Exploring the Optoelectronic Properties and Solar Cell Performance of Cs2SnI6−xBrx Lead-Free Double Perovskites: Combined DFT and SCAPS Simulation

 , , ,
, , ,  , and
, and
Abstract
1. Introduction
2. Details of Calculations
2.1. Material Study
2.2. SCAPS–1D
3. Results and Discussions
3.1. Material Properties
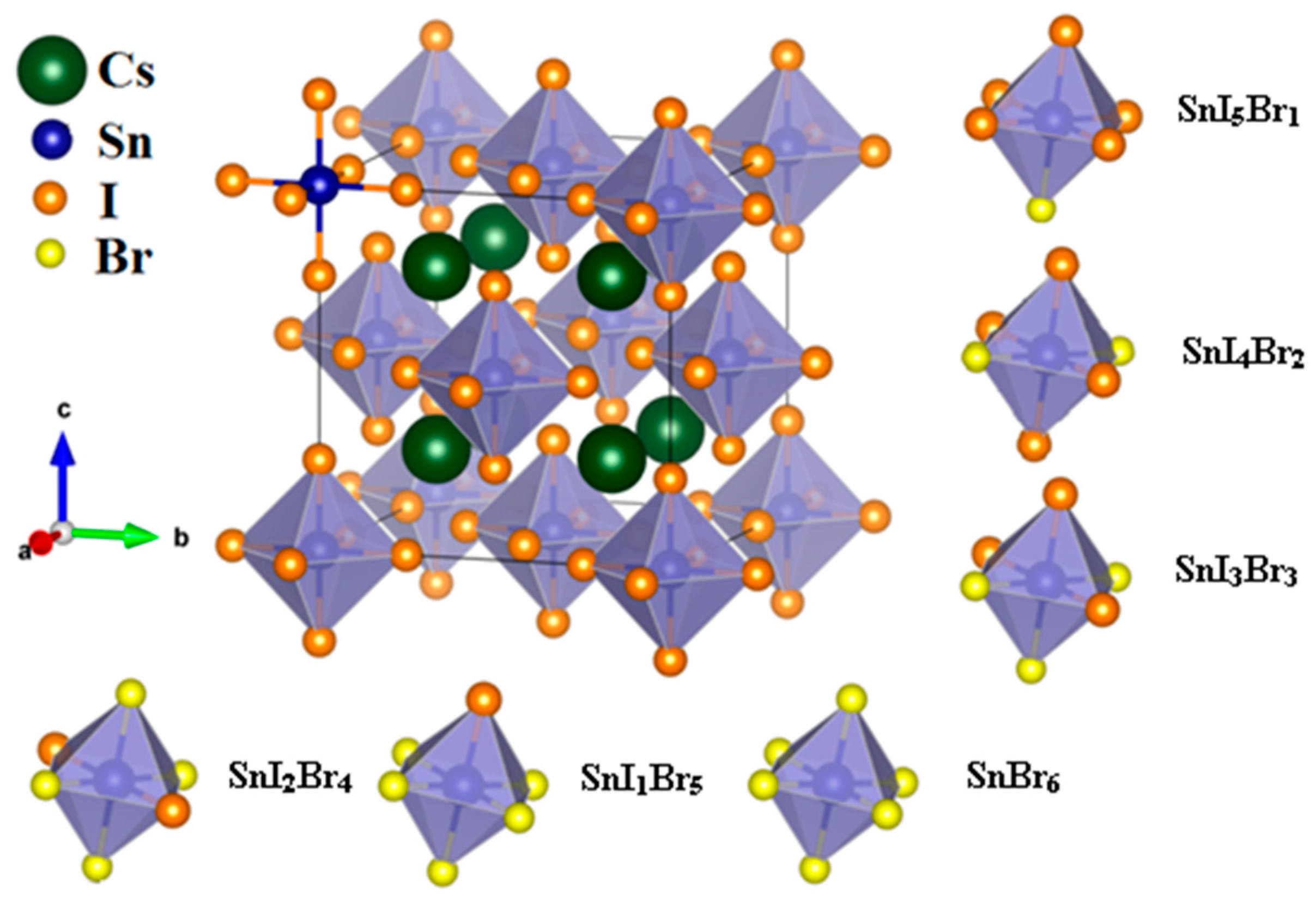
3.1.1. Structural Properties
3.1.2. Electronic Properties
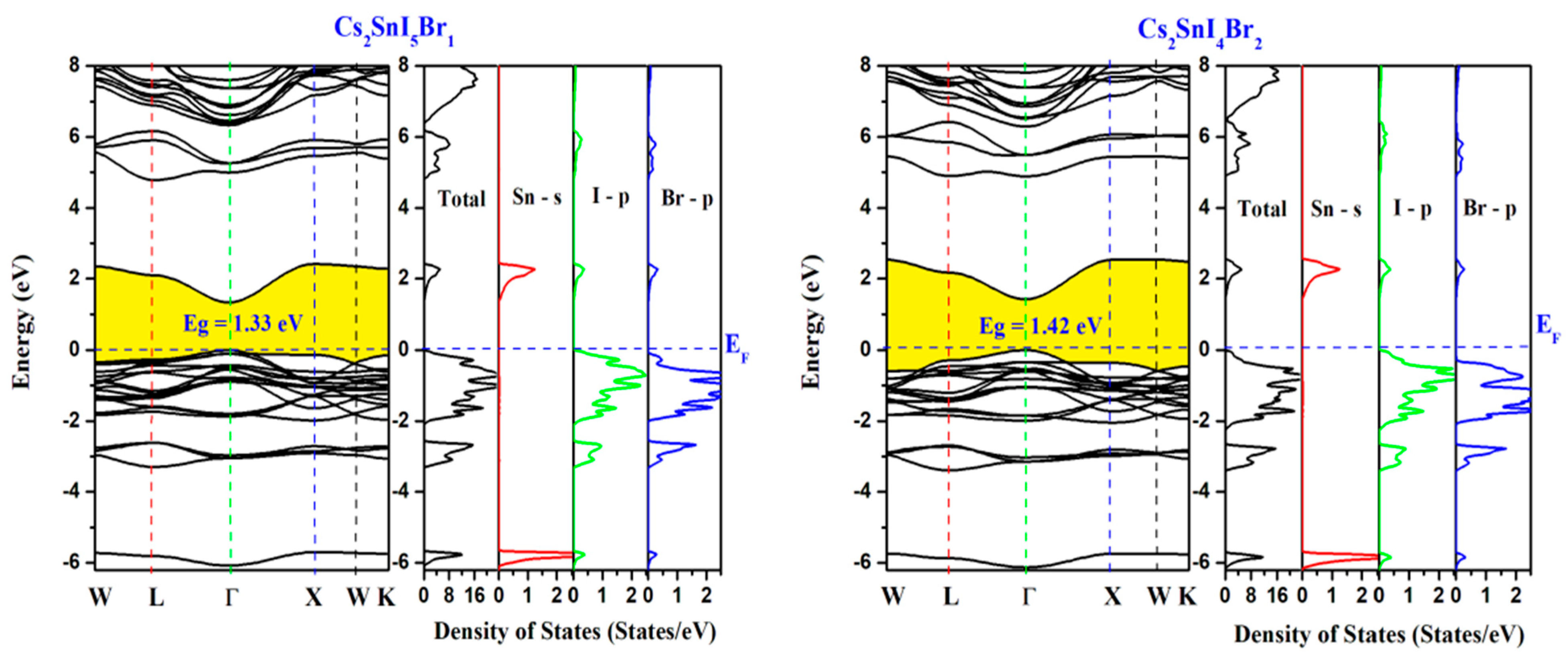
Electronic Band Structure and Densities of States
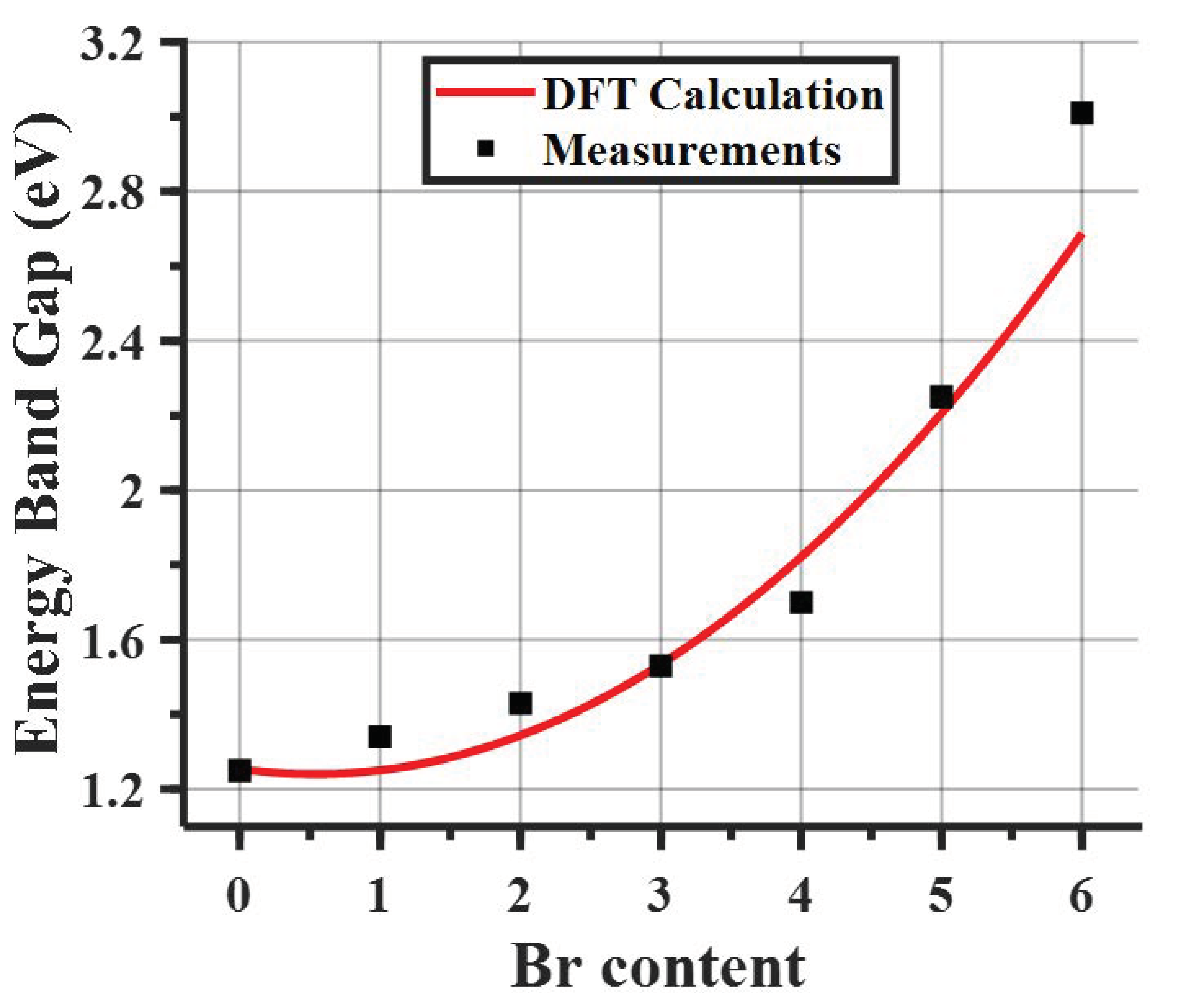
Effective Masses
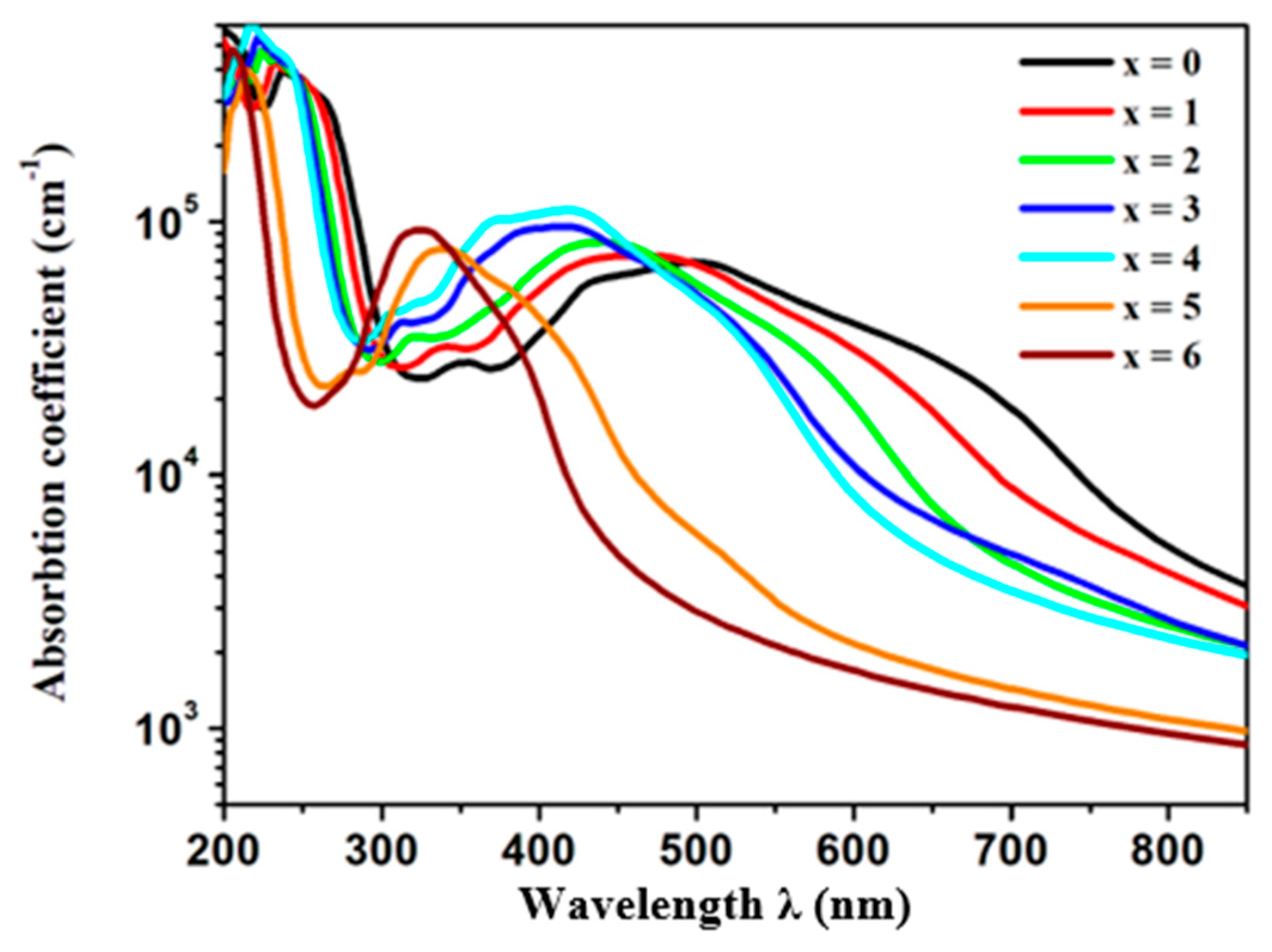
3.1.3. Optical Properties
3.2. Solar Cell Application
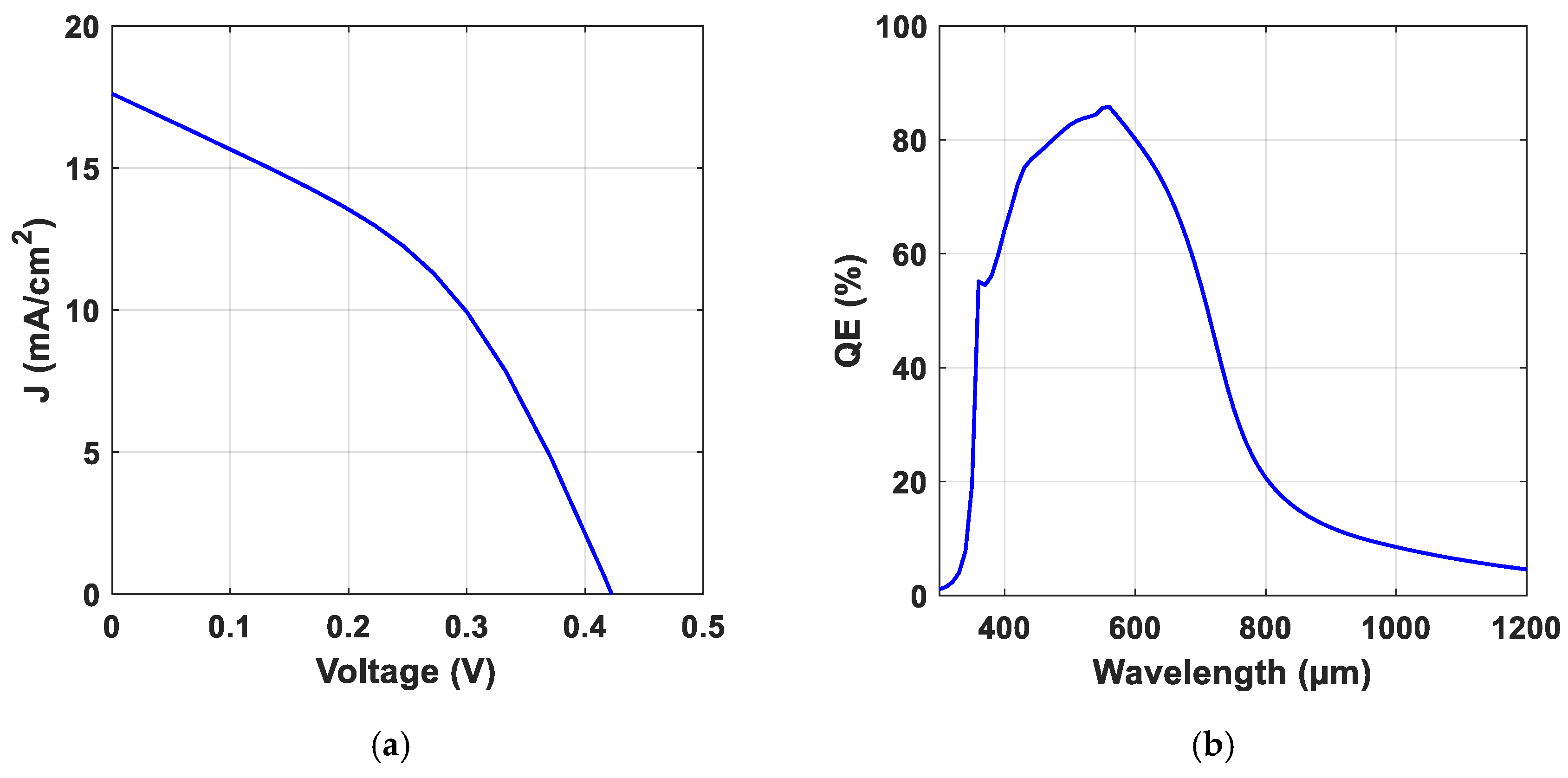
3.2.1. Structure and Calibration
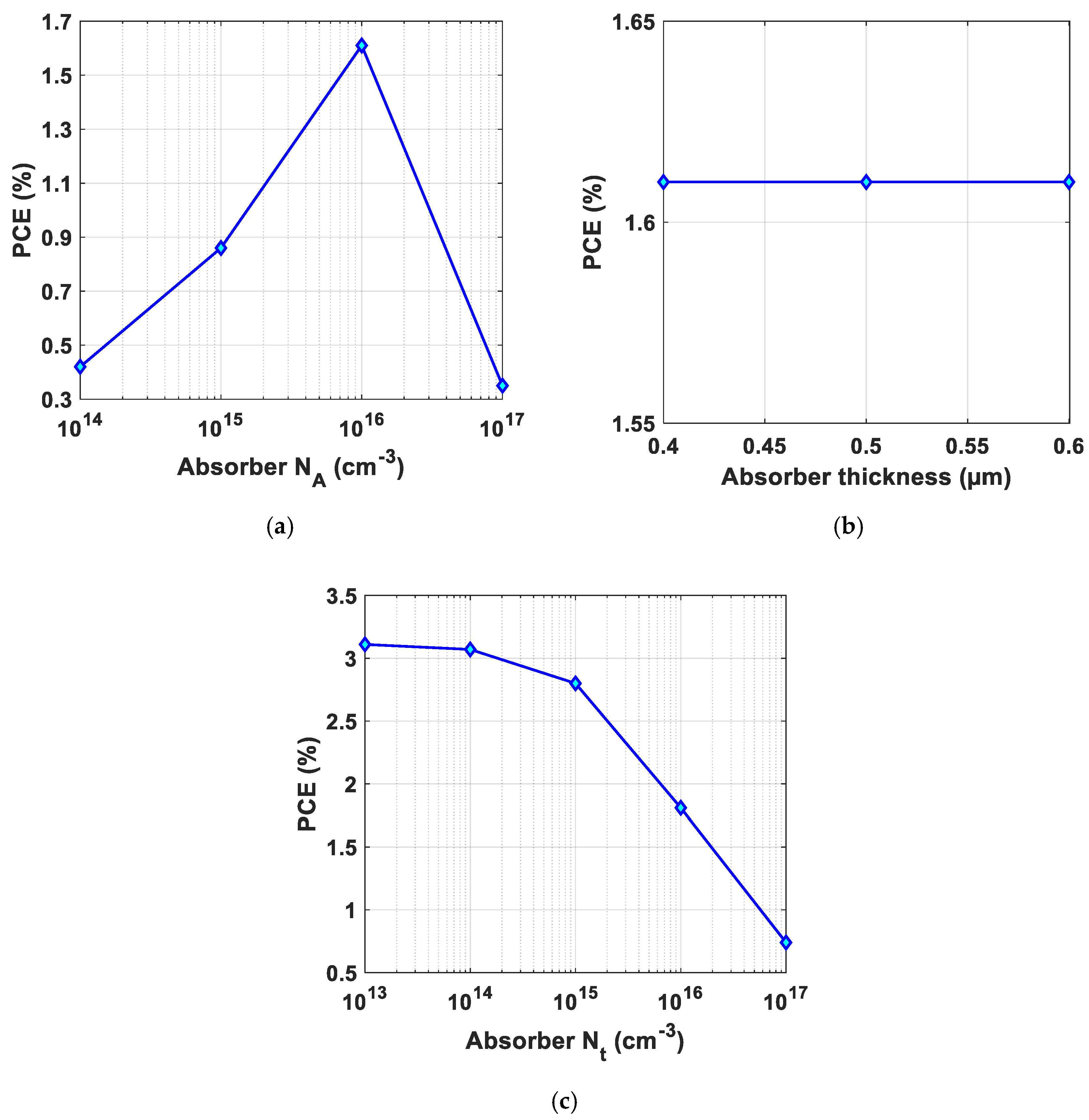
3.2.2. Solar Cell Optimization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| AM1.5G | Atmospheric Mass 1.5 Global (solar spectrum) |
| CB | conduction band |
| CF | capacitance–frequency |
| CV | capacitance–voltage |
| DFT | density functional theory |
| ETL | electron transport layer |
| Expt. | Experiment |
| FP–LAPW | full potential linearized augmented plane wave |
| FTO | fluoride-doped tin oxide |
| FF | fill factor |
| GGA–WC | Wu–Cohen generalized gradients approximation |
| HTL | hole transport layer |
| IB | intermediate band |
| IBZ | irreducible Brillouin zone |
| JV | current density-voltage |
| MT | muffinn-tin |
| mBJ | modified Becke–Johnson |
| OC | open circuit |
| PCE | power conversion efficiency |
| PSC | perovskite solar cell |
| PV | photovoltaic |
| QE | quantum efficiency |
| SC | solar cell |
| SCAPS | solar cell capacitance simulator |
| TB–mBJ | Tran and Blaha modified Becke–Johnson |
| TCO | transparent conducting oxide |
| UV | ultraviolet |
| VB | valence band |
| WD | Wemple–DiDomenico |
| 1D | one-dimensional |
References
- Shaheen, M.A.M.; Hasanien, H.M.; Turky, R.A.; Ćalasan, M.; Zobaa, A.F.; Aleem, S.H.E.A. OPF of modern power systems comprising renewable energy sources using improved chgs optimization algorithm. Energies 2021, 14, 6962. [Google Scholar] [CrossRef]
- Kojima, A.; Teshima, K.; Shirai, Y.; Miyasaka, T. Organometal halide perovskites as visible-light sensitizers for photovoltaic cells. J. Am. Chem. Soc. 2009, 131, 6050–6051. [Google Scholar] [CrossRef]
- Chung, I.; Song, J.-H.; Im, J.; Androulakis, J.; Malliakas, C.D.; Li, H.; Freeman, A.J.; Kenney, J.T.; Kanatzidis, M.G. CsSnI3: Semiconductor or metal? High electrical conductivity and strong near-infrared photoluminescence from a single material. high hole mobility and phase-transitions. ChemInform, 2012; 43, in print. [Google Scholar] [CrossRef]
- Gao, P.; Grätzel, M.; Nazeeruddin, M.K. Organohalide lead perovskites for photovoltaic applications. Energy Environ. Sci. 2014, 7, 2448–2463. [Google Scholar] [CrossRef]
- Colella, S.; Mosconi, E.; Fedeli, P.; Listorti, A.; Rizzo, A.; Gazza, F.; Orlandi, F.; Ferro, P.; Besagni, T.; Calestani, G.; et al. MAPbI3−xClx mixed halide perovskite for hybrid solar cells: The role of chloride as dopant on the transport and structural properties. MRS Proc. 2014, 1667, mrss14-1667-b03-02. [Google Scholar] [CrossRef]
- Eperon, G.E.; Paterno, G.M.; Sutton, R.J.; Zampetti, A.; Haghighirad, A.A.; Cacialli, F.; Snaith, H.J. Inorganic caesium lead iodide perovskite solar cells. J. Mater. Chem. A 2015, 3, 19688–19695. [Google Scholar] [CrossRef]
- NREL Photovaltic Research. Best Research-Cell Efficiency Chart. Available online: https://www.nrel.gov/pv/cell-efficiency.html (accessed on 25 November 2024).
- Liu, G.; Kong, L.; Gong, J.; Yang, W.; Mao, H.; Hu, Q.; Liu, Z.; Schaller, R.D.; Zhang, D.; Xu, T. Pressure-induced bandgap optimization in lead-based perovskites with prolonged carrier lifetime and ambient retainability. Adv. Funct. Mater. 2016, 27, 1604208. [Google Scholar] [CrossRef]
- Liu, H.; Siron, M.; Gao, M.; Lu, D.; Bekenstein, Y.; Zhang, D.; Dou, L.; Alivisatos, A.P.; Yang, P. Lead halide perovskite nanowires stabilized by block copolymers for Langmuir-Blodgett assembly. Nano Res. 2020, 13, 1453–1458. [Google Scholar] [CrossRef]
- Faheem, M.B.; Khan, B.; Feng, C.; Farooq, M.U.; Raziq, F.; Xiao, Y.; Li, Y. All-inorganic perovskite solar cells: Energetics, key challenges, and strategies toward commercialization. ACS Energy Lett. 2019, 5, 290–320. [Google Scholar] [CrossRef]
- Ju, M.-G.; Chen, M.; Zhou, Y.; Dai, J.; Ma, L.; Padture, N.P.; Zeng, X.C. Toward eco-friendly and stable perovskite materials for photovoltaics. Joule 2018, 2, 1231–1241. [Google Scholar] [CrossRef]
- Park, Y.; Jana, A.; Myung, C.W.; Yoon, T.; Lee, G.; Kocher, C.C.; Ying, G.; Osokin, V.; Taylor, R.A.; Kim, K.S. Enhanced photoluminescence quantum yield of MAPbBr3 nanocrystals by passivation using graphene. Nano Res. 2020, 13, 932–938. [Google Scholar] [CrossRef]
- Kaltzoglou, A.; Antoniadou, M.; Kontos, A.G.; Stoumpos, C.C.; Perganti, D.; Siranidi, E.; Raptis, V.; Trohidou, K.N.; Psycharis, V.; Kanatzidis, M.G.; et al. Optical-vibrational properties of the Cs2SnX6 (X = Cl, Br, I) defect perovskites and hole-transport efficiency in dye-sensitized solar cells. J. Phys. Chem. C 2016, 120, 11777–11785. [Google Scholar] [CrossRef]
- Wang, N.; Zhou, Y.; Ju, M.; Garces, H.F.; Ding, T.; Pang, S.; Zeng, X.C.; Padture, N.P.; Sun, X.W. Heterojunction-depleted lead-free perovskite solar cells with coarse-grained B-γ-CsSnI3 thin films. Adv. Energy Mater. 2016, 6, 1601130. [Google Scholar] [CrossRef]
- Hao, F.; Stoumpos, C.C.; Cao, D.H.; Chang, R.P.H.; Kanatzidis, M.G. Lead-free solid-state organic–inorganic halide perovskite solar cells. Nat. Photonics 2014, 8, 489–494. [Google Scholar] [CrossRef]
- Mastryukov, M.V.; Son, A.G.; Tekshina, E.V.; Vaimugin, L.A.; Kozyukhin, S.A.; Egorysheva, A.V.; Brekhovskikh, M.N. Effect of the purity of the precursor SnI2 on the optical properties of CsSnI3 perovskite thin films. Russ. J. Inorg. Chem. 2022, 67, 1652–1657. [Google Scholar] [CrossRef]
- Ran, C.; Gao, W.; Li, J.; Xi, J.; Li, L.; Dai, J.; Yang, Y.; Gao, X.; Dong, H.; Jiao, B.; et al. Conjugated organic cations enable efficient self-healing FASnI3 solar cells. Joule 2019, 3, 3072–3087. [Google Scholar] [CrossRef]
- Maughan, A.E.; Ganose, A.M.; Bordelon, M.M.; Miller, E.M.; Scanlon, D.O.; Neilson, J.R. Defect tolerance to intolerance in the vacancy-ordered double perovskite semiconductors Cs2SnI6 and Cs2TeI6. J. Am. Chem. Soc. 2016, 138, 8453–8464. [Google Scholar] [CrossRef] [PubMed]
- Combs, V.E.; Oswald, I.W.H.; Neilson, J.R. Hydrothermal crystal growth of mixed valence Cs2SbBr6. Cryst. Growth Des. 2019, 19, 4090–4094. [Google Scholar] [CrossRef]
- Ganesan, R.; Vinodhini, S.P.; Balasubramani, V.; Parthipan, G.; Sridhar, T.M.; Arulmozhi, R.; Muralidharan, R. Tuning the band gap of hybrid lead free defect perovskite nano crystals for solar cell applications. New J. Chem. 2019, 43, 15258–15266. [Google Scholar] [CrossRef]
- Liu, Y.; Gao, W.; Ran, C.; Dong, H.; Sun, N.; Ran, X.; Xia, Y.; Song, L.; Chen, Y.; Huang, W. All-inorganic Sn-based perovskite solar cells: Status, challenges, and perspectives. ChemSusChem 2020, 13, 6477–6497. [Google Scholar] [CrossRef]
- Rahim, W.; Cheng, A.; Lyu, C.; Shi, T.; Wang, Z.; Scanlon, D.O.; Palgrave, R.G. Geometric analysis and formability of the cubic A2BX6 vacancy-ordered double perovskite structure. Chem. Mater. 2020, 32, 9573–9583. [Google Scholar] [CrossRef]
- Faizan, M.; Khan, S.H.; Khachai, H.; Seddik, T.; Bin Omran, S.; Khenata, R.; Xie, J.; Al-Anazy, M.M. Electronic, optical, and thermoelectric properties of perovskite variants A2BX 6: Insight and design via first-principles calculations. Int. J. Energy Res. 2020, 45, 4495–4507. [Google Scholar] [CrossRef]
- Euvrard, J.; Wang, X.; Li, T.; Yan, Y.; Mitzi, D.B. Is Cs2TiBr6 a promising Pb-free perovskite for solar energy applications? J. Mater. Chem. A 2020, 8, 4049–4054. [Google Scholar] [CrossRef]
- Jong, U.-G.; Yu, C.-J.; Kye, Y.-H. Computational prediction of structural, electronic, and optical properties and phase stability of double perovskites K2SnX6 (X = I, Br, Cl). RSC Adv. 2019, 10, 201–209. [Google Scholar] [CrossRef]
- Bousahla, M.A.; Faizan, M.; Seddik, T.; Bin Omran, S.; Khachai, H.; Laref, A.; Khenata, R.; Znaidia, S.; Boukhris, I.; Khan, S.H. DFT study on the crystal structure, optoelectronic, and thermoelectric properties of lead-free inorganic A2PdBr6 (A = K, Rb, and Cs) perovskites. Mater. Today Commun. 2021, 30, 103061. [Google Scholar] [CrossRef]
- Cheghib, N.; Maizi, R.; Boudjahem, A.-G.; Ksouri, R.; Derdare, M. Theoretical study of the elastic, electronic, and thermodynamic properties of the Cs2MF6 (M = Cr and Mo) perovskites. Russ. J. Inorg. Chem. 2024; in print. [Google Scholar] [CrossRef]
- Ksouri, R.; Maizi, R.; Boudjahem, A.-G.; Cheghib, N.; Djaghout, I.; Derdare, M. DFT Study of structural, elastic, electronic, and thermodynamic properties of compounds Cs2TiCl6 and Cs2TiBr6. Russ. J. Inorg. Chem. 2024; in print. [Google Scholar] [CrossRef]
- Saparov, B.; Sun, J.-P.; Meng, W.; Xiao, Z.; Duan, H.-S.; Gunawan, O.; Shin, D.; Hill, I.G.; Yan, Y.; Mitzi, D.B. Thin-film deposition and characterization of a Sn-deficient perovskite derivative Cs2SnI6. Chem. Mater. 2016, 28, 2315–2322. [Google Scholar] [CrossRef]
- Barker, A.J.; Sadhanala, A.; Deschler, F.; Gandini, M.; Senanayak, S.P.; Pearce, P.M.; Mosconi, E.; Pearson, A.J.; Wu, Y.; Kandada, A.R.S.; et al. Defect-assisted photoinduced halide segregation in mixed-halide perovskite thin films. ACS Energy Lett. 2017, 2, 1416–1424. [Google Scholar] [CrossRef]
- Rezini, B.; Seddik, T.; Mouacher, R.; Vu, T.V.; Batouche, M.; Khyzhun, O.Y. Strain effects on electronic, optical properties and carriers mobility of Cs2SnI6 vacancy-ordered double perovskite: A promising photovoltaic material. Int. J. Quant. Chem. 2022, 122, e26977. [Google Scholar] [CrossRef]
- Yuan, G.; Huang, S.; Niu, J.; Qin, S.; Wu, X.; Ding, H.; Lu, A. Compressibility of Cs2SnBr6 by X-ray diffraction and Raman spectroscopy. Solid State Commun. 2018, 275, 68–72. [Google Scholar] [CrossRef]
- Tan, Z.; Li, J.; Zhang, C.; Li, Z.; Hu, Q.; Xiao, Z.; Kamiya, T.; Hosono, H.; Niu, G.; Lifshitz, E.; et al. Highly efficient blue-emitting bi-doped Cs2SnCl6 perovskite variant: Photoluminescence induced by impurity doping. Adv. Funct. Mater. 2018, 28, 1801131. [Google Scholar] [CrossRef]
- Qiu, X.; Cao, B.; Yuan, S.; Chen, X.; Qiu, Z.; Jiang, Y.; Ye, Q.; Wang, H.; Zeng, H.; Liu, J.; et al. From unstable CsSnI3 to air-stable Cs2SnI6: A lead-free perovskite solar cell light absorber with bandgap of 1.48 eV and high absorption coefficient. Sol. Energy Mater. Sol. Cells 2017, 159, 227–234. [Google Scholar] [CrossRef]
- Lee, B.; Stoumpos, C.C.; Zhou, N.; Hao, F.; Malliakas, C.; Yeh, C.-Y.; Marks, T.J.; Kanatzidis, M.G.; Chang, R.P.H. Air-stable molecular semiconducting iodosalts for solar cell applications: Cs2SnI6 as a hole conductor. J. Am. Chem. Soc. 2014, 136, 15379–15385. [Google Scholar] [CrossRef]
- Kapil, G.; Ohta, T.; Koyanagi, T.; Vigneshwaran, M.; Zhang, Y.; Ogomi, Y.; Pandey, S.S.; Yoshino, K.; Shen, Q.; Toyoda, T.; et al. Investigation of interfacial charge transfer in solution processed Cs2SnI6 thin films. J. Phys. Chem. C 2017, 121, 13092–13100. [Google Scholar] [CrossRef]
- Jung, H.S.; Park, N. Perovskite solar cells: From materials to devices. Small 2014, 11, 10–25. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Krenselewski, A.; Baik, S.I.; Seidman, D.N.; Chang, R.P.H. Solution processing of air-stable molecular semiconducting iodosalts, Cs2SnI6−xBrx, for potential solar cell applications. Sustain. Energy Fuels 2017, 1, 710–724. [Google Scholar] [CrossRef]
- Yuan, G.; Huang, S.; Qin, S.; Wu, X.; Ding, H.; Lu, A. Structural, optical, and thermal properties of Cs2SnI6–xBrx mixed perovskite solid solutions. Eur. J. Inorg. Chem. 2019, 2019, 2524–2529. [Google Scholar] [CrossRef]
- Han, X.; Liang, J.; Yang, J.; Soni, K.; Fang, Q.; Wang, W.; Zhang, J.; Jia, S.; Martí, A.A.; Zhao, Y.; et al. Lead-free double perovskite Cs2SnX6: Facile solution synthesis and excellent stability. Small 2019, 15, 1901650. [Google Scholar] [CrossRef] [PubMed]
- Qamar, S.; Sultan, M.; Akhter, Z.; Ela, S.E. A facile one-step solution synthesis of Cs2SnI6−xBrx using less-toxic methanol solvent for application in dye-sensitized solar cells. Int. J. Energy Res. 2022, 46, 13441–13452. [Google Scholar] [CrossRef]
- Nazir, S.; Noor, N.; Khan, M.A.; Mumtaz, S.; Elansary, H.O. DFT study of optoelectronic and thermoelectric properties of pure and doped double perovskite Cs2SnI6 for solar cell applications. Chem. Phys. Lett. 2024, 852, 141519. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, C.; Liao, Y.; Li, S.; Yang, P.; Xi, Y.; Cai, C.; Liu, W. Investigating optical adsorption properties of lead-free double perovskite semiconductors Cs2SnI6−xBrx (x = 0–6) via first principles calculation. Microw. Opt. Technol. Lett. 2021, 65, 1017–1023. [Google Scholar] [CrossRef]
- Guo, S.; Zhang, Q.; Sa, R.; Liu, D. Exploring the stability and physical properties of double perovskite Cs2SnI6 by doping the Cl/Br atom: A DFT study. Comput. Theor. Chem. 2022, 1209, 113609. [Google Scholar] [CrossRef]
- Madsen, G.K.H.; Blaha, P.; Schwarz, K.; Sjöstedt, E.; Nordström, L. Efficient linearization of the augmented plane-wave method. Phys. Rev. B 2001, 64, 195134. [Google Scholar] [CrossRef]
- Blaha, P.; Schwarz, K.; Madsen, G.; Kvasnicka, D.; Luitz, J. WIEN2k: An Augmented Plane Wave Plus Local Orbitals Program for Calculating Crystal Properties. User’s Guide, WIEN2k 14.2; Vienna University of Technology, Institute of Physical and Theoretical Chemistry: Vienna, Austria, 2014; Available online: https://indico.cells.es/event/53/attachments/541/841/WIEN2k_usersguide.pdf (accessed on 25 November 2024).
- Tran, F.; Blaha, P. Accurate band gaps of semiconductors and insulators with a semilocal exchange-correlation potential. Phys. Rev. Lett. 2009, 102, 226401. [Google Scholar] [CrossRef]
- Ketelaar, J.A.A.; Rietdijk, A.A.; van Staveren, C.H. Die Kristallstruktur von Ammonium-, Kalium-, Rubidium- und Cäsiumstannibromid. Recl. Trav. Chim. Pays-Bas 1937, 56, 907–908. [Google Scholar] [CrossRef]
- Stoumpos, C.C.; Malliakas, C.D.; Kanatzidis, M.G. Semiconducting tin and lead iodide perovskites with organic cations: Phase transitions, high mobilities, and near-infrared photoluminescent properties. Inorg. Chem. 2013, 52, 9019–9038. [Google Scholar] [CrossRef]
- Wang, G.; Wang, D.; Shi, X. Electronic structure and optical properties of Cs2AX2′X4 (A = Ge, Sn, Pb; X′, X = Cl, Br, I). AIP Adv. 2015, 5, 127224. [Google Scholar] [CrossRef]
- Yang, X.; Wang, Y.; Jiang, J.; Li, M.; Tang, Z.; Cai, H.; Zhang, F.; Wu, X. Composition effects on structure and optical properties in double perovskite derivatives semiconductors Cs2SnI6−xBrx (x = 0–6). APL Mater. 2020, 8, 021102. [Google Scholar] [CrossRef]
- Green, M.A. Multiple band and impurity photovoltaic solar cells: General theory and comparison to tandem cells. Prog. Photovolt. Res. Appl. 2001, 9, 137–144. [Google Scholar] [CrossRef]
- Huang, H.-M.; Jiang, Z.-Y.; Luo, S.-J. First-principles investigations on the mechanical, thermal, electronic, and optical properties of the defect perovskites Cs2SnX6 (X = Cl, Br, I). Chin. Phys. B 2017, 26, 096301. [Google Scholar] [CrossRef]
- Rasukkannu, M.; Velauthapillai, D.; Vajeeston, P. A first-principle study of the electronic, mechanical and optical properties of inorganic perovskite Cs2SnI6 for intermediate-band solar cells. Mater. Lett. 2018, 218, 233–236. [Google Scholar] [CrossRef]
- Cai, Y.; Xie, W.; Ding, H.; Chen, Y.; Thirumal, K.; Wong, L.H.; Mathews, N.; Mhaisalkar, S.G.; Sherburne, M.; Asta, M. Computational study of halide perovskite-derived A2BX6 inorganic compounds: Chemical trends in electronic structure and structural stability. Chem. Mater. 2017, 29, 7740–7749. [Google Scholar] [CrossRef]
- Ouerghui, W.; Alkhalifah, M.S. Density functional investigation of structural, electronic, optical and thermodynamic properties of Zn1−xBexO semiconductor. Appl. Phys. A 2019, 125, 374. [Google Scholar] [CrossRef]
- Ben Abdallah, H.; Ouerghui, W. Hybrid functional calculations of electro-optical properties of novel Ga1−xInxTe ternary chalcogenides. Appl. Phys. A 2020, 126, 1–12. [Google Scholar] [CrossRef]
- Ouerghui, W.; Alkhalifah, M.S.; Ben Abdallah, H. DFT calculations on ZnO1−x compounds for optoelectronic applications. J. Comput. Electron. 2021, 20, 467–479. [Google Scholar] [CrossRef]
- Ben Abdallah, H.; Ouerghui, W. Spin–orbit coupling effect on electronic, linear and nonlinear optical properties of Bi2S3 and the ternary bismuth sulfide Bi2S2.75Se0.25: Ab-initio calculations. Opt. Quant. Electron. 2021, 54, 20. [Google Scholar] [CrossRef]
- Ouerghui, W.; Gassoumi, M.; Beji, L.; Maaref, M. Optical properties of quaternary GaMnAsP thin layer grown by molecular beam epitaxy. Physica E Low-dim.l Syst. Nanostr. 2021, 131, 114733. [Google Scholar] [CrossRef]
- Basyoni, M.S.S.; Salah, M.M.; Mousa, M.; Shaker, A.; Zekry, A.; Abouelatta, M.; Alshammari, M.T.; Al-Dhlan, K.A.; Gontrand, C. On the investigation of interface defects of solar cells: Lead-based vs lead-free perovskite. IEEE Access 2021, 9, 130221–130232. [Google Scholar] [CrossRef]
- Srivastava, P.; Sadanand, S.; Rai, S.; Lohia, P.; Dwivedi, D.K.; Qasem, H.; Umar, A.; Akbar, S.; Algadi, H.; Baskoutas, S. Theoretical study of perovskite solar cell for enhancement of device performance using SCAPS-1D. Phys. Scr. 2022, 97, 125004. [Google Scholar] [CrossRef]
- Salem, M.S.; Shaker, A.; Almurayziq, T.S.; Alshammari, M.T. Prospective efficiency boosting of full-inorganic single-junction Sb2(S, Se)3 solar cell. Sol. Energy Mater. Sol. Cells 2022, 248, 112001. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhang, H.; Qiu, X.; Cao, B. The air and thermal stabilities of lead-free perovskite variant Cs2SnI6 powder. Mater. Lett. 2017, 199, 50–52. [Google Scholar] [CrossRef]
- Shockley, W.; Queisser, H.J. Detailed balance limit of efficiency of p-n junction solar cells. J. Appl. Phys. 1961, 32, 510–519. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alloys | Cs2SnI6 | Cs2SnI5Br1 | Cs2SnI4Br2 | Cs2SnI3Br3 | Cs2SnI2Br4 | Cs2SnI1Br5 | Cs2SnBr6 |
|---|---|---|---|---|---|---|---|
| This study | 11.590 | 11.462 | 11.322 | 11.187 | 11.029 | 10.869 | 10.837 |
| Expt. | 11.638 a | 11.528 e | 11.41 e | 11.274 e | 11.147 e | 10.961 e | 10.837 a |
| 11.652 b | - | - | - | - | - | - | |
| 11.627 c | - | - | - | - | - | 10.77 b | |
| 11.646 e | - | - | - | - | - | 10.859 e | |
| 11.67 f | - | - | - | - | - | 10.83 f | |
| Theory | 12.016 a | - | - | - | - | - | 11.218 a |
| 12.032 d | - | - | - | - | - | 11.243 d |
| Alloys | Eg | me* | mh* | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Present | Expt. | Theory | Present | Expt. | Theory | Present | Expt. | Theory | |
| Cs2SnI6 | 1.257 | 1.26 a, 1.24 c, 1.3 d, 1.25 e | 1.28 c, 0.36 f, 0.92 f, 0.883 b | 0.272 | 0.152 g, 0.306 g, 0.33 h | 1.851 | 1.4173 g 1.852 g, 1.5 h | ||
| Cs2SnI5Br1 | 1.33 | 1.34 e, 1.32 c, 1.375 d | 0.281 | 1.692 | |||||
| Cs2SnI4Br2 | 1.42 | 1.43 e, 1.34 c, 1.4 d | 0.285 | 0.967 | |||||
| Cs2SnI3Br3 | 1.52 | 1.53 e 1.46 c | 1.68 c | 0.292 | 1.108 | ||||
| Cs2SnI2Br4 | 1.69 | 1.7 e, 1.5 c, 1.63 d | 0.288 | 1.093 | |||||
| Cs2SnI1Br5 | 2.24 | 2.25 e, 1.75 c, 2.36 d | 0.361 | 2.213 | |||||
| Cs2SnBr6 | 2.69 | 2.7 b, 2.93 c, 2.85 d, 3.01 e | 2.9 c, 1.51 f, 2.36 f, 2.241 b | 0.39 | 2.574 | ||||
| Compound | R(0) (%) | ||||||
|---|---|---|---|---|---|---|---|
| Cs2SnI6 | 3.86 | 1.98 | 10.8 | 216 | 13.51 | 4.60 | 62.14 |
| Cs2SnI5Br | 3.85 | 1.96 | 10.6 | 221 | 13.86 | 4.80 | 66.52 |
| Cs2SnI4Br2 | 3.84 | 1.94 | 10.2 | 203 | 14.06 | 5.07 | 71.28 |
| Cs2SnI3Br3 | 3.77 | 1.85 | 9 | 221 | 14.3 | 5.82 | 83.22 |
| Cs2SnI2Br4 | 3.70 | 1.82 | 8.46 | 226 | 15.21 | 6.57 | 99.92 |
| Cs2SnI1Br5 | 3.24 | 1.77 | 7.77 | 402 | 15.88 | 7.40 | 117.51 |
| Cs2SnBr6 | 3.15 | 1.77 | 7.83 | 464 | 16.22 | 7.50 | 121.65 |
| FTO [61] | TiO2 [61] | Cs2SnI6 | P3HT [62] | |
|---|---|---|---|---|
| d (thickness, nm) | 500 | 30 | 650 | 100 |
| Eg (bandgap, eV) | 3.5 | 3.2 | 1.257 | 1.7 |
| χ (electron affinity, eV) | 4 | 4 | 4.01 | 3.5 |
| εr (Permittivity) | 9 | 9 | 7.2 | 3 |
| Nc (effective DOS at CB, 1/cm3) | 2.2 × 1018 | 2 × 1018 | 3.569 × 1018 | 2 × 1018 |
| Nv (effective DOS at VB, 1/cm3) | 1.8 × 1019 | 1.8 × 1019 | 1.21 × 1019 | 2 × 1019 |
| µn (electron mobility, cm2/V s) | 20 | 20 | 9 | 1.8 × 10−3 |
| µp (hole mobility, cm2/V s) | 10 | 10 | 9 | 1.8 × 10−2 |
| ND (doping density n-type, 1/cm3) | 2 × 1019 | 1 × 1017 | 0 | 0 |
| NA (doping density p-type, 1/cm3) | 0 | 0 | 1015 | 1019 |
| Nt (defect density, 1/cm3) | 1015 | 1015 | 1.68 × 1017 | 1015 |
| σn (capture cross-section electrons, cm2) | 10−15 | 2 × 10−14 | 2 × 10−14 | 10−15 |
| σp (capture cross-section holes, cm2) | 10−15 | 2 × 10−14 | 2 × 10−14 | 10−15 |
| Trap energy level (eV) | 0.6 | 0.6 | 0.6 | 0.8 |
| Energetic distribution | Single | Gaussian | Gaussian | Gaussian |
| HTL/Absorber Layer or Absorber Layer/ETL | |
|---|---|
| Type | Neutral |
| σn (cm2) | 10−15 |
| σp (cm2) | 10−15 |
| Distribution | Single |
| Energy level Et (eV) | 0.6 (above the highest Ev) |
| Total density (1/cm2) | 1016 |
| VOC (V) | JSC (mA/cm2) | FF (%) | PCE (%) | |
|---|---|---|---|---|
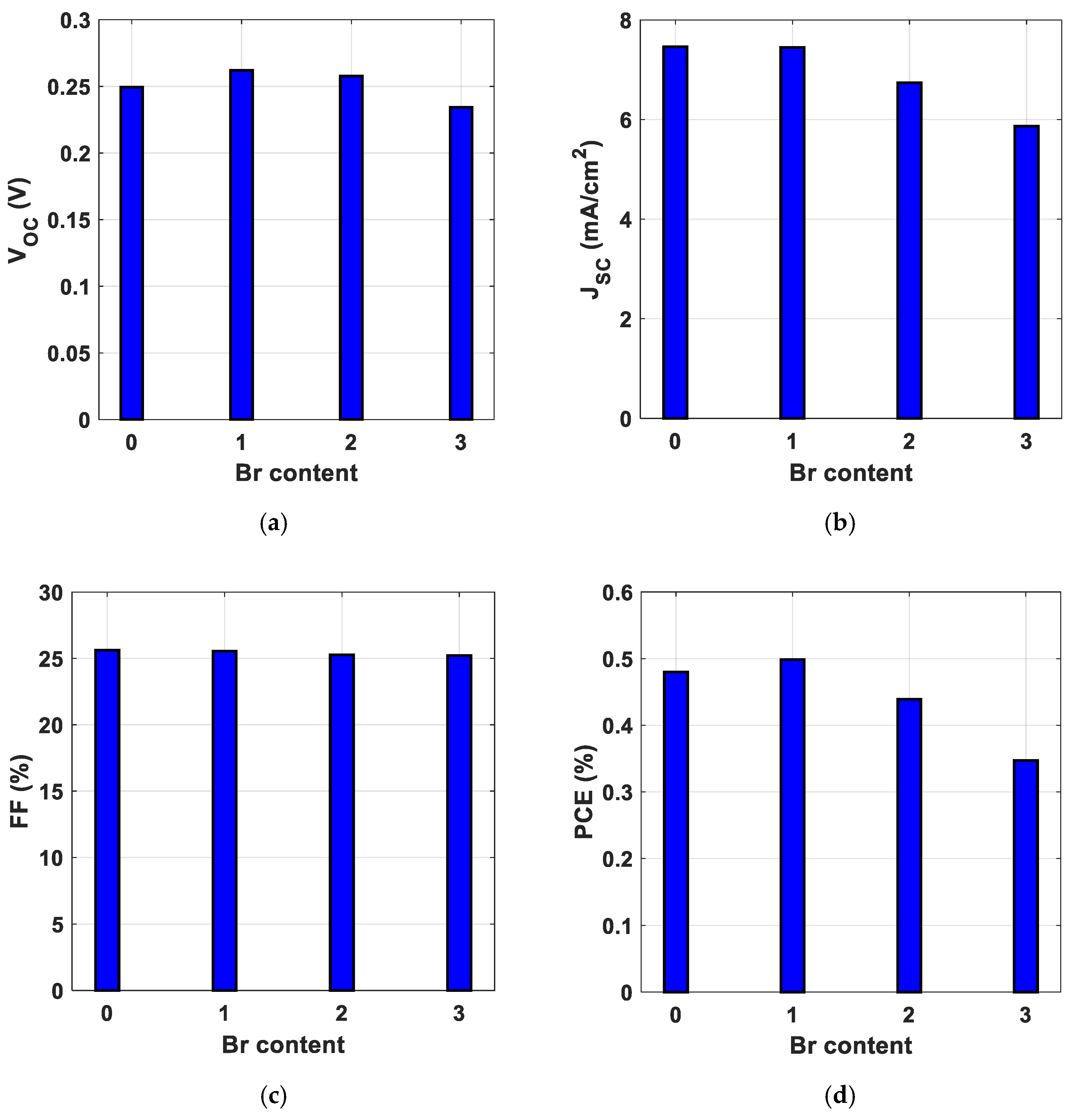
| Experiment [64] | 0.256 | 7.41 | 24.50 | 0.47 |
| Simulation | 0.249 | 7.46 | 25.63 | 0.48 |
| Cs2SnI5Br | Cs2SnI4Br2 | Cs2SnI3Br3 | |
|---|---|---|---|
| Eg (eV) | 1.339 | 1.428 | 1.528 |
| χ (eV) | 4.03 | 4.05 | 4.06 |
| εr (permittivity) | 7.13 | 7.06 | 7.05 |
| Nc (1/cm3) | 3.581 × 1018 | 3.652 × 1018 | 3.522 × 1018 |
| Nv (1/cm3) | 4.8 × 1019 | 2.08 × 1019 | 2.19 × 1019 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rezini, B.; Seddik, T.; Batouche, M.; Abdallah, H.B.; Ouerghui, W.; Salah, M.M.; Ahsan, M.; Shaker, A.; Al-Muhimeed, T.I.; Saeed, A.; et al. Exploring the Optoelectronic Properties and Solar Cell Performance of Cs2SnI6−xBrx Lead-Free Double Perovskites: Combined DFT and SCAPS Simulation. Physics 2025, 7, 3. https://doi.org/10.3390/physics7010003
Rezini B, Seddik T, Batouche M, Abdallah HB, Ouerghui W, Salah MM, Ahsan M, Shaker A, Al-Muhimeed TI, Saeed A, et al. Exploring the Optoelectronic Properties and Solar Cell Performance of Cs2SnI6−xBrx Lead-Free Double Perovskites: Combined DFT and SCAPS Simulation. Physics. 2025; 7(1):3. https://doi.org/10.3390/physics7010003
Chicago/Turabian StyleRezini, B., T. Seddik, M. Batouche, H. Ben Abdallah, W. Ouerghui, Mostafa M. Salah, Muhammad Ahsan, Ahmed Shaker, Tahani I. Al-Muhimeed, Ahmed Saeed, and et al. 2025. "Exploring the Optoelectronic Properties and Solar Cell Performance of Cs2SnI6−xBrx Lead-Free Double Perovskites: Combined DFT and SCAPS Simulation" Physics 7, no. 1: 3. https://doi.org/10.3390/physics7010003
APA StyleRezini, B., Seddik, T., Batouche, M., Abdallah, H. B., Ouerghui, W., Salah, M. M., Ahsan, M., Shaker, A., Al-Muhimeed, T. I., Saeed, A., & Mousa, M. (2025). Exploring the Optoelectronic Properties and Solar Cell Performance of Cs2SnI6−xBrx Lead-Free Double Perovskites: Combined DFT and SCAPS Simulation. Physics, 7(1), 3. https://doi.org/10.3390/physics7010003