Parker, S.C. Atomistic simulation of the surface structure of wollastonite. Chem. Phys. Lett. 2003, 377, 81–92. [Google Scholar] [CrossRef]
- Longo, R.C.; Cho, K.; Brüner, P.; Welle, A.; Gerdes, A.; Thissen, P. Carbonation of Wollastonite (001) Competing Hydration: Microscopic Insights from Ion Spectroscopy and Density Functional Theory. ACS Appl. Mater. Interfaces 2015, 7, 4706–4712. [Google Scholar] [CrossRef] [PubMed]
- Profeta, M.; Benoit, M.; Mauri, F.; Pickard, C.-J. First-Principles Calculation of the 17O NMR Parameters in Ca Oxide and Ca Aluminosilicates: The Partially Covalent Nature of the Ca-O Bond, a Challenge for Density Functional Theory. J. Am. Chem. Soc. 2004, 126, 12628–12635. [Google Scholar] [CrossRef] [PubMed]
- Kuganathan, N.; Iyngaran, P.; Vovk, R.; Chroneos, A. Defects, dopants and Mg diffusion in MgTiO3. Sci. Rep. 2019, 9, 4394. [Google Scholar] [CrossRef]
- Kuganathan, N.; Kordatos, A.; Chroneos, A. Defect Chemistry and Li-ion Diffusion in Li2RuO3. Sci. Rep. 2019, 9, 550. [Google Scholar] [CrossRef] [PubMed]
- Jay, E.E.; Rushton, M.J.D.; Chroneos, A.; Grimes, R.W.; Kilner, J.A. Genetics of superionic conductivity in lithium lanthanum titanates. Phys. Chem. Chem. Phys. 2015, 17, 178–183. [Google Scholar] [CrossRef]
- Islam, M.S.; Fisher, C.A.J. Lithium and sodium battery cathode materials: Computational insights into voltage, diffusion and nanostructural properties. Chem. Soc. Rev. 2014, 43, 185–204. [Google Scholar] [CrossRef]
- Kendrick, E.; Islam, M.S.; Slater, P.R. Atomic-scale mechanistic features of oxide ion conduction in apatite-type germanates. Chem. Commun. 2008, 715–717. [Google Scholar] [CrossRef] [PubMed]
- Gale, J.D. GULP: A computer program for the symmetry-adapted simulation of solids. J. Chem. Soc. Faraday Trans. 1997, 93, 629–637. [Google Scholar] [CrossRef]
- Gale, J.D.; Rohl, A.L. The General Utility Lattice Program (GULP). Mol. Simul. 2003, 29, 291–341. [Google Scholar] [CrossRef]
- Mott, N.F.; Littleton, M.J. Conduction in polar crystals. I. Electrolytic conduction in solid salts. Trans. Faraday Soc. 1938, 34, 485–499. [Google Scholar] [CrossRef]
- Grimes, R.W.; Busker, G.; McCoy, M.A.; Chroneos, A.; Kilner, J.A.; Chen, S.-P. The Effect of Ion Size on Solution Mechanism and Defect Cluster Geometry. Ber. Bunsenges. Phys. Chem. 1997, 101, 1204–1210. [Google Scholar] [CrossRef]
- Lewis, G.V.; Catlow, C.R.A. Potential models for ionic oxides. J. Phys. C Solid State Phys. 1985, 18, 1149–1161. [Google Scholar] [CrossRef]
- Kuganathan, N.; Islam, M.S. Li2MnSiO4 Lithium Battery Material: Atomic-Scale Study of Defects, Lithium Mobility, and Trivalent Dopants. Chem. Mater. 2009, 21, 5196–5202. [Google Scholar] [CrossRef]
- Hesse, K.F. Refinement of the crystal structure of wollastonite-2M (parawollastonite). Z. Krist. Cryst. Mater. 1984, 168, 93–98. [Google Scholar] [CrossRef]
- Kröger, F.A.; Vink, H.J. Relations between the Concentrations of Imperfections in Crystalline Solids. In Solid State Physics; Seitz, F., Turnbull, D., Eds.; Academic Press: Cambridge, MA, USA, 1956; Volume 3, pp. 307–435. [Google Scholar]
- Armstrong, A.R.; Kuganathan, N.; Islam, M.S.; Bruce, P.G. Structure and Lithium Transport Pathways in Li2FeSiO4 Cathodes for Lithium Batteries. J. Am. Chem. Soc. 2011, 133, 13031–13035. [Google Scholar] [CrossRef]
- Kempaiah Devaraju, M.; Duc Truong, Q.; Hyodo, H.; Sasaki, Y.; Honma, I. Synthesis, characterization and observation of antisite defects in LiNiPO4 nanomaterials. Sci. Rep. 2015, 5, 11041. [Google Scholar] [CrossRef]
- Politaev, V.V.; Petrenko, A.A.; Nalbandyan, V.B.; Medvedev, B.S.; Shvetsova, E.S. Crystal structure, phase relations and electrochemical properties of monoclinic Li2MnSiO4. J. Solid State Chem. 2007, 180, 1045–1050. [Google Scholar] [CrossRef]
- Kuganathan, N.; Kordatos, A.; Kelaidis, N.; Chroneos, A. Defects, Lithium Mobility and Tetravalent Dopants in the Li3NbO4 Cathode Material. Sci. Rep. 2019, 9, 2192. [Google Scholar] [CrossRef]
- Kuganathan, N.; Kordatos, A.; Anurakavan, S.; Iyngaran, P.; Chroneos, A. Li3SbO4 lithium-ion battery material: Defects, lithium ion diffusion and tetravalent dopants. Mater. Chem. Phys. 2019, 225, 34–41. [Google Scholar] [CrossRef]
- Gardiner, G.R.; Islam, M.S. Anti-Site Defects and Ion Migration in the LiFe0.5Mn0.5PO4 Mixed-Metal Cathode Material. Chem. Mater. 2010, 22, 1242–1248. [Google Scholar] [CrossRef]
- Islam, M.S.; Driscoll, D.J.; Fisher, C.A.J.; Slater, P.R. Atomic-Scale Investigation of Defects, Dopants, and Lithium Transport in the LiFePO4 Olivine-Type Battery Material. Chem. Mater. 2005, 17, 5085–5092. [Google Scholar] [CrossRef]
- Zhang, X.; Rui, X.; Chen, D.; Tan, H.; Yang, D.; Huang, S.; Yu, Y. Na3V2(PO4)3: An advanced cathode for sodium-ion batteries. Nanoscale 2019, 11, 2556–2576. [Google Scholar] [CrossRef] [PubMed]
- Eames, C.; Frost, J.M.; Barnes, P.R.F.; O’Regan, B.C.; Walsh, A.; Islam, M.S. Ionic transport in hybrid lead iodide perovskite solar cells. Nat. Commun. 2015, 6, 7497. [Google Scholar] [CrossRef] [PubMed]
- Malavasi, L.; Fisher, C.A.J.; Islam, M.S. Oxide-ion and proton conducting electrolyte materials for clean energy applications: Structural and mechanistic features. Chem. Soc. Rev. 2010, 39, 4370–4387. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.A.J.; Hart Prieto, V.M.; Islam, M.S. Lithium Battery Materials LiMPO4 (M = Mn, Fe, Co, and Ni): Insights into Defect Association, Transport Mechanisms, and Doping Behavior. Chem. Mater. 2008, 20, 5907–5915. [Google Scholar] [CrossRef]
- Nishimura, S.-I.; Kobayashi, G.; Ohoyama, K.; Kanno, R.; Yashima, M.; Yamada, A. Experimental visualization of lithium diffusion in LixFePO4. Nat. Mater. 2008, 7, 707. [Google Scholar] [CrossRef] [PubMed]
- Torres, A.; Luque, F.J.; Tortajada, J.; Arroyo-de Dompablo, M.E. Analysis of Minerals as Electrode Materials for Ca-based Rechargeable Batteries. Sci. Rep. 2019, 9, 9644. [Google Scholar] [CrossRef]
- Kuganathan, N.; Ganeshalingam, S.; Chroneos, A. Defect, transport, and dopant properties of andradite garnet Ca3Fe2Si3O12. AIP Adv. 2020, 10, 075004. [Google Scholar] [CrossRef]
- Kuganathan, N.; Chroneos, A. Defects and Dopants in CaFeSi2O6: Classical and DFT Simulations. Energies 2020, 13, 1285. [Google Scholar] [CrossRef]
- Dompablo, M.E.A.-D.; Krich, C.; Nava-Avendaño, J.; Biškup, N.; Palacín, M.R.; Bardé, F. A Joint Computational and Experimental Evaluation of CaMn2O4 Polymorphs as Cathode Materials for Ca Ion Batteries. Chem. Mater. 2016, 28, 6886–6893. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interaction | A/eV | ρ/Å | C/eV·Å6 | Y/e | K/eV·Å−2 |
|---|---|---|---|---|---|
| Ca2+–O2− | 1090.40 | 0.3372 | 0.00 | 1.260 | 34.0 |
| Si4+–O2− | 1283.91 | 0.32052 | 10.66 | 4.000 | 99999.0 |
| O2−–O2− | 22764.30 | 0.1490 | 27.89 | −2.86 | 74.92 |
| Parameter | Calculated | Experiment [35] | |∆|(%) |
|---|---|---|---|
| a (Å) | 15.23 | 15.41 | 1.17 |
| b (Å) | 7.30 | 7.32 | 0.24 |
| c (Å) | 7.01 | 7.06 | 0.76 |
| α (°) | 90.0 | 90.0 | 0.00 |
| β (°) | 91.5 | 95.3 | 3.96 |
| γ (°) | 90.0 | 90.0 | 0.00 |
| V (Å3) | 779.39 | 793.47 | 1.77 |
| Hop | Ca–Ca Distance (Å) | Activation Energy (eV) |
|---|---|---|
| A | 3.46 | 1.37 |
| B | 3.49 | 1.59 |
| C | 3.63 | 1.34 |
| D | 3.67 | 2.02 |
| Long-Range Path | Activation Energy (eV) |
|---|---|
| C↔D↔C↔D↔C | 2.02 |
| A↔B↔C↔A↔B | 1.59 |
| A↔B↔C↔D↔C | 2.02 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nimasha, S.; Ganeshalingam, S.; Kuganathan, N.; Davazoglou, K.; Chroneos, A. Defects and Calcium Diffusion in Wollastonite. Chemistry 2020, 2, 937-946. https://doi.org/10.3390/chemistry2040059
Nimasha S, Ganeshalingam S, Kuganathan N, Davazoglou K, Chroneos A. Defects and Calcium Diffusion in Wollastonite. Chemistry. 2020; 2(4):937-946. https://doi.org/10.3390/chemistry2040059
Chicago/Turabian StyleNimasha, Sumudu, Sashikesh Ganeshalingam, Navaratnarajah Kuganathan, Konstantinos Davazoglou, and Alexander Chroneos. 2020. "Defects and Calcium Diffusion in Wollastonite" Chemistry 2, no. 4: 937-946. https://doi.org/10.3390/chemistry2040059
APA StyleNimasha, S., Ganeshalingam, S., Kuganathan, N., Davazoglou, K., & Chroneos, A. (2020). Defects and Calcium Diffusion in Wollastonite. Chemistry, 2(4), 937-946. https://doi.org/10.3390/chemistry2040059