Abstract
One of the challenges in Fischer–Tropsch synthesis (FTS) is the high reduction temperatures, which cause sintering and the formation of silicates. These lead to pore blockages and the coverage of active metals, particularly in conventional catalyst promotion. To address the challenge, this article investigates the effects of the preparation method, specifically the inverse promotion of SiO2-supported Co catalysts with manganese (Mn), and their reduction in H2 for FTS. The catalysts were prepared using stepwise incipient wetness impregnation of a cobalt nitrate precursor into a promoted silica support. The properties of the catalysts were characterized using XRD, XPS, TPR, and BET techniques. The structure–performance relationship of the inversely promoted catalysts in FTS was studied using a fixed-bed reactor to obtain the best performing catalysts for heavy hydrocarbons (C5+). XRD and XPS results indicated that Co3O4 is the dominant cobalt phase in oxidized catalysts. It was found that with increase in Mn loading, the reduction temperature increased in the following sequence 10%Co/SiO2 < 10%Co/0.25%Mn-SiO2 < 10%Co/0.5%Mn-SiO2 < 10%Co/3.0%Mn-SiO2. The catalyst with the lowest Mn loading, 10%Co/0.25%Mn-SiO2, exhibited higher C5+ selectivity, which can be attributed to less MSI and higher reducibility. This catalyst showed the lowest CH4 selectivity possibly due to lower H2 uptake and higher CO chemisorption.
1. Introduction
Cobalt (Co)-based catalysts are an excellent choice for applying the Fischer–Tropsch synthesis (FTS) method to produce clean chemicals and fuels due to cobalt’s high activity in converting syngas. These catalysts are effective for FTS because they offer high selectivity, sustainable fuel production, better catalytic activity, and improved reducibility and dispersion. Cobalt is one of the metals that converts synthesis gas the most actively [1,2,3,4,5,6,7]. Given that cobalt is a highly expensive metal, it is necessary to synthesize a Co catalyst that is evenly distributed over the support to reduce the catalyst’s cost. Since Co alone cannot provide the appropriate final composition, oxide supports such as silica and alumina have been utilized for Co based catalysts in low-temperature Fischer–Tropsch (LTFT) [8,9,10,11]. The metal–support interaction, crystallite size, metal dispersion, mass transfer of the reactants and products, mechanical strength, and thermal stability of the catalyst are all influenced by the physicochemical and textural characteristics of the support material. Therefore, the key to creating an effective catalyst is choosing and synthesizing the right support material with a unique physicochemical composition, well-defined surface chemistry, a wide surface area, and the right pore size and volume [12,13]. To achieve the required catalyst qualities, catalyst preparation methods (i.e., preparation procedures, including impregnation, drying, and calcination), pH-controlled precipitation, evaporative deposition, and have been used [14,15,16].
Moreover, several tactics have been employed to adjust the cobalt–support interaction to improve the catalyst’s reduction capabilities. A modest quantity of promoter elements, which have a high capacity for dissociative hydrogen activation, could be added to enhance the cobalt dispersion and catalytic reducibility [17,18]. By grafting inactive or less active groups, deactivation or passivation of the chemically active oxygen groups on the support surface may also reduce the metal–support interaction, e.g., SiO2 has a several silanol (SiOH) groups on its surface, which can interact with cobalt oxides and affect catalyst structure and performance [12,19].
Both the catalyst’s composition and structure can be optimized by utilizing a certain the preparation technique, to increase product selectivity. The application of electronic promoters to enhance catalytic performance has been thoroughly studied in relation to co-based FTS catalysts [20]. It has been suggested that manganese (Mn) [21,22] is a potential promoter to increase both the hydrocarbon selectivity and catalytic activity [23]. Bell et al. discovered that a Mn promoter increased CO surface coverage and aided in CO dissociation, which caused a distribution of heavier hydrocarbon products due to reduced H availability and faster synthesis of C1 monomers [24].
It has been observed that adding trace quantities of Mn to cobalt-based catalysts increases their activity and C5+ selectivity while decreasing their selectivity for methane production [22,25,26,27,28]. The amount of Mn in the catalyst, as well as the reaction parameters (temperature, pressure, and CO conversion), determine how much activity increase occurs through Mn promotion [29,30]. Dinse et al. [24] and Johnson et al. [31] reported increased activity with Mn promotion at low concentrations of Mn (Mn/Co ratio < 0.125); however, activity decreased at Mn/Co ratios over 0.125. A higher rate with Mn promotion of Co/Carbon nanotubes (CNTs) was also found by Thiessen et al. [29], with a maximum at Mn/Co = 0.075 at 220 °C and 1 bar. Because the activation energy and the pre-exponential factor are reliant on the Mn content, the ideal Mn amount is temperature-dependent.
The enhanced dispersion of metallic Co, which is not usually observed [20,21,22,24,27], or a chemical promotion that modifies the bonding of CO on the catalytically active cobalt [25,27] has been suggested as the causes of the increase in activity with Mn promotion [24,31]. The latter was explained by the interaction between the adsorbed CO on the metallic cobalt surface and Mn2+ cations at the edges of MnO islands. This interaction strengthened CO adsorption and weakened the C-O bond in adsorbed CO, which has been hypothesized to be related to the observed increase in turnover frequency [32].
There is still little knowledge about the second metal’s and promoter’s activities and selectivities, despite a wealth of studies on catalyst loading and promotion. Typically, the metal salt precursor is impregnated first, followed by activation, and then the promoter is applied to the supported metal catalyst. The idea behind this method is to reduce at higher temperatures by absorbing the metal catalyst into the support material. The technique’s drawbacks include high reduction temperatures, which cause sintering and the creation of silicates and aluminates, as well as pore blockages when the promoter is introduced later (traditional catalyst promotion).
It is commonly known that conventional techniques for creating heterogeneous catalysts typically result in a mixture of various metal species and, as a result, provide limited control over the selectivity of the final product. One of the most significant areas of heterogeneous catalysis research in recent years has been the design and manufacturing of nanostructured metal catalysts with modified product selectivity or enhanced reaction activity for syngas conversion. According to Zhai et al. [33], those nanostructured materials appear to give fresh perspectives on the FTS reaction mechanism in addition to serving as a platform for product selectivity engineering. Using γ-alumina support Co catalysts, Goodwin et al. [34] investigated the support modification using titanium doping for FTS. No noble or nearly noble metal was used to boost the catalyst. To create titanium doped alumina, aluminum, and titanium alkoxide were co-hydrolyzed. This support was then impregnated with aqueous cobalt nitrate to achieve the required Co loading. According to their research, cobalt-on-alumina catalysts that have the proper amount of dopant loaded onto the support help to inhibit the synthesis of cobalt–alumina compounds, a result that is comparable to that of employing noble metal promoters. Using lanthanum (La) and barium (Ba), Alan H et al. [35] also looked into the impact of doping an alumina-supported Co catalyst. They found that, with little effect on product selectivity, the alumina support doped with either Ba or La enhanced FT activity and increased thermal stability.
Furthermore, transition metal oxides like Si, Zr, or Ti are frequently added to FT supports to improve structural stability [36]. In addition to changing the support surface’s structural makeup, the modifiers can affect the activity and selectivity of the FT. SiO2 modification of alumina supports has been demonstrated to lessen the amount of cobalt removed from the wax product and protect the support from the hydrothermal attack that is a natural part of the FT process [37]. Through the prevention of irreducible silicate production, zirconia (ZrO2) modification of silica catalysts was able to enhance cobalt reduction and improve catalyst performance [38].
In this work, we aimed to optimize the Co-Mn catalyst composition for inversely promoted Co-based catalysts (promoting the support before loading the metal catalyst). This article provides a methodical analysis of how Mn support modification affects Co-Mn-SiO2 catalysts for FTS, highlighting the significance of inverse support promotion for cobalt crystallite size, reduction degree, activity, and product (C5+) selectivity.
2. Materials and Methods
2.1. Catalysts Preparation
Two kinds of Co-based catalysts were prepared for this study.
- Non-promoted silica-supported Co catalyst (basis).
Commercially accessible SiO2 with a specific surface area of 317 m2/g and pore volume of 1.15 mL/g was the support utilized in these catalysts, serving as a baseline for comparison. The primary approach used to synthesize this Co/SiO2 was incipient wetness impregnation (IWI). Regarding the total amount of cobalt oxide precursor and SiO2 support, the cobalt weight loading was maintained at 10 wt.%. 4.98 g of Co(NO3)2·6H2O in an aqueous solution (10.35 mL) was used to saturate the first 9 g of SiO2. After being impregnated, the sample was dried inside the furnace for 12 h at 80 °C, and it was then calcined for 4 h at 400 °C, with a heating rate of 2 °C/min. This catalyst was labelled as 10% Co/SiO2. The best possible balance between metal dispersion and active site availability is achieved with a 10 wt.% Co loading on silica. High catalytic activity is produced by this loading because it guarantees enough surface area for CO adsorption [39,40]. High reproducibility in the preparation of 10 wt.% cobalt on silica catalysts guarantees consistent outcomes between batches. This uniformity is essential for consistent catalytic performance as shown by studies from our research institute [19,41,42].
- 2.
- Inverse promotion: promoting the support before loading the metal catalyst.
Using the same commercial support as mentioned above, a manganese loading of 0.25 wt.% was prepared. This involved impregnating the support with an aqueous solution containing 0.80g of manganese nitrate Mn(NO3)2 as a precursor. The resulting sample was dried at 80 °C for 12 h and then calcined at 400 °C for 4 h, yielding the support designated as 0.25%Mn-SiO2.
Subsequently, this support was further impregnated with a 10 wt.% loading of cobalt, considering the combined weight of cobalt oxide, manganese, and silica. The resulting sample underwent drying at 80 °C for 12 h followed by calcination at 400 °C for 4 h. This catalyst sample was labeled as 10%Co/0.25%Mn-SiO2. The same procedure was used to obtain the catalysts 10%Co/0.5%Mn-SiO2 and 10%Co/3.0%Mn-SiO2, and consequently having Mn/Co of 0.025, 0.05, and 0.3, respectively. Consideration of the Mn/Co ratios was dependent on literature reviews. The amount of Mn in the catalyst determines how much activity increase by manganese promotion occurs [24,29,31,32,43]. Dinse et al. [24] and Johnson et al. [31] reported increased activity with manganese promotion at low doses of Mn (Mn/Co ratio < 0.125); however, activity decreased at Mn/Co ratios over 0.125. A higher rate with Mn promotion of Co/CNT was also found by Thiessen et al. [29], with a maximum at Mn/Co = 0.075 at 220 °C and 1 bar.
2.2. Catalyst Characterization
X-ray powder diffraction (XRD)
X-ray powder diffraction spectra for the calcined catalysts were recorded on a Rigaku SmartLab X-ray Diffractometer using Cu Kα radiation (40 kV, 200 mA). The scan range was from 2θ = 5° to 90°. The average Co3O4 sizes were calculated using the Scherrer equation:
where d is the mean diameter of crystallites (nm); K is the Scherrer constant corresponding to the crystallite shape assumed 0.94; λ = 0.154 (nm) is X-ray wavelength; β is full width at half-max (FWHM) or integral breadth in radian [44].
The Co3O4 particle size was converted to the corresponding cobalt metal particle size according to the relative molar volumes of metallic cobalt and Co3O4. The resulting conversion factor for the diameter d(Co3O4) of a given Co3O4 particle being reduced to metallic cobalt is as follows:
d(Co0) = 0.75 × d(Co3O4)
H2 temperature programmed reduction (H2-TPR)
The TPR experiment was conducted to examine the catalyst’s reduction behavior using a CATLAB that was outfitted with an MS detector. A thermocouple was connected to the catalyst (0.2 g) in a quartz tubular reactor to enable continuous temperature monitoring. Before TPR measurement, water or impurities were removed from the calcined catalysts by purging them with Ar (flow rate: 50 mL/min) at 150 °C for two hours and then cooling them down to 50 °C. After that, 10%H2/Ar was turned on, and the temperature was increased from 50 to 900 °C at a rate of 10 °C/min.
CO and H2 chemisorption
To approximate the Co dispersion, metal surface area, and H2 uptake, CO and H2 adsorption isotherms were recorded at 35 °C using a CATLAB machine. After being evacuated at 150 °C for one hour, the 0.2 g samples were cooled to room temperature. Next, the catalysts were reduced in situ at 5000 C for 10 h at a ramping rate of 10 °C/min in flowing hydrogen or carbon monoxide (10%H2/Ar or 10%CO/Ar, respectively). Following reduction, the samples were cooled to 35 °C after being evacuated for 30 min at 400 °C. Assuming homogeneous, spherical cobalt metal particles, the cobalt metal particle size was computed from the cobalt metal dispersion [45].
These assumptions give the following formula:
d(Co0)(nm) = 96/D(%)
2.3. Catalyst Testing
A wide range of hydrocarbons, including olefins, oxygenates, and linear and branched paraffins, are produced using the Fischer–Tropsch process. Reaction conditions must be controlled in any study on the catalytic performance of comparable catalysts because of the complexity of the Fischer–Tropsch synthesis. The catalyst was tested in a fixed-bed microreactor for the Fischer–Tropsch process reactions. The 220 mm in length and 8 mm in diameter stainless steel FBR reactor was coated with a heating jacket to regulate and keep the temperature at the appropriate operating level. After loading a 1.0 g sample of the catalyst into the reactor, several parameters were monitored, including temperature, pressure, and space velocity. The catalyst was activated by reducing cobalt oxide to metallic cobalt, which is the active form for FTS, for 20 h using 5% H2 in Ar at 60 mL(NTP)/min before experiments.
For the FTS experiments, the fixed bed reactor was maintained at 220 °C and 20 bars until a steady state was attained. At a flow rate of 60 mL(NTP)/min, an FTS gas stream (H2/CO/N2 ratio = 6:3:1) was fed into the reactor. The Agilent gas chromatograph was used to conduct the online product analysis. Three phases make up the product stream: the gaseous phase, the oil phase, and the wax phase. An Agilent 7890B GC was used to examine the gaseous phase stream for the following gaseous components: CO, H2, N2, CO2, α-olefins C2–C5, and linear paraffins C1–C10. The Agilent GC has three detectors: the flame ionizing detector (FID), the thermal conductor detector (TCD)-A, and TCD-B. The FID was used to analyze the hydrocarbon products that were found, and the TCD-A was used to analyze the CO, CO2, and N2 components, while the TCD-B was used to detect H2. Argon 99.99% (AFROX Ltd., Johannesburg, South Africa,) was the carrier gas utilized in all detectors.
GC analysis: First, the oven is kept at 35 °C for five minutes. After that, it is gradually raised to 200 °C at a rate of 3 °C per minute. The temperature is then held at 200 °C for 60 min after. Each sample requires around 45 min to analyze. The GC is connected to a monitor, where The Agilent 7890B GC with 7693A ALS ChemStation edition is used to collect and store all of the data from the detectors about the sample.
3. Results and Discussions
3.1. Catalysts Textural Properties
The samples were degassed at 300 °C for 8 h. BET surface area, pore size, and volume were measured using nitrogen at 77 K. Instrument: Tristar II. Manufactured by Micromeritics (Atlanta, GA, USA).
Table 1 displays the physicochemical properties of Mn inversely boosted and non-promoted Co-based catalysts made by stepwise impregnation. When Mn is added, the BET surface area of the four catalysts decreases somewhat up to a Mn/Co ratio of 0.05 mol/mol, at which point it increases to a Mn/Co of 0.3. The latter can be explained by the creation of additional pores in addition to the possible removal of certain bigger organosilane groups than in Co. Furthermore, the interaction between a catalyst’s (precursor’s) BET surface area and the support material’s BET surface area is a complicated subject since the homogenous adsorption mechanism and uniform surface assumed by the BET theory might not apply to all materials. If there are interactions between the adsorbate molecules or if the surface is heterogeneous, this could result in inaccurate results. Depending on the relative particle size concerning their density, a physical combination of two materials with different densities may cause the BET surface area to rise or decrease. The complexity increases with the addition of a third component, and the loading of the third component and its average particle size are related to the observed change in the BET surface area for a physical mixture. Each of their pore volume pores diameters decreased as the Mn promoter level increased. This is explained by Mn and Co species partially filling the pores in the silica support.

Table 1.
Chemical composition and textural properties of the Mn inversely promoted and non-promoted catalysts.
3.2. Structure and Crystallite Size of Cobalt Species
X-ray Diffraction (XRD): Calcined Catalysts
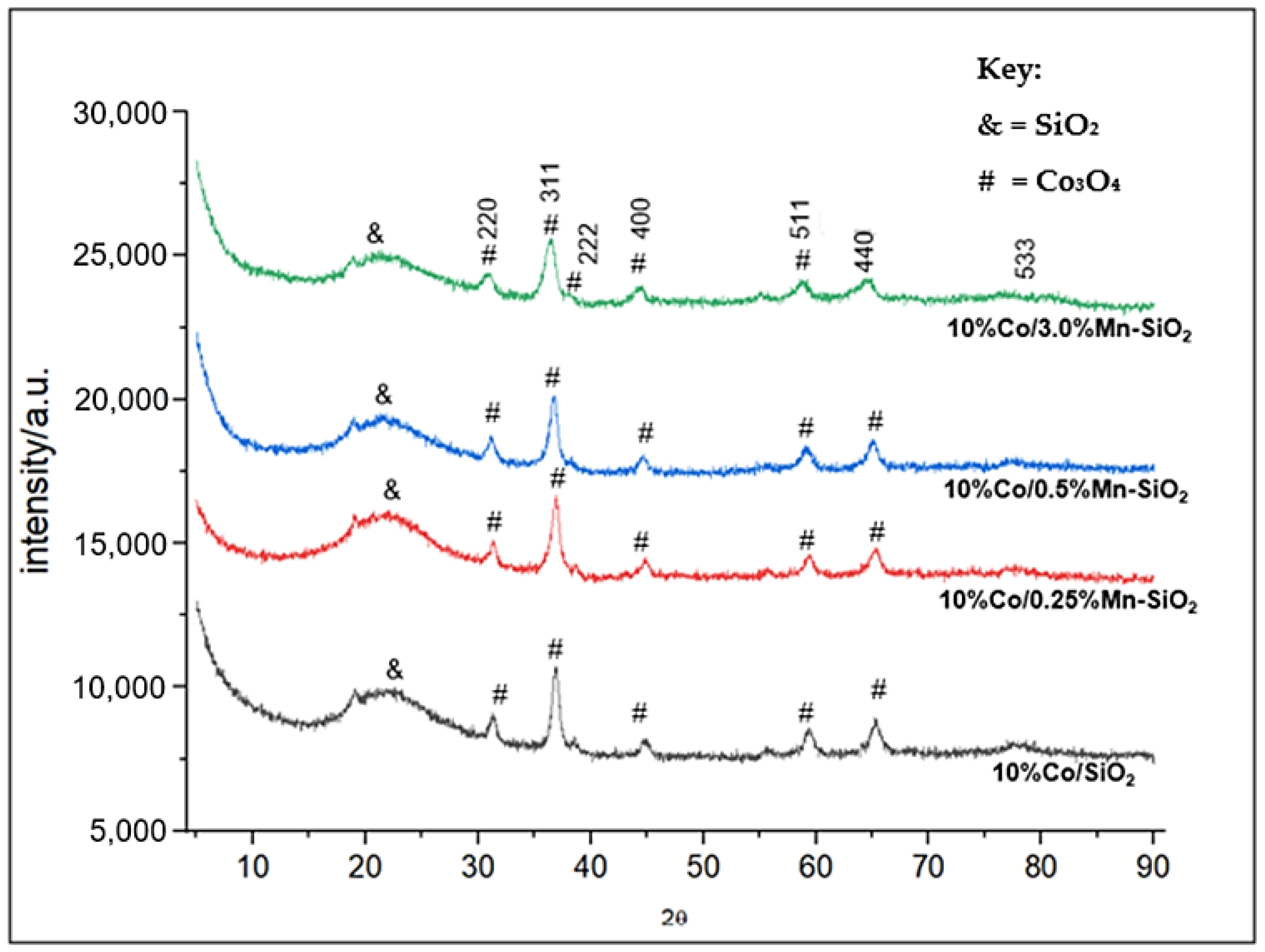
Figure 1 compiles the XRD patterns of the produced catalyst samples that were calcined in an air flow at 623 K. The JCPDS card no. 80-1538 reference indicates that the majority of the cobalt species are Co3O4 (2θ = 18.0°, 32.0°, 36.8°, 39.0°, 44.8°, 55.6°, 59.1°, 65.0°, and 77.0°). It should be noted that the XRD patterns contained no Mn-attributed diffraction peak which was expected at peaks 2θ = 12°, 24.7°, and 66.1° [46]. This suggests that the catalysts’ higher dispersion on their surfaces, which is impossible to assess using XRD as a result of the catalysts’ lower concentrations of Mn promoter. The breadth of the Co3O4 peaks was the sole variation observed in the X-ray diffraction patterns of the calcined samples. Therefore, the amount of silica support and promoter applied determined the average size of the Co3O4 crystallites. The particle sizes of the calcined samples were then determined using the Scherrer equation. Table 1 indicates a clear rise in Co3O4 particle size as support pore size increased. Comparable results have been documented for cobalt supported by silica [47,48,49,50,51]. Additionally, Co3O4 crystallite size increased with increasing γ-Al2O3 pore size, as reported by Xiong et al. [52]. It should be noted that no discernible relationship was discovered between the Co3O4 particle sizes and the BET surface areas of the supports.
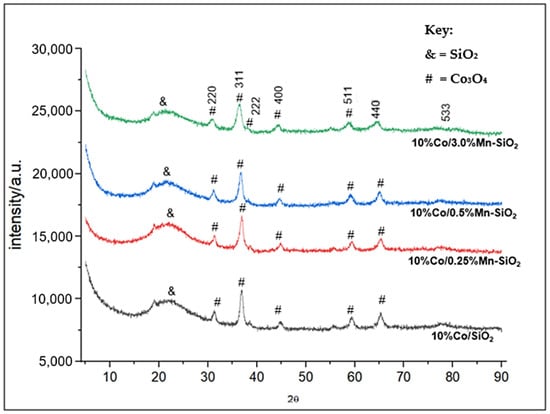
Figure 1.
XRD patterns of the Mn inversely promoted and non-promoted catalysts.
3.3. X-ray Photoelectron Spectroscopy: Calcined Catalysts
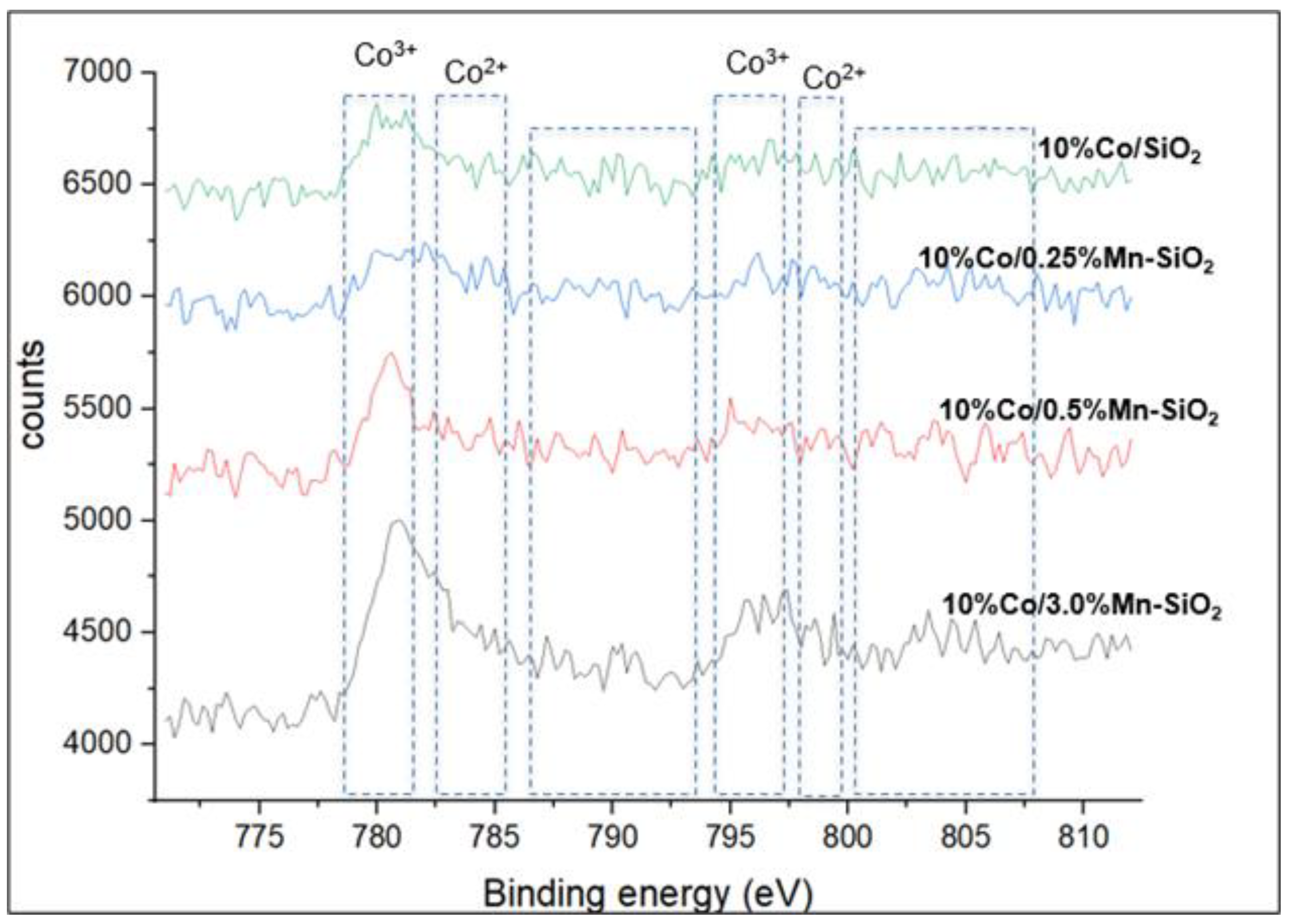
XPS was used to establish that the Co3O4 structure was present in all calcined samples. The deconvoluted peaks corresponding to the Co3+ 2p3/2 and Co2+ 2p3/2 were located approximately at 780.1 and 781.4 eV. At 794.8 and 796.8 eV, the corresponding 2p1/2 spin-orbit component for Co3+ and Co2+ is visible, and on the high binding energy sides, a low strong shake-up satellite structure was noted. Consequently, Co3O4 is the predominant cobalt phase in oxidized catalysts, as demonstrated by the XP spectra obtained for Co2p. This result is in line with the Co3O4 XRD patterns that have been seen in every catalyst. As seen in Figure 2, the change of Mn causes the binding energies of Co 2p3/2 to move to lower values. The XPS data show that with Mn modification, the electron density surrounding the Co and Mn atoms declined. When Mn is present, inversely promoted catalysts’ Co2+ peak intensity is lower than it is in the Mn-free sample. For the Mn-promoted catalysts, the Co3+/Co2+ ratio rises, indicating a preferred replacement of Mn2+ ions in the mixed oxide spinel structure by Co2+ ions. Additionally, Table 2 illustrates the reduced atomic percentage of the Co 2p3/2 peaks for the 0.5%Mn promoted catalyst, indicating a reduced interaction between SiO2 and the other Co2+ species. This is also evident on the BE of approximately 780.9 eV, which is lower than 781.1 eV in the 10%Co/SiO2 non-promoted sample.
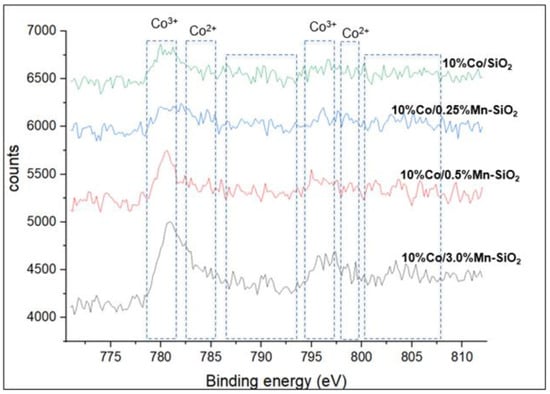
Figure 2.
The Co 2p XP spectra with the two spin orbit components Co 2p3/2 and Co 2p1/2.
Table 2.
XPS atomic percentages for the different catalysts.
Following the addition of MnOx to Co3O4, the binding energy of Co 2p3/2 increased. This observation points to a rise in the Mn–Co–O catalysts’ Co3+/Co2+ fraction. According to published research, silicas impregnated and then calcined may result in an enrichment of Co3O4 particles on the outside of SiO2 grains [48]. A greater concentration of Co3O4 in the vicinity of the catalyst grains’ outer surface may cause the Co2p to be more intense. Furthermore, a stronger shake-up satellite structure emerges with the addition of Mn, and the location of the XPS Co 2p peak moves to the region with greater binding energies. These findings suggest that isolated surface Co2+ species may also be present in some concentration in promoted catalysts close to the outer surface of catalyst particles, in addition to the predominant Co3O4 phase. According to earlier reports, these isolated Co2+ ions may be located in the amorphous phases of cobalt silicate/hydrosilicate [36,53,54,55,56]. The TPR data (Figure 3) show that these cobalt species have substantially lower reducibility in hydrogen than the Co3O4 crystalline phase.
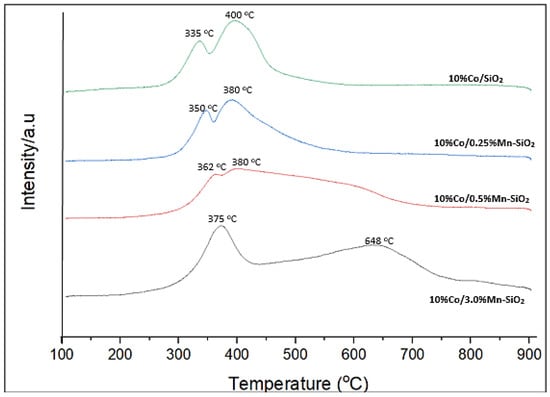
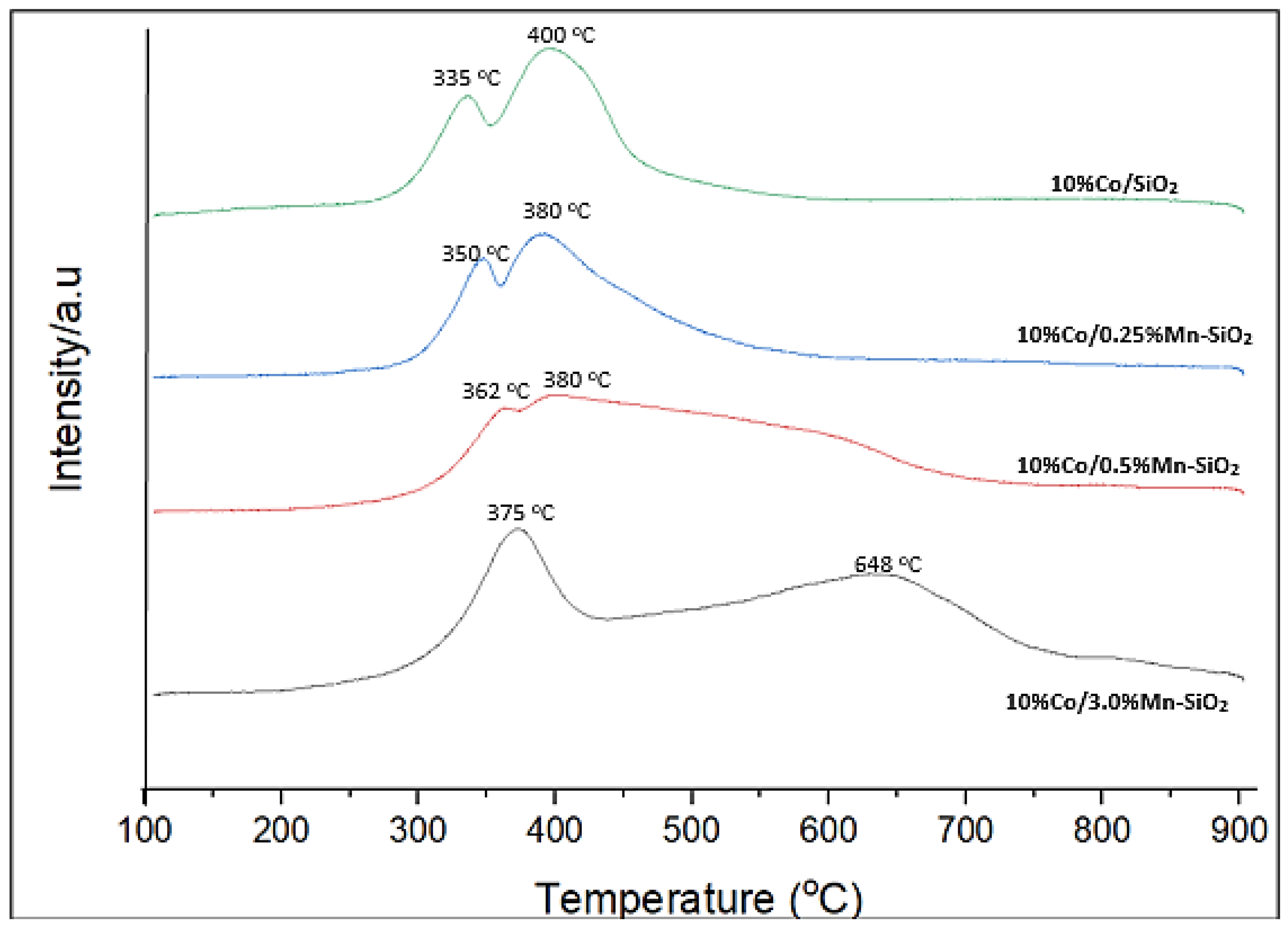
Figure 3.
H2-TPR profiles for the synthesized Mn inversely promoted and non-promoted catalysts.
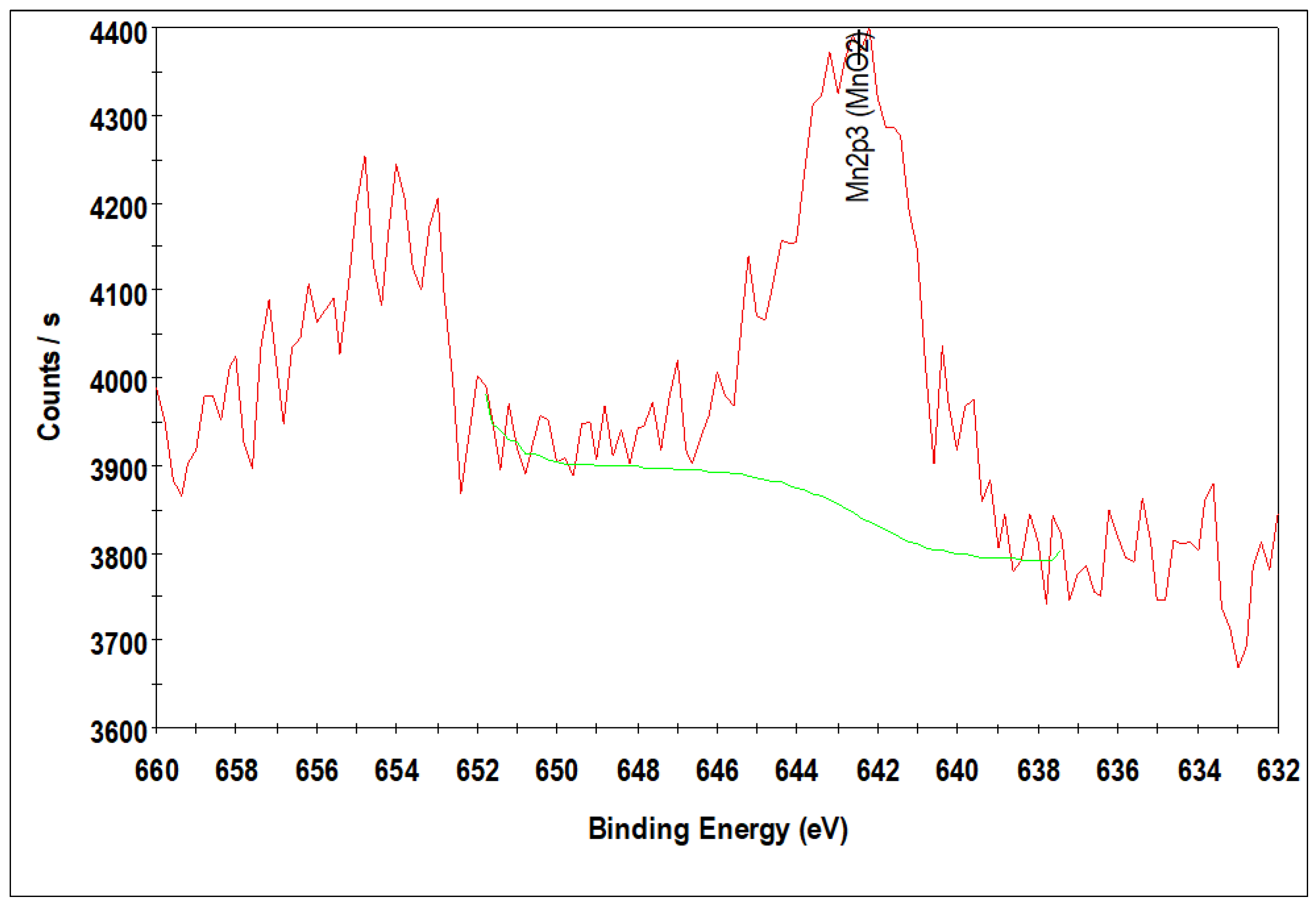
Due to the lower loading of Mn 2p peaks corresponding to MnO for 10%Co/0.25%Mn-SiO2 and 10%Co/0.5%Mn-SiO2 could not be observed because they were below the XPS sensitivity or resolution limit, making it difficult to validate the chemical environment of the manganese species in the Mn inversely promoted catalysts. Thus, Figure 4 solely displays the Mn 2p3/2 photoelectron spectra of 10%Co/3.0%Mn-SiO2. With a spin orbit level energy spacing of 11.5 eV, the deconvoluted Mn 2p displayed two peaks at 642.2 eV for Mn 2p3/2 and 653.7 eV for Mn 2p1/2. This is common for Mn3+-based materials [57,58]. This suggests that Mn is substituted preferentially in spinel oxide’s Oh sites.
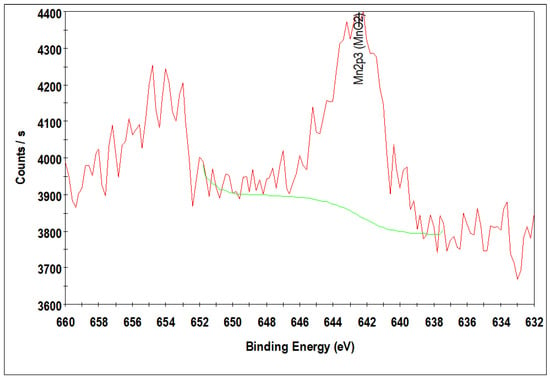
Figure 4.
XPS spectra of the Mn 2p level 10%Co/3.0%Mn-SiO2.
The average particle size determined by averaging the Co3O4 (220), (311), (400), (511), and (440) peaks were found to be greater than the average support pore diameter as determined by Scherrer’s equation, as shown in Figure 1 and Equation (1). As a result, it appeared that most of the particles were found outside the support pores. It is important to compare the Co3O4 particle size and pore diameter carefully when determining the position of the particles. The authors think that the particles are inside the pore system, and they attribute the contradictory findings in Figure 1 to the selection of the pore shape model. The BJH model oversimplifies the pore geometry since it presumes that there are only cylindrical pores and that pore networks do not exist [51]. When the loading of Mn increased, the average particle sizes shrank. This is consistent with the outcomes of H2 chemisorption results. An increase in dispersion caused by a rise in Mn loading subsequently increased metal surface area. Notably, with 10%Co/SiO2, there is a discernible variation in the particle sizes measured by XRD and H2 chemisorption. This could be explained by the movement and agglomeration of particles in the chemisorption reaction during reduction.
3.4. Reducibility of Co Species
H2 Programmed Reduction (TPR)
Using H2-TPR, the reduction behavior of the unpromoted and Mn inversely promoted catalysts was investigated as shown in Figure 3. An initial peak at 335–350 °C for 10%Co/SiO2 and 10%Co/0.25%Mn-SiO2, and 362–375 °C for 10%Co/0.5%Mn-SiO2 and 10%Co/3.0%Mn-SiO2, was noted. Subsequent peaks occurred at about 400 °C, as opposed to 648 °C for 10%Co/3.0%Mn-SiO2. The TPR profile for Co catalysts supported by silica has been thoroughly studied, and the reduction peaks located at 200–400 °C and 400–800 °C, respectively, indicate a two-step reduction process. The reduction of Co3O4 to CoO is frequently attributed to the first stage, and the subsequent reduction of CoO to Co0 is attributed to the second step [18,59]. With an increase in Mn loading, the positions of the reduction peaks of Co3O4 to CoO and CoO to Co changed to higher temperatures, suggesting a strong interaction between Si species and cobalt nanoparticles. Equation (3) was utilized to compute the reduction degrees of each catalyst, which are displayed in Table 3. The reduction degrees of the 10%Co/SiO2, 10%Co/0.25%Mn-SiO2, 10%Co/0.5%Mn-SiO2, and 10%Co/3.0%Mn-SiO2 catalysts were 96.0, 94.3, 94.0, and 92.3%, respectively. For catalysts 10%Co/0.5%Mn-SiO2 and 10%Co/3.0%Mn-SiO2 the second peak appeared at 362–375 °C, i.e., the highest temperature peak area increased in this case and further addition of Mn reduced the Co reducibility. This suggests once more that these catalysts have a Co-Mn interaction, which reduces the amount of Co reducibility [27].
Table 3.
XRD and chemisorption data for the Mn inversely promoted and non-promoted catalysts.
3.5. Fischer Tropsch Tests
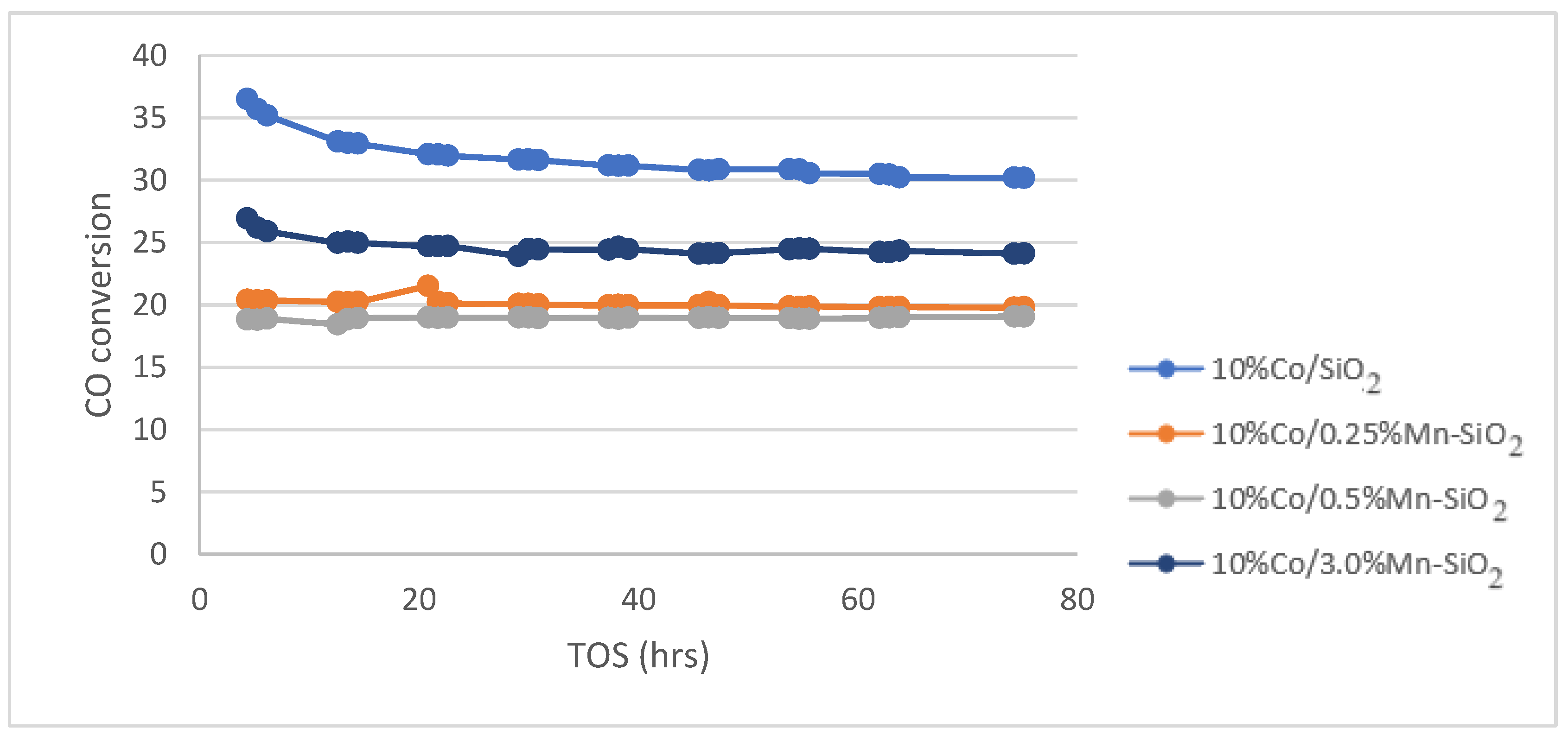
The calcined catalysts were activated with an H2 flow at a rate of 60 mL/g-cat/min for 20 h at 350 °C (as obtained from H2 TPR) before the reaction. Following the in situ catalyst’s activation, the reaction was cooled to the target reaction temperature of 220 °C and flushed with helium for 30 min to get rid of any excess H2. The Fischer–Tropsch reaction test was run at 20 bar pressure, 220 °C, 60 mL/g-cat/min, and H2/CO (v/v) = 2/1. For the Mn inversely promoted and non-promoted silica-supported catalysts, the percentage of CO conversion, CH4 selectivity, rate of CO conversion, and C5+ selectivity is shown for a 75 h time on stream (TOS) in Figure 5, Figure 6 and Figure 7.
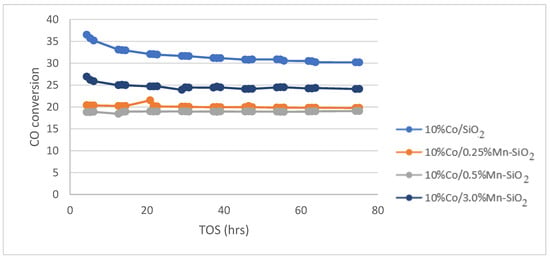
Figure 5.
CO conversion for the synthesized Mn inversely promoted and non-promoted catalysts. Reaction conditions: T = 220 °C; P = 20 bar; H2/CO2 = 3/1, for TOS = 75 h.
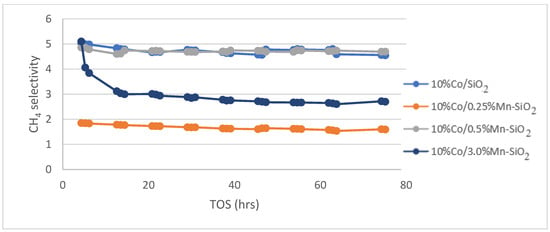
Figure 6.
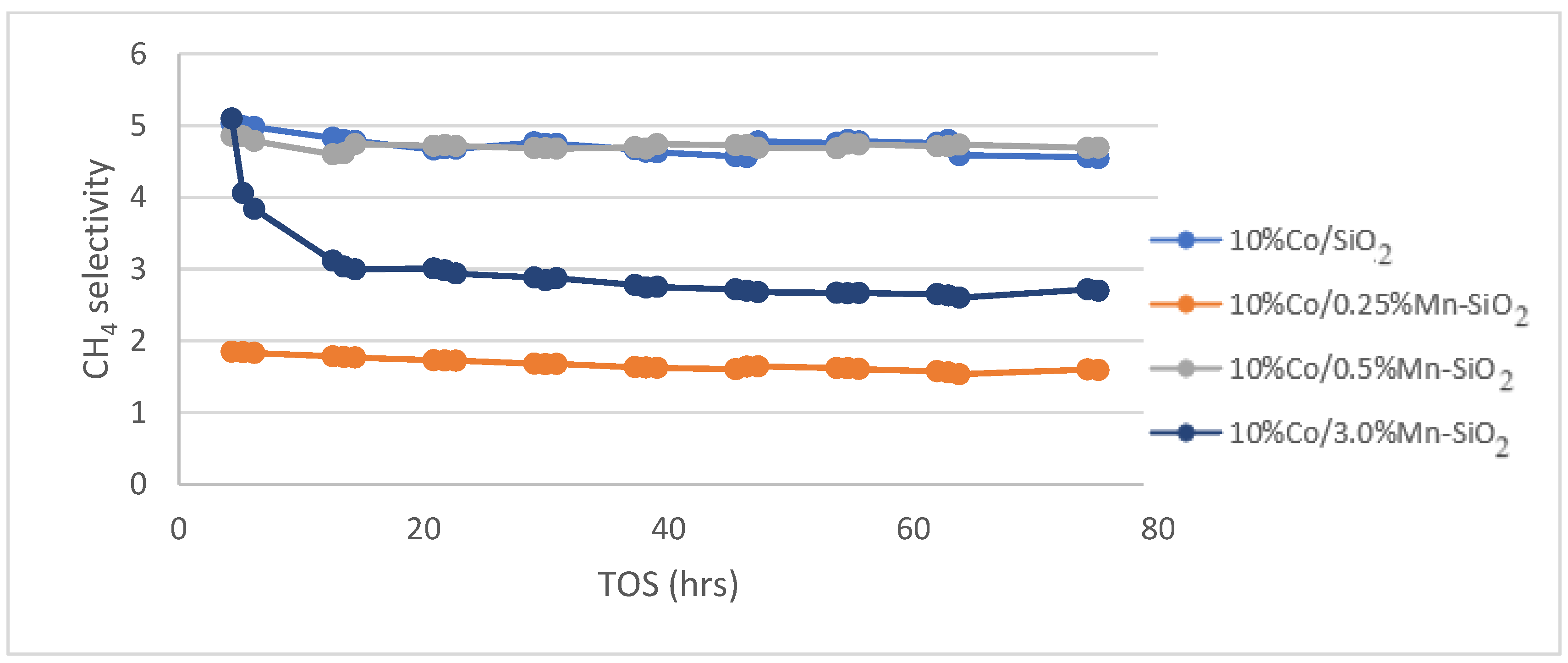
Methane selectivity for the synthesized Mn inversely promoted and non-promoted catalysts. Reaction conditions: T = 220 °C; P = 20 bar; H2/CO2 = 3/1, for TOS = 75 h.
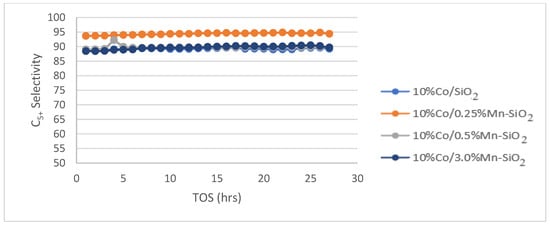
Figure 7.
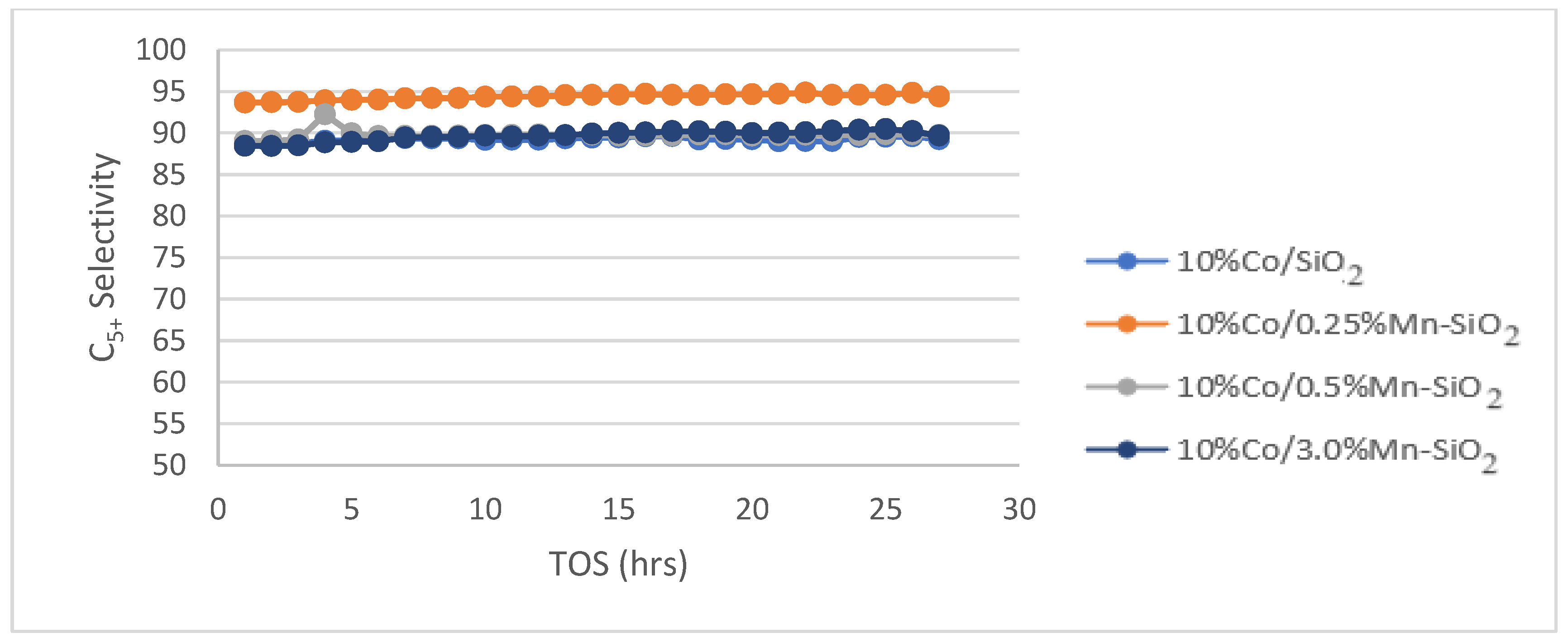
C5+ selectivity for the synthesized Mn inversely promoted and non-promoted catalysts. Reaction conditions: T = 220 °C; P = 20 bar; H2/CO2 = 3/1; for TOS = 75 h.
FTS Mass Balance Calculations
The experimental setup utilized in this investigation allows for the input of the inlet volumetric flow rate, which is necessary to compute the output flow rate. As an internal standard, N2 was added together with the reactor feed. N2 is only present in the feed stream and the reactor output gas stream since it is an inert gas during the FT reaction. As a result, the N2 balance throughout the reactor can be written as follows:
CO conversion, the rate of CO conversion, and product selectivity were defined as follows [4]:
where Fin = inlet total molar flow rate in mol/min.
- Fout = outlet total molar flow rate in mol/min.
- XN2, in = inlet molar fraction of N2.
- XN2,out = outlet molar fraction of N2.
- Xco2,in = CO molar fraction in the gas feed.
- Xco2,out = molar fraction of CO in the gas stream leaving the reactor.
- mcat = mass of the catalyst (in grams).
- = moles of carbons in the product stream.
The CO conversion for each catalyst at a TOS of 75 h is shown in Table 4. The inclusion of Mn caused the conversion of 10%Co/0.25%Mn-SiO2 and 10%Co/0.5%Mn-SiO2 to decrease from 31.8% to 20.1 and 18.9%, respectively, before slightly increasing to 24.0% for 10%Co/3.0%Mn-SiO2. The preparation method for Mn promotion, that is, inverse promotion as opposed to conventional promotion, may be the reason for this study’s decline in activity. The H2 TPR and H2 chemisorption measurement reveals a reduction in the level of active metal reduction as shown in Table 3 (DOR) in Co-prompted catalysts, which might be the justification for lower activity for promoted catalysts.
Table 4.
Catalyst activity and product selectivity for the non-promoted and inversely promoted catalysts. Reaction conditions: T = 220 °C; P = 20 bar; H2/CO2 = 3/1, for TOS = 75 h.
The CO conversion trend is comparable to the hydrogen intake pattern found in Table 3’s H2 chemisorption data. H2 absorption was decreased by the addition of Mn to the cobalt, mainly in the 10%Co/0.25%Mn-SiO2 and 10%Co/0.5%Mn-SiO2 catalysts. Dinse et al. [24] and Johnson et al. [31] reported increased activity with manganese promotion at low doses of Mn (Mn/Co ratio < 0.125), however activity decreased at Mn/Co ratios over 0.125. Therefore, the divergence in the results may be ascribed to the amount of manganese in the catalyst, the preparation procedure, and the reaction parameters [24,29,31]. Moreover, the observed disparities in accessible Co surface area are the main reason for the conversion differences between the unpromoted and Mn-promoted catalysts. It has previously been proposed that Mn decorates the surfaces of the Co particles and that increased activity sites close to the Co-Mn interface are the basis of this effect’s promotion [31]. When a catalyst is unpromoted and the Mn/Co ratio is low, the crystal structure changes from hexagonal close-packed (hcp) to face-centered cubic (fcc), where product selectivities are strongly dependent on promoter loading. Therefore, it is necessary to consider the likelihood that the promoting effects were brought on by a Mn-induced shift from hcp to fcc structure. According to reports, hcp Co nanoparticles have a greater FTS turnover frequency than fcc Co nanoparticles [60,61]. Regarding product selectivity, reports indicate that the product distributions for the two crystal structures differ not significantly [62,63]. These results lead to a prediction that, in contrast to the effects of Mn that have been observed, a shift from the hcp to the fcc crystal structure would result in a decrease in site activity.
The effects of Mn promoter and non-promoted catalysts on CH4 products on FTS selectivity are shown in Figure 6. It unequivocally demonstrates a decrease in methane selectivity for catalysts supported by Mn. Methane selectivity dropped for 10%Co/0.25%Mn-SiO2 and 10%Co/3.0%Mn-SiO2 catalysts, respectively, from 4.7% to 1.7 and 3.0%. The selectivity towards the desired C5+ products is significantly increased as a result of the notable effects of manganese stimulation on the selectivity of undesirable by-products, such as methane. The increasing trend in chain growth probability indicates a shift to higher molecular weight hydrocarbons by increasing the amount of Mn promoter. On the surface of the cobalt crystallite, Mn enrichment for Co–Mn bimetallic catalysts may be the cause of the increased C5+ selectivity when compared to the unpromoted catalyst. Reduced CO conversion rates are the result of increased CO uptake decreasing H2’s mobility on the catalyst surface.
The product selectivities (C5+) of the non-promoted and Mn inversely promoted Co catalysts are displayed in Figure 7. 10%Co/SiO2, 10%Co/0.25%Mn-SiO2, 10%Co/0.5%Mn-SiO2, and 10%Co/3.0%Mn-SiO2 have C5+ selectivities of 89.2%, 94.4%, 89.8%, and 89.7%, respectively. There is a decline in the selectivity of C5+ hydrocarbons as the loading on Mn is increased. This is because Mn starts to cover the Co surface once the percentage of promoted interface sites exceeds Mn/Co 0.025. As a result, there would be fewer active sites and a fall in CO consumption rates per gram of Co. These results align with Østbye, et al. [64], who realized a CO conversion of ≈20% for the Mn-promoted catalyst which was less than the unpromoted Co catalyst of ≈37%.
The formation of an alloy of metallic Mn with Co on the nanoparticle surface, which might happen at low Mn loadings where selectivity is extremely sensitive to promoter loading, was one theory that was taken into consideration to explain the trends in catalyst performance as a function of promoter loading. It seems sense that Mn would preferentially reside on the surfaces of the nanoparticles because its surface free energy is significantly lower than that of Co [65]. Moreover, the Mn loading (Mn/Co = 0.1) at which C5+ selectivity hits a plateau is about equal to the amount required to create a monolayer on the surfaces of the nanoparticles. The reason for the lack of improvement in C5+ selectivity above Mn/Co atomic ratios of 0.1 could be attributed to the saturation of the nanoparticle surfaces by Mn. For the Co-Mn catalysts examined in this work, no indication of metallic Mn was found, even though these tendencies are phenomenologically coherent [66].
The results shown above agree with those of Breejen et al. [67]. They discovered that slightly greater C5+ selectivity values were found for bigger levels of MnO (Mn/Co > 0.13) but at a significant cost to the activity. This could be caused by a cobalt surface obstruction [22] or a reduced degree of reduction as a result of MnO’s reduction-retarding impact [27]. It was discovered that adding a small amount of MnO to the AC catalyst reduced its activity; the value declined even more at larger Mn/Co ratios. This could be because MnO is obstructing the cobalt surface [21]. However, the positive effect of MnO is demonstrated by a rise in Mn/Co atomic number and C5+ selectivity. However, the rise in C5+ selectivity indicates that MnO is favorable. Consequently, this study shows that 10%Co/0.25%Mn-SiO2 is the most effective catalyst among all those examined in this study and that it is capable of both increasing the much-desired C5+ selectivity and decreasing the CH4 selectivity.
4. Conclusions
By using Mn as a promoter element, the production of CH4 may be reduced and selectivity may be improved. But only by employing a proper preparation technique that results in a sufficient Mn-Co interaction in the active catalyst can this promoting effect be realized. By limiting pore blockages and active Co sites, inverse promotion can lead to improved catalyst product selectivity and stability. Co-based catalysts are prone to deactivation due to carbon buildup (coking) and sintering. Inverse promotion can help mitigate these issues by altering the surface chemistry and morphology of the catalyst. In all oxidized catalysts, Co3O4 crystallites are the most prevalent cobalt phase, according to the XRD analysis of the calcined samples. For Mn inversely promoted catalysts, the XRD-derived crystallite sizes are marginally larger than the support pore width, indicating that the particles were primarily on the support’s outer surface. When the catalysts are reduced in situ under identical FT pretreatment conditions, XPS measurements verify that the cobalt is more difficult to reduce when supported over the Mn modified silica. The H2 TPR profiles, which show significant hydrogen consumption as Mn loading rises from 0.25% to 3.0%, are likewise compatible with this feature. With the addition of Mn to the catalysts, CO conversion demonstrated a decline, which is consistent with the reduced surface area. When comparing the catalysts’ catalytic performance in the FT process at 600 h−1 space velocity, it is found that the Mn inversely promoted catalysts had higher C5+ selectivities, with 10%Co/0.25%Mn-SiO2 having the highest at 94.4%. This may be explained by the Co oxide’s strong interaction with the Mn-modified silica substrate, which prevents particle mobility and, as a result, reduces H2 chemisorption and increases CO chemisorption.
Author Contributions
Conceptualization, N.T. and J.G.; methodology, N.T. and J.G.; validation, Y.Y.; formal analysis, J.G.; writing—original draft preparation, N.T.; writing—review and editing, N.T.; visualization, J.G.; supervision, J.G. and Y.Y. All authors have read and agreed to the published version of the manuscript.
Funding
The authors of this manuscript would like to acknowledge the financial support from the University of South Africa (UNISA), the Institute of Catalysis and Energy Solutions (ICES), and the National Research Foundation (NRF).
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors on request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Potgieter, J.H.; Moodley, D.; Botha, T.; Visagie, J.; Manong, T.; Frank, M.; Claeys, M.; van Steen, E.; Böltken, T.; Pfeifer, P. Development of promoted cobalt/alumina Fischer-Tropsch catalysts for increased activity and selectivity: Micro-reactor to piloting scale. Catal. Today 2024, 432, 114554. [Google Scholar] [CrossRef]
- Moodley, D.J. On the Deactivation of Cobalt-based Fischer-Tropsch Synthesis Catalysts. Ph.D. Thesis, Chemical Engineering and Chemistry, Technische Universiteit Eindhoven, Eindhoven, The Netherlands, 2008. [Google Scholar]
- Gupta, S.; Fernandes, R.; Patel, R.; Spreitzer, M.; Patel, N. A review of cobalt-based catalysts for sustainable energy and environmental applications. Appl. Catal. A Gen. 2023, 661, 119254. [Google Scholar] [CrossRef]
- Shiba, N.C.; Liu, X.; Yao, Y. Insight into the Physicochemical Properties of Co-Based Catalysts in Fischer—Tropsch Synthesis. Reactions 2023, 4, 420–431. [Google Scholar] [CrossRef]
- Okoye-Chine, C.G.; Moyo, M.; Hildebrandt, D. The effect of hydrophobicity on SiO2–supported Co catalysts in Fischer-Tropsch synthesis. Fuel 2021, 296, 120667. [Google Scholar] [CrossRef]
- Ma, W.; Dalai, A.K. Effects of Structure and Particle Size of Iron, Cobalt and Ruthenium Catalysts on Fischer–Tropsch Synthesis. Reactions 2021, 2, 62–77. [Google Scholar] [CrossRef]
- Li, Z.; Si, M.; Xin, L.; Liu, R.; Liu, R.; Lü, J. Cobalt catalysts for Fischer-Tropsch synthesis: The effect of support, precipitant and pH value. Chin. J. Chem. Eng. 2018, 26, 747–752. [Google Scholar] [CrossRef]
- Wolf, M.; Fischer, N.; Claeys, M. Review Formation of metal-support compounds in cobalt-based Fischer-Tropsch synthesis: A review. Chem. Catal. 2021, 1, 1014–1041. [Google Scholar] [CrossRef]
- Adeleke, A.A.; Liu, X.; Lu, X.; Moyo, M.; Hildebrandt, D. Cobalt hybrid catalysts in Fischer-Tropsch synthesis. Rev. Chem. Eng. 2020, 36, 437–457. [Google Scholar] [CrossRef]
- Maximov, A.L.; Kulikova, M.V.; Dementyeva, O.S.; Ponomareva, A.K. Cobalt-Containing Dispersion Catalysts for Three-Phase Fischer—Tropsch Synthesis. Front. Chem. 2020, 8, 567848. [Google Scholar] [CrossRef]
- Wentrup, J.; Pesch, G.R.; Thöming, J. Dynamic operation of Fischer-Tropsch reactors for power-to-liquid concepts: A review. Renew. Sustain. Energy Rev 2022, 162, 112454. [Google Scholar]
- Gholami, Z.; Tišler, Z.; Rubáš, V. Recent advances in Fischer-Tropsch synthesis using cobalt-based catalysts: A review on supports, promoters, and reactors Recent advances in Fischer-Tropsch synthesis using. Catal. Rev. 2021, 63, 512–595. [Google Scholar] [CrossRef]
- Smith, D.F.; Hawk, C.O.; Golden, P.L. The mechanism of the formation of higher hydrocarbons from water gas. J. Am. Chem. Soc. 1930, 52, 3221–3232. [Google Scholar] [CrossRef]
- Niemel, M.K.; Krause, A.O.I.; Vaara, T.; Lahtinen, J. Preparation and characterization of Co/SiO2. Top. Catal. 1995, 2, 45–57. [Google Scholar]
- Bartholomew, C.H. Effects of Support and Dispersion on the CO Hydrogenation Activity/Selectivity Properties of Cobalt. J. Catal. 1984, 88, 78–88. [Google Scholar]
- Bartholomew, C.H. The Stoichiometries of H2 and CO Adsorptions Support and Preparation on Cobalt: Effects of support and preparation. J. Catal. 1984, 77, 63–77. [Google Scholar]
- Peng, X.; Cheng, K.; Kang, J.; Gu, B.; Yu, X.; Zhang, Q.; Wang, Y. Impact of Hydrogenolysis on the Selectivity of the Fischer—Tropsch Synthesis: Diesel Fuel Production over Mesoporous Zeolite-Y-supported cobalt nanoparticles. Angew. Comun. 2015, 54, 4553–4556. [Google Scholar]
- Pei, Y.; Ding, Y.; Zhu, H.; Zang, J.; Song, X.; Dong, W. Effect of Al2O3 Promoter on a Performance of C1—C 14 a -Alcohols Direct Synthesis over Co/AC Catalysts via Fischer—Tropsch Synthesis. Catal. Lett. 2014, 144, 1433–1442. [Google Scholar] [CrossRef]
- Okoye-chine, C.G.; Mbuya, C.O.L.; Ntelane, T.S.; Moyo, M.; Hildebrandt, D. The effect of silanol groups on the metal-support interactions in silica- supported cobalt Fischer-Tropsch catalysts. A temperature programmed surface reaction. J. Catal. 2020, 381, 121–129. [Google Scholar] [CrossRef]
- Li, Z.; Zhong, L.; Yu, F.; An, Y.; Dai, Y.; Yang, Y.; Lin, T.; Li, S.; Wang, H.; Gao, P.; et al. Effects of Sodium on the Catalytic Performance of CoMn Catalysts for Fischer-Tropsch to Olefin Reactions. ACS Catal. 2017, 7, 3622–3631. [Google Scholar] [CrossRef]
- Colley, S.; Copperthwaite, R.G.; Hutchings, G.J.; Van der Riet, M. Carbon Monoxide Hydrogenation Using Cobalt Manganese Oxide Catalysts: Initial Catalyst Optimization Studies. Ind. Eng. Chem. Res. 1988, 27, 1339–1344. [Google Scholar] [CrossRef]
- Bezemer, G.L.; Radstake, P.B.; Falke, U.; Oosterbeek, H.; Kuipers, H.P.C.E.; Van Dillen, A.J. Investigation of promoter effects of manganese oxide on carbon nanofiber-supported cobalt catalysts for Fischer—Tropsch synthesis. J. Catal. 2006, 237, 152–161. [Google Scholar] [CrossRef]
- Khodakov, A.Y.; Chu, W.; Fongarland, P. Advances in the development of novel cobalt Fischer-Tropsch catalysts for synthesis of long-chain hydrocarbons and clean fuels. Chem. Rev. 2007, 107, 1692–1744. [Google Scholar] [CrossRef]
- Dinse, A.; Aigner, M.; Ulbrich, M.; Johnson, G.R.; Bell, A.T. Effects of Mn promotion on the activity and selectivity of Co/SiO2 for Fischer—Tropsch Synthesis. J. Mol. Catal. A Chem. 2012, 288, 104–114. [Google Scholar] [CrossRef]
- Morales, F.; Desmit, E.; Degroot, F.; Visser, T.; Weckhuysen, B.M. Effects of manganese oxide promoter on the CO and H 2 adsorption properties of titania-supported cobalt Fischer—Tropsch catalysts. J. Catal. 2007, 246, 91–99. [Google Scholar] [CrossRef]
- Morales, F.; De Groot, F.M.F.; Gijzeman, O.L.J.; Mens, A.; Stephan, O. Mn promotion effects in Co/TiO2 Fischer—Tropsch catalysts as investigated by XPS and STEM-EELS. J. Catal. 2005, 230, 301–308. [Google Scholar] [CrossRef]
- Cano, F.M.; Gijzeman, O.L.J.; De Groot, F.M.F.; Weckhuysen, B.M. Manganese promotion in cobalt-based Fischer-Tropsch catalysis. Stud. Surf. Sci. Catal. 2004, 147, 271–276. [Google Scholar]
- Feltes, T.E.; Espinosa-Alonso, L.; de Smit, E.; D’Souza, L.; Meyer, R.J.; Weckhuysen, B.M.; Regalbuto, J.R. Selective adsorption of manganese onto cobalt for optimized Mn/Co/TiO2 Fischer—Tropsch catalysts. J. Catal. 2010, 270, 95–102. [Google Scholar] [CrossRef]
- Thiessen, J.; Rose, A.; Meyer, J.; Jess, A.; Curulla-ferré, D. Microporous and Mesoporous Materials Effects of manganese and reduction promoters on carbon nanotube supported cobalt catalysts in Fischer—Tropsch synthesis. Microporous Mesoporous Mater. 2012, 164, 199–206. [Google Scholar] [CrossRef]
- Werner, S.; Johnson, G.R.; Bell, A.T. Synthesis and Characterization of Supported Cobalt—Manganese Nanoparticles as Model Catalysts for Fischer—Tropsch Synthesis. J. ChemCatChem 2014, 6, 2881–2888. [Google Scholar] [CrossRef]
- Johnson, G.R.; Werner, S.; Bell, A.T. An Investigation into the Effects of Mn Promotion on the Activity and Selectivity of Co/SiO2 for Fischer-Tropsch Synthesis: Evidence for Enhanced CO Adsorption and Dissociation. ACS Catal. 2015, 5, 5888–5903. [Google Scholar] [CrossRef]
- Tucker, C.L.; Ragoo, Y.; Mathe, S.; Macheli, L.; Bordoloi, A.; Rocha, T.C.; Govender, S.; Kooyman, P.J.; van Steen, E. Manganese promotion of a cobalt Fischer-Tropsch catalyst to improve operation at high conversion. J. Catal. 2022, 411, 97–108. [Google Scholar] [CrossRef]
- Zhai, P.; Sun, G.; Zhu, Q.; Ma, D. Fischer-tropsch synthesis nanostructured catalysts: Understanding structural characteristics and catalytic reaction. Nanotechnol. Rev. 2013, 2, 547–576. [Google Scholar] [CrossRef]
- Panpranot, J.; Goodwin, J.G.; Sayari, A. CO hydrogenation on Ru-promoted Co/MCM-41 catalysts. J. Catal. 2002, 211, 530–539. [Google Scholar] [CrossRef]
- Jean-Marie, A.; Griboval-Constant, A.; Khodakov, A.Y.; Diehl, F. Cobalt supported on alumina and silica-doped alumina: Catalyst structure and catalytic performance in Fischer-Tropsch synthesis. Comptes Rendus Chim. 2009, 12, 660–667. [Google Scholar] [CrossRef]
- Oukaci, R.; Singleton, A.H.; Goodwin, J.G. Comparison of patented Co F–T catalysts using fixed-bed and slurry bubble column reactors. Appl. Catal. A Gen. 1999, 186, 129–144. [Google Scholar] [CrossRef]
- Barradas, S.; Caricato, E.A.; Van Berge, P.J.; Van de Loosdrecht, J. Support modification of cobalt based slurry phase Fischer-Tropsch catalyst. Stud. Surf. Sci. Catal. 2000, 143, 55–65. [Google Scholar]
- Feller, A.; Claeys, M.; Van Steen, E. Cobalt Cluster Effects in Zirconium Promoted Co/SiO2 Fischer—Tropsch Catalysts. J. Catal. 1999, 185, 120–130. [Google Scholar] [CrossRef]
- Zhang, H.; Lancelot, C.; Chu, W.; Hong, J.; Khodakov, A.Y.; Chernavskii, P.A.; Zheng, J.; Tong, D. The nature of cobalt species in carbon nanotubes and their catalytic performance in Fischer–Tropsch reaction. J. Mater. Chem. 2009, 19, 9241–9249. [Google Scholar]
- Davis, B.H. Fischer—Tropsch Synthesis: Comparison of Performances of Iron and Cobalt Catalysts. Ind. Eng. Chem. Res. 2007, 46, 8938–8945. [Google Scholar] [CrossRef]
- Yao, Y.; Hildebrandt, D.; Glasser, D.; Liu, X. Fischer—Tropsch Synthesis Using H2/CO/CO2 Syngas Mixtures over a Cobalt Catalyst. Ind. Eng. Chem. Res. 2010, 49, 11061–11066. [Google Scholar] [CrossRef]
- Yao, Y.; Liu, X.; Hildebrandt, D.; Glasser, D. The effect of CO2 on a cobalt-based catalyst for low temperature Fischer—Tropsch synthesis. Chem. Eng. J. 2012, 193–194, 318–327. [Google Scholar] [CrossRef]
- Warayanon, W.; Tungkamani, S.; Sukkathanyawat, H.; Phongaksorn, M.; Ratana, T.; Sornchamni, T. Effect of Manganese Promoter on Cobalt Supported Magnesia Catalyst for Fischer-Tropsch Synthesis. Energy Procedia 2015, 79, 163–168. [Google Scholar] [CrossRef]
- Vosoughi, V.; Badoga, S.; Dalai, A.K.; Abatzoglou, N. Effect of Pretreatment on Physicochemical Properties and Performance of Multiwalled Carbon Nanotube Supported Cobalt Catalyst for Fischer − Tropsch Synthesis. Ind. Eng. Chem. Res. 2016, 55, 6049–6059. [Google Scholar] [CrossRef]
- Monshi, A.; Foroughi, M.R.; Monshi, M.R. Modified Scherrer Equation to Estimate More Accurately Nano-Crystallite Size Using XRD. World J. Nano Sci. Eng. 2012, 2, 154–160. [Google Scholar]
- Šťastný, M.; Issa, G.; Popelková, D.; Ederer, J.; Kormunda, M.; Kříženecká, S.; Henych, J. Nanostructured manganese oxides as highly active catalysts for enhanced hydrolysis of bis(4-nitrophenyl)phosphate and catalytic decomposition of methanol. Catal. Sci. Technol. 2021, 11, 1766–1779. [Google Scholar] [CrossRef]
- Saib, A.; Claeys, M.; van Steen, E. Silica supported cobalt Fischer-Tropsch catalysts: Effect of pore diameter of support. Catal. Today 2001, 71, 395–402. [Google Scholar] [CrossRef]
- Khodakov, A.Y.; Bechara, R.; Griboval-Constant, A. Fischer—Tropsch synthesis over silica supported cobalt catalysts: Mesoporous structure versus cobalt surface density. Appl. Catal. A Gen. 2003, 254, 273–288. [Google Scholar] [CrossRef]
- Intarasiri, S.; Ratana, T.; Sornchamni, T.; Tungkamani, S. Pore size effect of mesoporous support on metal particle size of Co / SiO2 catalyst in Fischer-Tropsch synthesis. Int. J. Adv. Appl. Sci. 2018, 5, 80–85. [Google Scholar]
- Song, D.; Li, J. Effect of catalyst pore size on the catalytic performance of silica supported cobalt Fischer—Tropsch catalysts. J. Mol. Catal. A Chem. 2006, 247, 206–212. [Google Scholar] [CrossRef]
- Borg, Ø.; Eri, S.; Blekkan, E.A.; Storsæter, S.; Wigum, H.; Rytter, E.; Holmen, A. Fischer-Tropsch synthesis over γ-alumina-supported cobalt catalysts: Effect of support variables. J. Mol. Catal. A Chem. 2006, 248, 206–212. [Google Scholar] [CrossRef]
- Xiong, H.; Zhang, Y.; Wang, S.; Li, J. Fischer—Tropsch synthesis: The effect of Al2O3 porosity on the performance of Co/Al2O3 catalyst. Catal. Commun. 2005, 6, 512–516. [Google Scholar] [CrossRef]
- Puskas, I. Metal-Support Interactions in Precipitated, Magnesium-Promoted Cobalt-Silica Catalysts. J. Catal. 1992, 135, 615–628. [Google Scholar] [CrossRef]
- Okamoto, Y.; Nagata, K.; Adachi, T.; Imanaka, T.; Inamura, K. Preparation and Characterization of Highly Dispersed Cobalt Oxide and Sulfide Catalysts Supported on SiO2. J. Phys. Chem. 1991, 95, 310–319. [Google Scholar] [CrossRef]
- Ming, H.; Baker, B.G. A: Characterization of cobalt Fischer-Tropsch catalysts I. Unpromoted cobalt-silica gel catalysts. Appl. Catal. A Gen. 1995, 123, 23–36. [Google Scholar] [CrossRef]
- Khodakov, A.; Ducreux, O.; Lynch, J.; Rebours, B.; Chaumette, P. Structural Modification of Cobalt Catalysts: Effect of Wetting Studied by X-Ray and Infrared Techniques. Oil Gas Sci. Technol.—Rev. d’IFP Energies Nouv. 1999, 54, 525–536. [Google Scholar] [CrossRef]
- Stranick, M.A.; Stranick, M.A. Mn2O3 by XPS. J. Surf. Sci. Spectra 1999, 6, 39–46. [Google Scholar] [CrossRef]
- Jahan, M.; Tominaka, S.; Henzie, J. Phase pure α- Mn2O3 prisms and their bifunctional electrocatalytic activity in oxygen evolution and reduction reactions. J. Roy Soc. Chem. 2016, 45, 18494–18501. [Google Scholar] [CrossRef]
- Das, T.K.; Jacobs, G.; Davis, B.H. Fischer—Tropsch synthesis: Deactivation of promoted and unpromoted cobalt—Alumina catalysts. Catal. Lett. 2005, 101, 187–190. [Google Scholar] [CrossRef]
- Sadeqzadeh, M.; Karaca, H.; Safonova, O.; Fongarland, P.; Chambrey, S.; Roussel, P.; Griboval-Constant, A.; Lacroix, M.; Curulla-Ferré, D.; Luck, F.; et al. Identification of the active species in the working alumina-supported cobalt catalyst under various conditions of Fischer—Tropsch synthesis. Catal. Today 2011, 164, 62–67. [Google Scholar] [CrossRef]
- Enache, D.I.; Rebours, B.; Roy-Auberger, M.; Revel, R. In Situ XRD Study of the Influence of Thermal Treatment on the Characteristics and the Catalytic Properties of Cobalt-Based Fischer—Tropsch Catalysts. J. Catal. 2002, 205, 346–353. [Google Scholar] [CrossRef]
- Ducreux, O.; Rebours, B.; Lynch, J.; Roy-Auberger, M.; Bazin, D. Microstructure of Supported Cobalt Fischer-Tropsch Catalysts. Oil Gas Sci. Technol.—Rev. d’IFP Energies Nouv. 2008, 64, 49–62. [Google Scholar] [CrossRef]
- Gnanamani, M.K.; Jacobs, G.; Shafer, W.D.; Davis, B.H. Fischer—Tropsch synthesis: Activity of metallic phases of cobalt supported on silica. Catal. Today 2013, 215, 13–17. [Google Scholar] [CrossRef]
- Pedersen, E.; Svenum, I.-H.; Blekkan, A. Mn promoted Co catalysts for Fischer-Tropsch production of light olefins—An experimental and theoretical study. J. Catal. 2018, 361, 23–32. [Google Scholar] [CrossRef]
- Tyson, W.R. Surface free energies of solid metals” estimation from liquid. J. Surf. Sci. 1977, 62, 267–276. [Google Scholar] [CrossRef]
- Akbarzadeh, O.; Mohd Zabidi, N.A.; Aljunid Merican, Z.M.; Sagadevan, S.; Kordijazi, A.; Das, S.; Amani Babadi, A.; Ab Rahman, M.; Hamizi, N.A.; Abdul Wahab, Y.; et al. Effect of Manganese on Co—Mn/CNT Bimetallic Catalyst Performance in Fischer—Tropsch Reaction. J. Symmetry 2019, 11, 1328. [Google Scholar] [CrossRef]
- Breejen, J.P.D.; Frey, A.M.; Yang, J.; Holmen, A.; van Schooneveld, M.M.; de Groot, F.M.F.; Stephan, O.; Bitter, J.H.; de Jong, K.P. A Highly Active and Selective Manganese Oxide Promoted Cobalt-on-Silica Fischer—Tropsch Catalyst. Top. Catal. 2011, 54, 768–777. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).