Abstract
Bis(diphenylphosphino)methane dioxide (dppmO2) forms eight-coordinate cations [M(dppmO2)4]Cl3 (M = La, Ce, Pr, Nd, Sm, Eu, Gd) on reaction in a 4:1 molar ratio with the appropriate LnCl3 in ethanol. Similar reaction in a 3:1 ratio produced seven-coordinate [M(dppmO2)3Cl]Cl2 (M = Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb), whilst LuCl3 alone produced six-coordinate [Lu(dppmO2)2Cl2]Cl. The complexes have been characterised by IR, 1H and 31P{1H}-NMR spectroscopy. X-ray structures show that [M(dppmO2)4]Cl3 (M = Ce, Sm, Gd) contain square antiprismatic cations, whilst [M(dppmO2)3Cl]Cl2 (M = Yb, Dy, Lu) have distorted pentagonal bipyramidal structures with apical Cl. The [Lu(dppmO2)2Cl2]Cl has a cis octahedral cation. The structure of [Yb(dppmO2)3(H2O)]Cl3·dppmO2 is also reported. The change in coordination numbers and geometry along the series is driven by the decreasing lanthanide cation radii, but the chloride counter anions also play a role.
1. Introduction
Early work viewed the chemistry of the lanthanides (Ln) (Ln = La–Lu, ≠ Pm unless otherwise indicated) in oxidation state III as very similar and often only two or three elements were examined, and the results were assumed to apply to all. More recent work has shown this to be a very unreliable approach and detailed studies of all fourteen elements (excluding only the radioactive Pm) are required to establish properties and trends [1,2]. Sometimes yttrium is also included since it is similar in size to holmium. The main changes along the series are due to the lanthanide contraction, the reduction in the radius of the M3+ ions between La (1.22 Å) and Lu (0.85 Å), and at some point a reduction in coordination number may be driven by steric effects, especially with bulky ligands. However, the decrease in radius also results in an increase in the charge/radius ratio along the series and this can lead to significant electronic effects on the ligand preferences. This interplay of steric and electronic effects means that changes in coordination number or ligand donor set can occur at different points along the series with different ligands. The effects are very nicely demonstrated in a recent article, which examined the changes which occurred in the series of lanthanide nitrates with complexes of 2,2′-bipyridyl, 2,4,6-tri-α-pyridyl-1,3,5-triazine and 2,2′; 6′,2”-terpyridine [2]. Tertiary phosphine oxides have proved popular ligands to explore lanthanide chemistry and the area has been the subject of a comprehensive review [3], and several detailed studies of trends along the series La-Lu have been reported [4,5,6,7]. We reported bis(diphenylphosphino)methane dioxide (dppmO2) formed square-antiprismatic cations [La(dppmO2)4]3+ with Cl, I or [PF6] counter ions, but lutetium gave only octahedral [Lu(dppmO2)2X2]+ (X = Cl, I) and [Lu(dppmO2)2Cl(H2O)]2+ [8]. Other dppmO2 complexes reported include several types with Ln(NO3)3 [4], [Dy(dppmO2)4][CF3SO3]3 [9], [Eu(dppmO2)4][ClO4]3 [10], [La(dppmO2)4][CF3SO3]3 and [Lu(dppmO2)3(H2O)][CF3SO3]3 [11]. Here, we report a systematic study of the systems LnCl3-dppmO2 for all fourteen accessible lanthanides.
2. Materials and Methods
Infrared spectra were recorded as Nujol mulls between CsI plates using a Perkin-Elmer Spectrum 100 spectrometer over the range 4000–200 cm−1. 1H and 31P{1H}-NMR spectra were recorded using a Bruker AV–II 400 spectrometer and are referenced to the protio resonance of the solvent and 85% H3PO4, respectively. Microanalyses were undertaken by London Metropolitan University or Medac. Hydrated lanthanide trichlorides and anhydrous LnCl3 (Ln–Nd, Pr, Gd, Ho) were from Sigma-Aldrich and used as received. The Ph2PCH2PPh2 (Sigma-Aldrich) in anhydrous CH2Cl2 was converted to Ph2P(O)CH2P(O)Ph2 by air oxidation catalysed by SnI4 [12].
X-Ray Experimental. Details of the crystallographic data collection and refinement parameters are given in Table 1. Many attempts were made to grow crystals for X-ray examination from a variety of solvents including EtOH and CH2Cl2, either by slow evaporation or layering with hexane or pentane. The crystal quality was often rather poor, and all of the structures have disordered co-solvent, either water or ethanol. No attempt was made to locate the protons on the co-solvent. Several showed disorder in one or more of the phenyl rings. Good-quality crystals used for single crystal X-ray analysis were grown from [Lu(dppmO2)4}Cl2]Cl (CH2Cl2/hexane), [Ce(dppmO2)4]Cl3, [Sm(dppmO2)4]Cl3, [Gd(dppmO2)4]Cl3 (EtOH), [Yb(dppmO2)3Cl]Cl2, [Yb(dppmO2)3(H2O)]Cl3·dppmO2 (EtOH), [Lu(dppmO2)3Cl]Cl2 (CH2Cl2).

Table 1.
X-ray crystallographic data a.
Data collections used a Rigaku AFC12 goniometer equipped with a HyPix-600HE detector mounted at the window of an FR-E+ SuperBright molybdenum (λ = 0.71073 Å) rotating anode generator with VHF Varimax optics (70 µm focus) with the crystal held at 100 K (N2 cryostream). Structure solution and refinements were performed with either SHELX(S/L)97 or SHELX(S/L)2013 [13,14]. The crystallographic data in cif format have been deposited as CCDC 2033611-2033618.
All samples were dried in high vacuum at room temperature for several hours, but this treatment does not remove lattice water or alcohol. Heating the samples in vacuo is likely to cause some decomposition of the complexes [7] and was not applied.
[La(dppmO2)4]Cl3·4H2O and [Lu(dppmO2)2Cl2]Cl·H2O were made as described [8]. The individual new complexes were isolated as described below, with yields of 50–80%.
[Ce(dppmO2)4]Cl3·6H2O—CeCl3·7H2O (0.025 g, 0.067 mmol) and dppmO2 (0.112 g, 0.268 mmol) afforded colourless crystals of [Ce(dppmO2)4]Cl3·4H2O, by concentrating the ethanolic solution and layering with n-hexane (1 mL). Required for C100H100CeCl3O12P8 (2020.1): C, 59.46; H, 4.99%. Found: C, 59.50; H, 4.50%. 1H-NMR (CD2Cl2): δ = 1.52 (s, H2O) 3.60 (vbr, [8H], PCH2P), 7.10 (s, [32H], Ph), 7.35 (m, [16H], Ph), 7.70 (m, [32H], Ph). 31P{1H}-NMR (CD2Cl2): δ = 48.6 (s). IR (Nujol mull)/cm−1: 3500 br, 1630 (H2O), 1158, 1099s (P=O).
[Pr(dppmO2)4]Cl3·6H2O—To a solution of PrCl3·6H2O (0.025 g, 0.070 mmol) in ethanol (5 mL) was added a solution of dppmO2 (0.117 g, 0.281 mmol) in ethanol (10 mL). A white powdered solid formed on slow evaporation of the ethanol. Required for C100H100Cl3O14P8Pr (2020.9): C, 59.43; H, 4.99%. Found: C, 59.06; H, 4.62% 1H-NMR (CD2Cl2): δ = 4.63 (m, [8H], PCH2P), 7.19 (s, [32H], Ph), 7.44 (m, [16H], Ph), 8.19 (m, [32H], Ph). 31P{1H}-NMR (CD2Cl2): δ = 64.0 (s). IR (Nujol mull)/cm−1: 3500 br, 1630 (H2O), 1161, 1102 (P=O).
[Nd(dppmO2)4]Cl3·4H2O—To a solution of NdCl3·6H2O (0.025 g, 0.070 mmol) in ethanol (5 mL) was added a solution of dppmO2 (0.116 g, 0.279 mmol) in ethanol (10 mL). A white powdered solid formed on slow evaporation of the ethanol. Required for C100H96Cl3NdO12P8 (1988.2): C, 60.41; H, 4.87%. Found: C, 60.41; H, 4.62%. 1H-NMR (CD2Cl2): δ = 1.52 (s, H2O) 3.66 (m, [8H], PCH2P), 7.14 (s, [32H], Ph), 7.35 (m, [16H], Ph), 7.76 (m, [32H], Ph). 31P{1H}-NMR (CD2Cl2): δ = 62.9 (s). IR (Nujol mull)/cm−1: 3500 br, 1630 (H2O), 1159 s, 1101 s (P=O).
[Sm(dppmO2)4]Cl3·4H2O—To a solution of SmCl3·6H2O (0.025 g, 0.069 mmol) in ethanol (5 mL) was added a solution of dppmO2 (0.114 g, 0.274 mmol) in ethanol (10 mL). Colourless crystals were formed via slow evaporation of the ethanol. Required for C100H96Cl3O12P8Sm (1994.3): C, 60.22; H, 4.85%. Found: C, 60.05; H, 4.50%. 1H-NMR (CD2Cl2): δ = 2.10 (s, H2O), 5.08 (br, [8H], PCH2P), 7.20 (s, [32H], Ph), 7.39 (m, [16H], Ph), 7.83 (m, [32H], Ph). 31P{1H}-NMR (CD2Cl2): δ = 35.6. IR (Nujol mull)/cm−1: 3500 br, 1630 (H2O), 1162 s, 1101 (P=O).
[Eu(dppmO2)4]Cl3·4H2O—To a solution of EuCl3·6H2O (0.025 g, 0.068 mmol) in ethanol (5 mL) was added a solution of dppmO2 (0.114 g, 0.274 mmol) in ethanol (10 mL) and the solution was stirred for 20 min. The solution was then concentrated, and colourless crystals were formed through layering with n-hexane (1 mL). Required for C100H96Cl3EuO12P8 (1995.9): C, 60.41; H, 4.87%. Found: C, 60.73; H, 4.71%. 1H-NMR (CD2Cl2): δ = 2.15 (s, H2O) 3.12 (br, [8H] PCH2P), 7.18 (s, [32H], Ph), 7.38 (m, [16H], Ph), 7.83 (m, [32H], Ph). 31P{1H}-NMR (CD2Cl2): δ = 25.0 (br, “free” dppmO2), −13.4. IR (Nujol mull)/cm−1: 3500 br, 1630 (H2O), 1159, 1099 (P=O).
[Gd(dppmO2)4]Cl3·4H2O—To a solution of GdCl3·6H2O (0.025 g, 0.067 mmol) in ethanol (5 mL) was added a solution of dppmO2 (0.112 g, 0.269 mmol) in ethanol (10 mL). Colourless crystals were formed through slow evaporation of the solvent. Required for C100H96Cl3GdO12P8 (2001.2): C, 60.02; H, 4.83%. Found: C, 60.05; H, 4.86%. 1H-NMR (CD2Cl2): δ = no resonance. 31P{1H}-NMR (CD2Cl2): δ = no resonance. IR (Nujol mull)/cm−1: 3500 br, 1630 (H2O), 1160, 1099 (P=O).
[Sm(dppmO2)3Cl]Cl2—To a solution of SmCl3·6H2O (0.025 g, 0.069 mmol) in ethanol (5 mL) was added a solution of dppmO2 (0.086 g, 0.206 mmol) in ethanol (10 mL). The solvent was removed in vacuo and the resulting white solid was washed with cold ethanol. Colourless crystals were obtained via slow evaporation of an ethanolic solution of the product. Required for C75H66Cl3O6P6Sm (1505.9): C, 59.80; H, 4.42%. Found: C, 59.62; H, 4.55%. 1H-NMR (CD2Cl2): δ = 3.67 (br m, [6H], PCH2P), 7.15 (br, [24H], Ph), 7.35 (m, [12H], Ph), 8.05 (m, [24H], Ph). 31P{1H}-NMR (CD2Cl2): δ = 38.15 (s). IR (Nujol mull)/cm−1: 1153 s, 1097 s (P=O).
[Eu(dppmO2)3Cl]Cl2—To a solution of EuCl3·6H2O (0.025 g, 0.068 mmol) in ethanol (5 mL) was added a solution of dppmO2 (0.085 g, 0.205 mmol) in ethanol (10 mL). The solvent was removed in vacuo and the resulting white solid was washed with cold ethanol. Required for C75H66EuCl3O6P6 (1507.49): C, 59.76; H, 4.41%. Found: C, 59.71; H, 4.56%. 1H-NMR (CDCl3): δ = 3.66 (br, [6H], PCH2P), 7.03 (br m, [36H], Ph), 7.87 (br, [24H], Ph). 31P{1H}-NMR (CDCl3): δ = −14.8 (s). IR (Nujol mull)/cm−1: 1153 s, 1098 s (P=O).
[Gd(dppmO2)3Cl]Cl2·3H2O—To a solution of GdCl3·6H2O (0.025 g, 0.067 mmol) in ethanol (5 mL) was added a solution of dppmO2 (0.084 g, 0.201 mmol) in ethanol (10 mL). The solvent was removed in vacuo and the resulting white solid was washed with cold ethanol. Required for C75H66Cl3O6P6Gd (166.8): C, 57.49, H, 4.63%; Found: C, 57.17; H, 4.43%. 1H-NMR (CD2Cl2): no resonance. 31P{1H}-NMR (CD2Cl2): no resonance. IR (Nujol mull)/cm−1: 3500 br, 1630 (H2O), 1155 s, 1098 s (P=O).
[Tb(dppmO2)3Cl]Cl2·H2O—To a solution of TbCl3·6H2O (0.025 g, 0.067 mmol) in ethanol (5 mL) was added a solution of dppmO2 (0.084 g, 0.201 mmol) in ethanol (10 cm3). The solvent was removed in vacuo and the resulting white solid was washed with cold ethanol. Required for C75H68Cl3O7P6Tb (1532.5): C, 58.78; H, 4.47%. Found: C, 59.41; H, 4.54%. 1H-NMR (CD2Cl2): δ = 1.9 (br H2O), 3.50 (br m, [6H], PCH2P), 5.89 (br, [36H], Ph), 7.46 (br, [24H], Ph). 31P{1H}-NMR (CD2Cl2): δ = −29.2 (s). IR (Nujol mull)/cm−1: 3500 br, 1630 (H2O), 1153 s, 1097 s (P=O).
[Dy(dppmO2)3Cl]Cl2·H2O—To a solution of TbCl3·6H2O (0.025 g, 0.066 mmol) in ethanol (5 mL) was added a solution of dppmO2 (0.083 g, 0.199 mmol) in ethanol (10 cm3). The solution was filtered then concentrated and layered with hexane (1 mL) yielding a white powdered product. Colourless crystals were formed by layering a CH2Cl2 solution of the product with hexane. Required for C75H68Cl3DyO7P6 (1536.0): C, 58.64; H, 4.46%. Found: C, 58.21; H, 4.63%. 1H-NMR (CD2Cl2): δ = 1.9 (vbr H2O), 3.66 (br m, [6H], PCH2P), 7.33 (br, [36H], Ph), 8.66 (br, [24H], Ph). 31P{1H}-NMR (CD2Cl2): δ = 18 (vbr, s). IR (Nujol mull)/cm−1: 3500 br, 1630 (H2O), 1156 s, 1099 s (P=O).
[Ho(dppmO2)3Cl]Cl2·H2O—To a solution of HoCl3 (0.050 g, 0.124 mmol) in ethanol (5 mL) was added a solution of dppmO2 (0.230 g, 0.55 mmol) in ethanol (10 mL). The solvent was removed in vacuo and the resulting pale pink solid was washed with cold ethanol. Required for C75H68Cl3HoO7P6 (1538.5): C, 58.66; H, 4.55%. Found: C, 59.41; H, 4.52%. 1H-NMR (CD2Cl2): δ = 2.1 (br, H2O), 3.72 (br s, [6H], PCH2P), 6.78 (br, [36H], Ph), 7.68 (br, [24H], Ph)]. 31P{1H}-NMR (CD2Cl2): δ = −13.5 (s). IR (Nujol mull)/cm−1: 3500 br, 1630 (H2O), 1154 s, 1097 s (P=O).
[Er(dppmO2)3Cl]Cl2·3H2O—To a solution of ErCl3·6H2O (0.025 g, 0.065 mmol) in ethanol (5 mL) was added a solution of dppmO2 (0.082 g, 0.196 mmol) in ethanol (10 mL). The solvent was removed in vacuo and the resulting white solid was washed with cold ethanol. Required for C75H72Cl3ErO9P6 (1576.8): C, 57.13; H, 4.60%. Found: C, 57.08; H, 4.54%. 1H-NMR (CD2Cl2): δ = δ = 1.2 (br, H2O), 3.25 (br s, [6H], PCH2P), 5.52 (vbr, [12H], Ph), 7.15 (br s, [24H], Ph)], 7.28 (br s, [24H], Ph)]. 31P{1H}-NMR (CD2Cl2): δ = −60.8 (s). IR (Nujol mull)/cm−1: 3500 br, 1630 (H2O), 1155 s, 1097 s (P=O).
[Tm(dppmO2)3Cl]Cl2·3H2O—To a solution of TmCl3·6H2O (0.025 g, 0.065 mmol) in ethanol (5 mL) was added a solution of dppmO2 (0.081 g, 0.195 mmol) in ethanol (10 mL). The solvent was removed in vacuo and the resulting white solid was washed with cold ethanol. Required for C75H72Cl3O9P6Tm (1578.5): C, 57.07; H, 4.60%. Found: C, 56.61; H, 4.45%. 1H-NMR (CD2Cl2): δ = 3.48 (m, [6H], PCH2P), 7.11 (br, [24H], Ph), 7.68 (br, [36H], Ph)]. 31P{1H}-NMR (CD2Cl2): δ = −54.8 (s). IR (Nujol mull)/cm−1: 3500 br, 1630 (H2O), 1156 s, 1096 s (P=O).
[Yb(dppmO2)3Cl]Cl2·H2O—To a solution of YbCl3·6H2O (0.025 g, 0.065 mmol) in ethanol (5 mL) was added a solution of dppmO2 (0.080 g, 0.194 mmol) in ethanol (10 mL). The solvent was removed in vacuo and the resulting white powder was washed with cold ethanol. Required for C75H68Cl3O7P6Yb (1546.58): C, 58.24; H, 4.43%. Found: C, 58.73; H, 4.45%. 1H-NMR (CD2Cl2): δ = 3.50 (m, [6H], PCH2P), 6.64 (br, [24H], Ph), 7.15 (br, [36H], Ph). 31P{1H}-NMR (CD2Cl2): δ = +9.2 (s). IR (Nujol mull)/cm−1: 3500 br, 1630 (H2O), 1154 s, 1097 s (P=O).
3. Results
The reaction of LnCl3·nH2O (Ln = La [8], Ce, Pr, Nd, Sm, Eu or Gd; n = 6 or 7) with four mol. equivalents of dppmO2 in ethanol gave good yields of tetrakis-dppmO2 complexes, [Ln(dppmO2)4]Cl3. The IR and 1H-NMR spectra show the the isolated complexes retain significant amounts of lattice water, and sometimes EtOH, which is not removed by prolonged drying of the bulk powders in vacuo. The high molecular weights make the microanalyses rather insensitive to the amount of water, but are generally consistent with a formulation [Ln(dppmO2)4]Cl3·nH2O (n = 6: Ce, Pr; n = 4: Nd, Sm, Eu, Gd), although the amount of lattice solvent probably varies with the sample and is unlikely to be stoichiometric. The presence of significant amounts of lattice solvent is common in lanthanide phosphine oxide systems [7,8,9,10], and although evident in X-ray crystal structures, it is often disordered and difficult to model. Obtaining good quality crystals of the complexes proved difficult, but crystals of the Ce, Sm and Gd salts were obtained from various organic solvents and the compositions are shown in Table 1. The crystals contain different amounts of solvent of crystallisation to the bulk samples as they were grown from different media (and crystals were not dried in vacuo). The IR spectra (Table 2) show that the υ(PO) stretch in dppmO2 at 1187 cm−1 has been lost and replaced by a new very strong and broad band ~1160 cm−1 and a second band at ~ 1100 cm−1, which are due to the coordinated phosphine oxide groups. The frequencies appear invariant with the lanthanide present, which may be due to small differences being obscurred by the width of the bands. In [LnCl3(OPPh3)3] and [LnCl2(OPPh3)4]+ the frequency of the υ(PO) stretch increases by ~ 10 cm−1 between La and Lu [7]. The 31P{1H}-NMR chemical shift of dppmO2 at δ = +25.3 shows a high frequency shift to +33.1 in [La(dppmO2)4]Cl3, whilst the corresponding spectra of the Ce, Pr, Nd and Sm complexes show larger shifts due to the presence of the paramagnetic lanthanide ion (Table 2). In contrast, although the solid [Eu(dppmO2)4]Cl3 complex was isolated without difficulty, the 31P{1H}-NMR spectrum shows a strong feature at δ ~ +25 (“free” dppmO2), along with a second resonance at δ = −13.4, which may be assigned to [Eu(dppmO2)3Cl]Cl2 (see below), indicating substantial dissociation of one dppmO2 in solution; the broad resonance of the free dppmO2 is indicative of exchange on the NMR timescale. [Gd(dppmO2)4]Cl3 was isolated, and its constitution confirmed by its X-ray crystal structure, but no 1H or 31P{1H}-NMR resonances were observed, an effect seen in other gadolinium systems [6,7] and ascribed to fast relaxation by the f7 configuration of the metal. Attempts to isolate [Ln(dppmO2)4]Cl3 complexes for Ln = Dy-Lu were unsuccessful. We note that [Dy(dppmO2)4][CF3SO3]3 [9] was isolated with triflate counter ions, but with chloride only [Dy(dppmO2)3Cl]Cl2 was produced (below). An in situ 31P{1H}-NMR spectrum of CeCl3·7H2O + 2 dppmO2 in CH2Cl2 showed a single resonance at δ = +48, which is consistent with formation of [Ce(dppmO2)4]3+, confirming the preference for formation of the tetrakis complexes early in the series, even when there is a deficit of ligand.
Table 2.
IR and 31P{1H}-NMR spectroscopic data.
The X-ray structures of [Ce(dppmO2)4]Cl3 (Figure 1), [Sm(dppmO2)4]Cl3 (Figure 2) and [Gd(dppmO2)4]Cl3 (Figure 3) show distorted square antiprismatic cations, very similar to those in [La(dppmO2)4][PF6]3 [8] and [Nd(dppmO2)4]Cl3 [15]. The average Ln-O distances in this series are: La = 2.514 Å, Ce = 2.486 Å, Nd = 2.465 Å, Sm = 2.429 Å and Gd = 2.420 Å, correlating well with the decreasing Ln3+ radii (La = 1.216 Å, Ce =1.196 Å, Nd = 1.163 Å, Sm = 1.132 Å, Gd = 1.107 Å). The P = O bond lengths and the O-Ln-O chelate angles do not vary significantly along the series. The Ce-O(P) distances in [Ce(dppmO2)4]Cl3 are markedly longer than those in [Ce(Me3PO)4(H2O)4]Cl3 (2.372(2)-2.423(2) Å) [16], which has a distorted dodecahedral geometry with a CeO8 donor set.
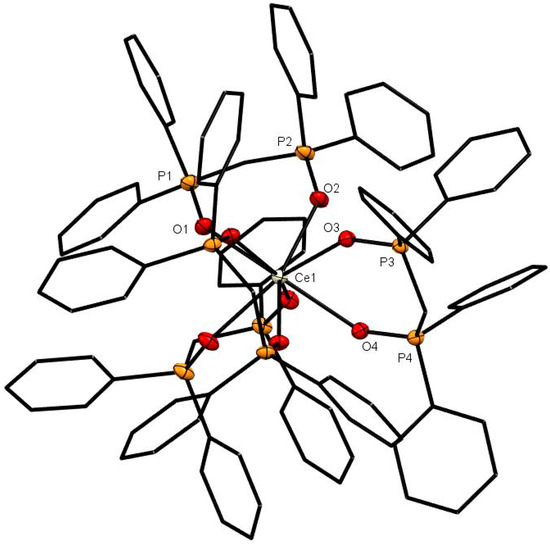
Figure 1.
The cation in [Ce(dppmO2)4]Cl3. The chloride anions and solvate molecules are omitted. Selected bond lengths (Å): Ce1–O1 = 2.4874(14), Ce1–O2 = 2.4790(14), Ce1–O3 = 2.4967(13), Ce1–O4 = 2.4803(14), P1–O1 = 1.5031(14), P2–O2 = 1.5021(14), P3–O3 = 1.5018(14), P4–O4 = 1.5031(14). Chelate angle O-Ce-O = 73.1° (av).
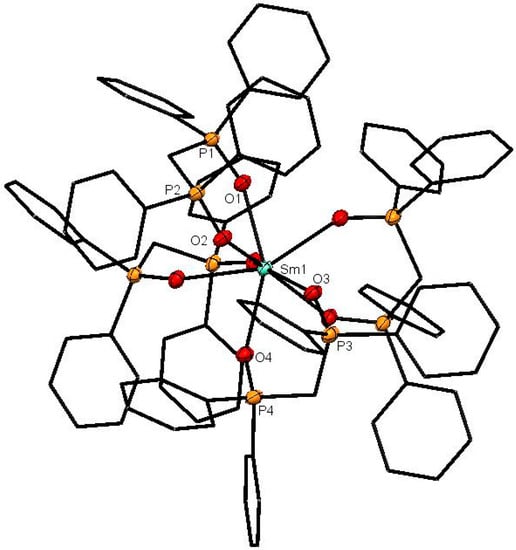
Figure 2.
The cation in [Sm(dppmO2)4]Cl3. The chloride anions and solvate molecules are omitted. Selected bond lengths (Å): Sm1–O1 = 2.4160(14), Sm1–O2 = 2.4400(15), Sm1–O3 = 2.4358(14), Sm1–O4 = 2.4268(15), P1–O1 = 1.5025(15), P2–O2 = 1.5019(15), P3–O3 = 1.4961(15), P4–O4 = 1.4961(16). Chelate angle O-Sm-O = 72.9° (av).
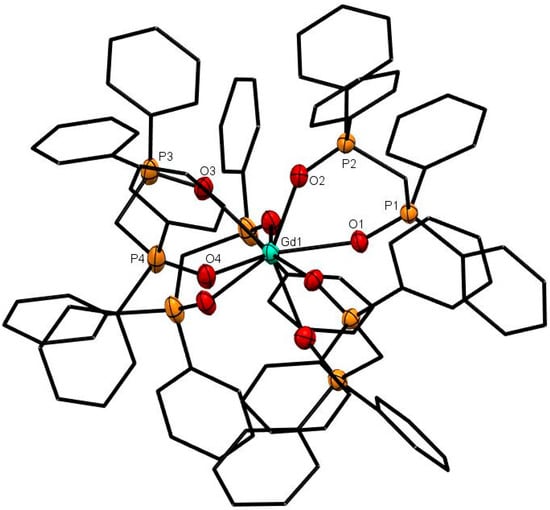
Figure 3.
The cation in [Gd(dppmO2)4]Cl3. The chloride anions and solvate molecules are omitted. Selected bond lengths (Å): Gd1–O1 = 2.420(2), Gd1–O2 = 2.409(3), Gd1–O3 = 2.415(2), Gd1–O4 = 2.398(2), P1–O1 = 1.504(2), P2–O2 = 1.501(3), P3–O3 = 1.501(3), P4–O4 = 1.501(3). Chelate angle O-Sm-O = 73.1° (av).
The reaction of LnCl3·6H2O (Ln =Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb) with 3 mol. equivalents of dppmO2 in EtOH, followed by concentration of the solution or precipitation with hexane, afforded [Ln(dppmO2)3Cl]Cl2 complexes. Examination of the IR and 1H-NMR spectra indicated these incorporated less water or ethanol lattice solvent molecules than the [Ln(dppmO2)4]Cl3, and this was confirmed by the microanalyses. The Sm and Eu complexes appear largely free of solvent of crystallisation, whilst the Tb, Ho and Yb approximate to [Ln(dppmO2)3Cl]Cl2·H2O, and the Gd, Er and Tm complexes are [Ln(dppmO2)3Cl]Cl2·3H2O; again, this is likely to vary from sample to sample and with the isolation method. The IR spectra (Table 2) show the two υ(PO) bands as in the tetrakis complexes, but the higher energy bands of the tris complexes are ~ 5–10 cm−1 lower in frequency than in the former. We were unable to identify υ(Ln-Cl) vibrations in the far IR spectra. The 31P{1H}-NMR spectra of the [Ln(dppmO2)3Cl]Cl2 show single resonances to high or low frequency of dppmO2 depending on the fn configuration of the Ln ion present (Table 2) and are generally similar to those found in other systems [5,6,7], although the magnitude of the shifts varies widely with the specific fn configuration. The line broadening is also highly variable between complexes of different Ln ions. The addition of dppmO2 to a solution of [Ln(dppmO2)3Cl]Cl2 (Ln = Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb) in CH2Cl2 showed 31P{1H}-NMR resonances assignable to “free” dppmO2 and [Ln(dppmO2)3Cl]Cl2, but no new resonances that could be attributed to the formation of significant amounts of [Ln(dppmO2)4]3+. Although the resonances are broad in some cases, the observed chemical shifts are identical to those in pure [Ln(dppmO2)3Cl]Cl2. For [Sm(dppmO2)3Cl]Cl2 δ(31P{1H}) = 38, the resonance shifts to δ = 35.6 upon addition of dppmO2, attributable to the formation of [Sm(dppmO2)4]Cl3, showing that both tris- and tetrakis-dppmO2 complexes exist in solution for samarium in the presence of the appropriate amount of ligand.
The X-ray structures of [Er(dppmO2)3Cl]Cl2 (Er-O = 2.28 Å av.) [17], [Yb(dppmO2)3Cl]Cl2 (Figure 4; Yb-O = 2.28 Å av.) and [Dy(dppmO2)3Cl]Cl2 (Figure S43) show pentagonal bipyramidal cations with an apical chloride. The Ln-O distances are rather variable (Er-O = 2.244(6)–2.328(6) Å; Yb-O = 2.250(2)–2.269(3) Å), but are shorter than those in the tetrakis-dppmO2 cations, reflecting both the reduced coordination number and the smaller metal ion radii (Er = 1.062, Yb = 1.042 Å). The contraction in ionic radii is also evident in the Ln-Cl distances of 2.598(2) Å (Er) and 2.5829(9) Å (Yb). Crystals of [Dy(dppmO2)3Cl]Cl2 were also obtained and show the same cation type, but during refinement, several of the phenyl rings exhibited severe disorder and the data are therefore not included here (Figure S43).
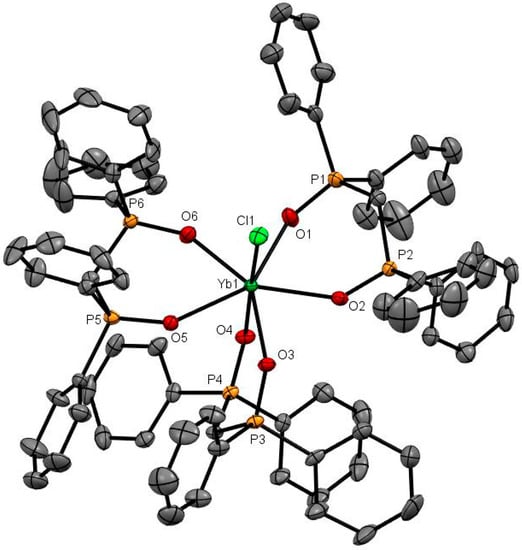
Figure 4.
The X-ray structure of [Yb(dppmO2)3Cl]Cl2. The chloride anions and solvate molecules are omitted. Selected bond lengths (Å) and angles (°): Yb1–Cl1 = 2.5834(9), Yb1–O1 = 2.298(3), Yb1–O2 = 2.282(3), Yb1–O3 = 2.250(2), Yb1–O4 = 2.248(2), Yb1–O5 = 2.338(2), Yb1–O6 = 2.269(3), P–O = 1.494(3)-1.509(3), Cl1–Yb1–O4 = 173.89(9), O1–Yb1–O2 = 73.79(9), O3–Yb1–O4 = 80.64(9), O5–Yb1–O6 = 73.95(9).
Lutetium was previously reported to form the only bis-dppmO2 complex, [Lu(dppmO2)2Cl2]Cl, in this series [8], and this has now been confirmed by the X-ray crystal structure which shows a cis-octahedral geometry (Figure 5). The Lu-O distance of 2.230 Å (av) is shorter than the Ln-O distances in the seven- or eigth-coordinate complexes, and correlates both with the reduced coordination number and the smaller radius of Lu3+ (1.032 Å). Treatment of a CH2Cl2 solution of [Lu(dppmO2)2Cl2]Cl with dppmO2 caused the 31P{1H}-NMR resonance to shift from +40 to +38.3, which suggests that [Lu(dppmO2)3Cl]Cl2 forms in solution. A few crystals of this product were isolated from a mixture containing excess dppmO2. These showed a pentagonal bipyramidal dication (Figure 6). As expected, the Lu-Cl and Lu-O bond lengths are slightly longer than in the six-coordinate cation, but are shorter than the corresponding bonds in [Yb(dppmO2)3Cl]Cl2, showing that the expected contraction continues along the series. The complex, [Lu(dppmO2)3(H2O)][CF3SO3]3, is known and its X-ray crystal structure showed seven-coordinate lutetium [11]. Although not confirmed by an X-ray structure, yttrium is reported to form a six-coordinate complex, [Y(dppmO2)2Cl2]Cl [18].
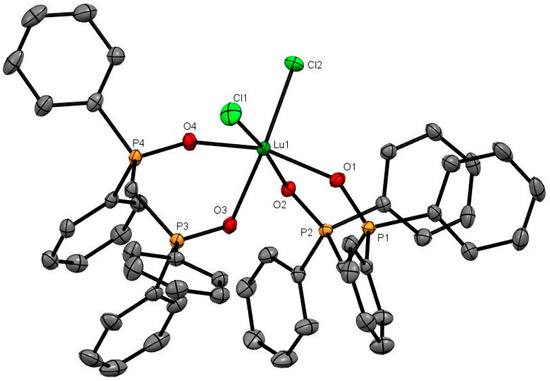
Figure 5.
The cation in [Lu(dppmO2)2Cl2]Cl. Selected bond lengths (Å) and angles (°): Lu1–Cl1 = 2.5581(8), Lu1–Cl2 = 2.5163(7), Lu1–O1 = 2.227(2), Lu1–O2 = 2.227(2), Lu1–O3 = 2.274(2), Lu1–O4 = 2.200(2), P1–O1 = 1.510(2), P2–O2 = 1.506(2), P3–O3 = 1.513(2), P4–O4 = 1.507(2), Cl2–Lu1–Cl1 = 95.97(3), O1–Lu1–Cl1 = 97.27(6), O1–Lu1–Cl2 = 99.68(6), O1–Lu1–O2 = 81.56(8), O1–Lu1–O3 = 85.65(7), O2–Lu1–Cl2 = 91.94(5), O2–Lu1–O3 = 84.89(7), O3–Lu1–Cl1 = 87.22(6), O4–Lu1–Cl1 = 94.66(6), O4–Lu1–Cl2 = 92.55(6), O4–Lu1–O2 = 84.74(8), O4–Lu1–O3 = 81.36(7).
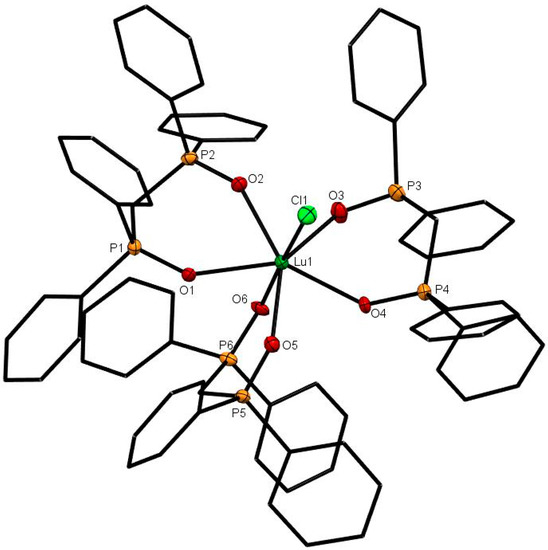
Figure 6.
The X-ray structure of [Lu(dppmO2)3Cl]Cl2. The chloride anions and solvate molecules are omitted. Selected bond lengths (Å) and angles (°): Lu1–Cl1 = 2.5604(7), Lu1–O1 = 2.341(5), Lu1–O2 = 2.268(5), Lu1–O3 = 2.268(5), Lu1–O4 = 2.297(5), Lu1–O5 = 2.354(5), Lu1–O6 = 2.227(5), P–O = 1.497(5)-1.510(5), Cl1–Lu1–O6 = 176.35(14), O1–Lu1–O2 = 73.37(17), O3–Lu1–O4 = 73.37(17), O5–Lu1–O6 = 83.45(18).
A different crystal isolated from the YbCl3-dppmO2 reaction proved, on structure solution, to be [Yb(dppmO2)3(H2O)]Cl3·dppmO2·12H2O (Figure 7), which contains a seven-coordinate Yb centre coordinated to three dppmO2 and a water molecule, with the Lu-coordinated water hydrogen-bonded to an adjacent uncoordinated dppmO2 molecule. The geometry is best described as a very distorted pentagonal bipyramid with the water occupying an equatorial position and is similar to the geometry found in [Lu(dppmO2)3(H2O)][CF3SO3]3 [11]. The Yb-OH2 distance of 2.3263(14) Å is ~ 0.05 Å longer than the Yb-O(P).
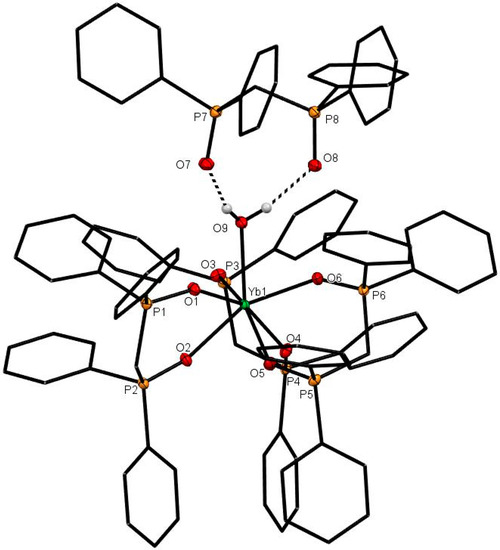
Figure 7.
The cation in [Yb(dppmO2)3(H2O)]Cl3·dppmO2·12H2O also showing the hydrogen-bonded dppmO2 molecule. Selected bond lengths (Å): Yb1–O3 = 2.2341(14), Yb1–O2 = 2.2899(13), Yb1–O9 = 2.3263(14), Yb1–O4 = 2.2683(13), Yb1–O6 = 2.2208(13), Yb1–O1 = 2.2328(13), Yb1–O5 = 2.2696(14), Pn–On (n = 1–6) = 1.5034(14)–1.5072(14), P7–O7 = 1.4924(15), P8–O8 = 1.4926(15).
A large number of disordered solvate water molecules were also present, which proved very difficult to model, but the geometry of the ytterbium cation is clearly defined.
4. Discussion
The chemistry of dppmO2 with lanthanides described in the previous section proves to be very systematic along the series La–Lu. For La–Gd, it was possible to isolate [Ln(dppmO2)4]Cl3. Although it could be isolated in the solid state, the solution 31P-NMR spectroscopic data indicate that [Eu(dppmO2)4]Cl3 was largely dissociated in CH2Cl2 solution into [Eu(dppmO2)3Cl]2+ and dppmO2; the isolation of the tetrakis-dppmO2 complex no doubt resulting from it being the least soluble species in an exchanging mixture in solution, although present in very minor amounts. The case of [Gd(dppmO2)4]Cl3 is likely to be similar, although the fast relaxation of the f7 ion precluded 31P-NMR study. For the elements Sm-Yb, the complexes [Ln(dppmO2)3Cl]Cl2 were readily isolated, but only for samarium was it possible to convert [Ln(dppmO2)3Cl]Cl2 to [Ln(dppmO2)4]Cl3 in CH2Cl2 solution by treatment with more dppmO2. Similarly, at the end of the series, the complex isolated was [Lu(dppmO2)2Cl2]Cl, for which treatment with dppmO2 afforded a new species in solution, identified as [Lu(dppmO2)3Cl]Cl2 by a structure determination from a few crystals obtained in the presence of excess dppmO2, although a bulk sample could not be isolated [8]. The change from eight-coordination in [Ln(dppmO2)4]Cl3 at the beginning of the series, to seven-coordination from Sm onwards, and finally to six-coordination at Lu, parallels the reduction in Ln3+ radii. Isolation of both the eight- and seven-coordinate complexes was possible only for Sm, Eu and Gd. However, one should note that the chloride counter ions also have some role, in that whilst in the LnCl3/dppmO2 series tetrakis-dppmO2 species did not form beyond Gd, the complex [Dy(dppmO2)4][CF3SO3]3 [9] has been isolated from dmf solution with triflate counter ions. The role that anions and solvents play in lanthanide chemistry is often overlooked [2], but can be critical in determining which complex is isolated from solution. For example, the reaction of LnCl3 with Ph3PO results in isolation of [Ln(Ph3PO)3Cl3] from acetone, but [Ln(Ph3PO)4Cl2]Cl from ethanol [7]. On further examination by 31P-NMR spectroscopy, both species were found to be present in either solvent (in varying amounts), and the form isolated reflected the least soluble complex in the particular solvent, which then precipitated from the mixture of rapidly interconverting species.
5. Conclusions
Through this synthetic, structural and spectroscopic study of the coordination of dppmO2 to the lanthanide trichlorides, we have established where the switch from eight-, to seven-, to six-coordination at the Ln(III) centre occurs along the lanthanide series, with X-ray crystallographic authentication for representative examples. The data also reveal subtle, but systematic, variations in the spectroscopic (e.g., ν(PO)) and structural parameters across the series, reflecting the change in ionic radii, the charge:radius ratio and also the influence of the presence of the competitive chloride ions.
Supplementary Materials
The following are available online at https://www.mdpi.com/2624-8549/2/4/60/s1; Figure S1–1H-NMR spectrum of [Ce(dppmO2)4]Cl3 in CD2Cl2; Figure S2–31P{1H} spectrum of Ce(dppmO2)4]Cl3 in CD2Cl2; Figure S3–Infrared spectrum of [Ce(dppmO2)4]Cl3 (Nujol mull); Figure S4–1H-NMR spectrum of [Pr(dppmO2)4]Cl3 in CD2Cl2; Figure S5–31P{1H}-NMR spectrum of [Pr(dppmO2)4]Cl3 in CD2Cl2; Figure S6–Infrared spectrum of [Pr(dppmO2)4]Cl3 (Nujol mull); Figure S7—1H-NMR spectrum of [Nd(dppmO2)4]Cl3 in CD2Cl2; Figure S8—31P{1H}-NMR spectrum of [Nd(dppmO2)4]Cl3 in CD2Cl2; Figure S9—Infrared spectrum of [Nd(dppmO2)4]Cl3 (Nujol mull); Figure S10—1H-NMR spectrum of [Sm(dppmO2)4]Cl3 in CD2Cl2; Figure S11—31P{1H}-NMR spectrum of [Sm(dppmO2)4]Cl3 in CD2Cl2; Figure S12—Infrared spectrum of [Sm(dppmO2)4]Cl3 (Nujol mull); Figure S13—1H-NMR spectrum of [Eu(dppmO2)4]Cl3 in CD2Cl2 ; Figure S14—31P{1H}-NMR spectrum of [Eu(dppmO2)4]Cl3 in CD2Cl2; Figure S15—Infrared spectrum of [Eu(dppmO2)4]Cl3 (Nujol mull); Figure S16—Infrared spectrum of [Gd(dppmO2)4]Cl3 (Nujol mull); Figure S17—1H-NMR spectrum of [SmCl(dppmO2)3]Cl2 in CD2Cl2 (* = EtOH); Figure S18—31P{1H}-NMR spectrum of [SmCl(dppmO2)3]Cl2 in CD2Cl2; Figure S19—Infrared spectrum of [SmCl(dppmO2)3]Cl2 (Nujol mull); Figure S20—1H-NMR spectrum of [EuCl(dppmO2)3]Cl2 in CDCl3; Figure S21—31P{1H}-NMR spectrum of [EuCl(dppmO2)3]Cl2 in CDCl3; Figure S22—31P{1H}-NMR spectrum of [EuCl(dppmO2)3]Cl2 + excess dppmO2 in CDCl3; Figure S23—Infrared spectrum of [EuCl(dppmO2)3]Cl2 (Nujol mull); Figure S24—Infrared spectrum of [GdCl(dppmO2)3]Cl2 (Nujol mull); Figure S25—1H-NMR spectrum of [TbCl(dppmO2)3]Cl2 in CD2Cl2 (* = EtOH); Figure S26—31P{1H}-NMR spectrum of [TbCl(dppmO2)3]Cl2 in CD2Cl2; Figure S27—Infrared spectrum of [TbCl(dppmO2)3]Cl2 (Nujol mull); Figure S28—1H-NMR spectrum of [DyCl(dppmO2)3]Cl2 in CD2Cl2; Figure S29—31P{1H}-NMR spectrum of [DyCl(dppmO2)3]Cl2 in CD2Cl2; Figure S30—Infrared spectrum of [DyCl(dppmO2)3]Cl2 (Nujol mull); Figure S31—1H-NMR spectrum of [HoCl(dppmO2)3]Cl2 in CD2Cl2 (* = EtOH); Figure S32—31P{1H}-NMR spectrum of [HoCl(dppmO2)3]Cl2 in CD2Cl2; Figure S33—Infrared spectrum of [HoCl(dppmO2)3]Cl2 (Nujol mull); Figure S34—1H-NMR spectrum of [ErCl(dppmO2)3]Cl2 in CD2Cl2 (* = EtOH); Figure S35—31P{1H}-NMR spectrum of [ErCl(dppmO2)3]Cl2 in CD2Cl2; Figure S36—Infrared spectrum of [ErCl(dppmO2)3]Cl2 (Nujol mull); Figure S37—1H-NMR spectrum of [TmCl(dppmO2)3]Cl2 in CD2Cl2 (* = EtOH); Figure S38—31P{1H}-NMR spectrum of [TmCl(dppmO2)3]Cl2 in CD2Cl2; Figure S39—Infrared spectrum of [TmCl(dppmO2)3]Cl2 (Nujol mull); Figure S40—1H-NMR spectrum of [YbCl(dppmO2)3]Cl2 in CD2Cl2 (* = EtOH); Figure S41—31P{1H}-NMR spectrum of [YbCl(dppmO2)3]Cl2 in CD2Cl2; Figure S42—Infrared spectrum of [YbCl(dppmO2)3]Cl2 (Nujol mull); Figure S43—The cation in [DyCl(dppmO2)3]Cl2. The chloride anions and solvate molecules are omitted.
Author Contributions
Conceptualization, R.D.B., W.L. and G.R.; formal analysis, R.D.B.; investigation, R.D.B.; data curation, R.D.B.; writing—original draft preparation, R.D.B. and W.L.; writing—review and editing, R.D.B., W.L. and G.R.; supervision, W.L. and G.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cotton, S.A. Lanthanide and Actinide Chemistry; John Wiley: Chichester, UK, 2006. [Google Scholar]
- Cotton, S.A.; Raithby, P.R. Systematics and surprises in lanthanide coordination chemistry. Coord. Chem. Rev. 2017, 340, 220–231. [Google Scholar] [CrossRef]
- Platt, A.W.G. Lanthanide phosphine oxide complexes. Coord. Chem. Rev. 2017, 340, 62–78. [Google Scholar] [CrossRef]
- Lees, A.M.J.; Platt, A.W.G. Complexes of lanthanide nitrates with bis(diphenylphosphino)methane dioxide. Inorg. Chem. 2003, 42, 4673–4679. [Google Scholar] [CrossRef] [PubMed]
- Levason, W.; Newman, E.H.; Webster, M. Tetrakis(triphenylphosphine oxide) complexes of the lanthanide nitrates; synthesis, characterization and crystal structures of [La(Ph3PO)4(NO3)3] and [Lu(Ph3PO)4(NO3)2]NO3. Polyhedron 2000, 19, 2697–2705. [Google Scholar] [CrossRef]
- Hill, N.J.; Leung, L.-S.; Levason, W.; Webster, M. Homoleptic octahedral hexakis(trimethylphosphine oxide)lanthanide hexafluorophosphates, [Ln(Me3PO)6][PF6]3; synthesis, structures and properties. Inorg. Chim. Acta 2003, 343, 169–174. [Google Scholar] [CrossRef]
- Glazier, M.J.; Levason, W.; Matthews, M.L.; Thornton, P.L.; Webster, M. Synthesis, properties and solution speciation of lanthanide chloride complexes of triphenylphosphine oxide. Inorg. Chim. Acta 2004, 357, 1083–1091. [Google Scholar] [CrossRef]
- Bannister, R.D.; Levason, W.; Reid, G. Diphosphine dioxide complexes of lanthanum and lutetium—The effects of ligand architecture and counter-anion. Polyhedron 2017, 133, 264–269. [Google Scholar] [CrossRef]
- Jin, Q.-H.; Wu, J.-Q.; Zhang, Y.-Y.; Zhang, C.-L. Crystal structure of tetra(bis(diphenylphosphino)methane dioxide-O,O]dysprosium(III) tri(trifluoromethanesulfonate)-N,N-dimethylformamide (1:2) [Dy(C25H22P2O2)4][CF3SO3]3 ·2C3H7NO. Z. Krystallogr. NCS 2009, 224, 428–432. [Google Scholar] [CrossRef]
- Huang, L.; Ma, B.-Q.; Huang, C.-H.; Makk, T.C.W.; Yao, G.-Q.; Xu, G.-X. Synthesis, characterisayion and crystal structure of a complex of europium perchlorate with methylenebis(diphenylphosphine oxide). J. Coord. Chem. 2001, 54, 95–103. [Google Scholar] [CrossRef]
- Fawcett, J.; Platt, A.W.G. Structures and catalytic properties of complexes of bis(diphenylphosphino)methane dioxide with scandium and lanthanide trifluoromethanesulfonates. Polyhedron 2003, 22, 967–973. [Google Scholar] [CrossRef]
- Levason, W.; Patel, R.; Reid, G. Catalytic air oxidation of tertiary phosphines in the presence of tin(IV) iodide. J. Organomet. Chem. 2003, 688, 280–282. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallog. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELX. Acta Crystallog. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Grachova, E.V.; Linti, G.; Vologzhanina, A.V. Personal communication CCDC 860683: Experimental Crystal Structure Determination; The Cambridge Crystallographic Data Centre (CCDC): Cambridge, UK, 2016. [Google Scholar] [CrossRef]
- Hill, N.J.; Leung, L.-S.; Levason, W.; Webster, M. Tetraaquatetrakis(trimethylphosphineoxide-κ-O)cerium(III) trichloride trihydrate. Acta Crystallog. Sect. C 2002, 58, m295. [Google Scholar] [CrossRef] [PubMed]
- Grachova, E.V.; Linti, G.; Vologzhanina, A.V. Personal communication CCDC 860682: Experimental Crystal Structure Determination; The Cambridge Crystallographic Data Centre (CCDC): Cambridge, UK, 2016. [Google Scholar] [CrossRef]
- Hill, N.J.; Levason, W.; Popham, M.C.; Reid, G.; Webster, M. Yttrium halide complexes of phosphine- and arsine-oxides: Synthesis, multinuclear NMR and structural studies. Polyhedron 2002, 21, 445–455. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).