Abstract
In this study, methyl/tbutyl salicylate-bearing zirconium complexes (C1–C8) were prepared by the reaction of zirconium (IV) propoxide/butoxide with salicylic acid, 3-methylsalicylic acid, 4-methylsalicylic acid, and 3,5-di-tert-butylsalicylic acid in alcohols, respectively. All these complexes (C1–C8) were characterized by 1H NMR, 13C NMR, FTIR, mass spectroscopy (MS), elemental, and thermogravimetric analyses (TGA). These complexes were utilized as catalysts in the ring-opening polymerization (ROP) of Ɛ-caprolactone and were very effective. Polycaprolactone (PCL) was characterized by 1H-NMR, 13C NMR, and gel permeation chromatography (GPC). In this study, perhaps for the first time, the effects of electron-donating substituents (Me and tBu) on Ɛ-caprolactone polymerization reactions on salicylate ligands linked to zirconium atoms were investigated.
1. Introduction
Polycaprolactone (PCL) has attracted a great deal of interest from industry and academia due to a number of different chemical and physical properties. In addition, its biodegradability and biocompatibility are also remarkable, which is why it is used in medicine for drug delivery, biomaterial production, and many more applications [1,2,3,4]. In this context, the synthesis of biodegradable PCL with a certain molecular weight is of great interest, whether for the mechanical support of PCL in a timeframe compatible with the restoration of injured tissue or for the controlled release of drugs [5,6]. For the synthesis of PCL under mild conditions, modified single-ended metal alcoholates have an important place. For this purpose, modifications of metal alkoxides using pentenoate, perfluoroheptanoate, pivalate, phthalate, picolinate or similar ligands have been carried out to adjust the reactivity of starting metal alkoxides and to reduce the number of bound alkoxy groups [7,8,9,10]. With such complexations, new molecular compounds exhibiting a different molecular structure and reactivity have been obtained and used as catalysts. But the need for catalysts with new properties is increasing day by day. Zirconium carboxylates prepared using the sol–gel method were utilized as starting materials in the synthesis of inorganic–organic hybrid materials employed in many fields [11,12,13,14]. Despite the wide range of applications of zirconium–organic compounds, there is not enough structural information on substituted salicylate zirconium compounds containing electron donor groups. Such compounds are used as selective catalysts with high activity in ring-opening polymerization.
When carboxylate-zirconium-alkoxide is used as a catalyst, only alkoxy groups take an active role as the polymerization-initiating group in ring-opening polymerization since carboxylate groups are more stably bound to the metal as chelates. The second advantage of the carboxylate ligand is that it gives a substitution reaction with alkoxy groups, which is an important parameter in adjusting the number of alkoxy groups bound to the metal. In studies involving many zirconium derivatives, the open structures of the complexes, the binding mode of the ligands, and the composition of the complexes depending on the experimental conditions have not been determined. Therefore, there is a need to develop new active and stable catalysts to be used in the field of ROP. As it is known, the synthesis of single-site catalysts is an important criterion for the growth of polymers from a single end and the formation of linear and uniform polymers [15,16,17,18,19].
The first aim of this study is to synthesize new compounds between salicylate ligands containing electron donor groups and zirconium propoxide/butoxide starting materials, and to characterize the resulting compounds. The second aim is to use these complexes as ROP catalysts for the polymerization of a Ɛ-CL monomer under mild ambient conditions. The third objective was to study the effects of the substituted groups (Me and tBu) and their position (3, 4, and 5) on the salicylate ligand over the polymerization reaction of Ɛ-CL.
2. Experimental Section
2.1. Materials and Measurements
Zirconium(IV) n-propoxide (70% in 1-propanol, Aldrich, Darmstadt, Germany), Zirconium(IV) n-butoxide (80% in 1-butanol, Aldrich), salicylic acid (SAH, 98%, Aldrich), 3-methylsalicylic acid (3-Me-SAH, 97%, Aldrich), 4-methylsalicylic acid (4-Me-SAH, 99%, Aldrich), 3,5-di-tert-butylsalicylic acid (3,5-tBu2-SAH, 99%, Aldrich), Ɛ-caprolactone (Ɛ-CL, 99%, Alfa Aesar, Germany), and tetrahydrofuran (THF, 99.9%, Merck, Darmstadt, Germany) were used as received. 1-Propanol (99%, Merck) and 1-butanol (99%, Merck) were dried over activated 4A° molecular sieves. All syntheses without polymers were carried out in closed vessels under ambient atmosphere.
1H and 13C{1H}NMR samples were measured by a Bruker 400 MHz spectrometers. Infrared spectra of zirconium complexes (C1–C8) were carried out with a Brucker Tensor 27 Fourier-transform infrared spectrophotometer (FTIR) equipped with single reflection ATR using diamond crystal in wavenumber from 400 to 4000 cm−1 at a resolution of 4 cm−1. The elemental analyses were performed on a LECO CHNS-932 elemental analyzer. Mass spectrometry (Waters SYNAPT, HRMS) was employed to measure the molecular masses of zirconium complexes (C1–C8) using the electrospray ionization (ESI±) method. Thermogravimetric analysis was carried out with a Perkin Elmer Pyris 1 TGA. All samples (C1–C8) were heated between 25 °C and 790 °C at a heating rate of 10 °C/min in synthetic air flow. Colorimetric measurements for PCL were performed by differential scanning calorimetry (PerkinElmer DSC 8000). The PCL sample was heated in N2 atmosphere from −80 °C to +80 °C at a heating rate of 10 °C per minute and thus measurements were performed. Gel permeation chromatographic (GPC) analysis was performed on a Shimadzu prominence GPC system equipped with a refractive index detector (RID-10A), a solvent delivery unit (LC-20AD), a column oven (CTO-10AS), and a set of two columns, PSS SDV 5 µL 1000 A° and PSS SDV 5 µL 50 A°. THF (99.9%) was used as the mobile phase at 1.0 mL/min. PCL concentration and the injection volume were ~2 mg/mL and 50 µL, respectively. The calibration curve was plotted with several polystyrene standards including the molecular weight range 162 to 67,000 Da.
2.2. Preparation of [(SA-OH)2(SA)Zr2(OnPr)2O] (C1)
Salicylic acid (1.05 g, 7.60 mmol) was added to the solution of zirconium n-propoxide (1.78 g, 3.81 mmol) in 30 mL n-propanol. The reaction mixture was stirred at room temperature (RT) for 3 h. Then, the volatiles were removed from the solution by vacuum evaporator at 50 °C for 3 h. The resulting white solid product was then washed three times with hexane and dried in an evaporator under reduced pressure. Elemental analysis (EA) (C27H28O12Zr2, (SA-OH)2(SA)Zr2(OnPr)2O, Mw = 726.95 g/mol): Calc. C 44.61, H 3.88%. Found: C 42.84, H 3.16%. MS: C27H28O12Zr2 = 723.97, C27H28O12Zr2 = 713.42 Da. 1H NMR (CDCl3) δ/ppm: 0.91 (t, 6H, J = 7.4 Hz, CH3, OnPr), 1.61 (sext, 4H, J = 7.4 Hz, CH2, OnPr), 3.35 (t, 4H, J = 6.6 Hz, OCH2, OnPr), 7.03 (brd, 2H, H-3, ph), 7.06 (t, 2H, J = 5.6 Hz, H-5, ph), 7.33 (brd, 2H, H-4, ph), 7.90 (d, 2H, J = 6.32 Hz, H-6, ph), 10.5 (s, 2H, 2OH). 13C NMR (CDCl3) δ/ppm: 10.35 (CH3, OnPr), 25.57 (CH2, OnPr), 62.43 (OCH2, OnPr), 111.7 (C-1, ph), 117.6 (C-3, ph), 119.5 (C-5, ph), 130.78 (C-6, ph), 162.1 (C-2, ph), 173.28 (COO). FTIR (cm−1): 3238 (OH), 3057 (Csp2-H), 2958 (Csp3-H), 1580 (COO, asym), 1523, 1463, 1399 (COO, sym), 1241 (Csp2-O), 1147, 1101, 1031 (Csp3-O), 954, 888, 864, 806, 755.
2.3. Preparation of [(3-Me-SA-OH)2(3-Me-SA)Zr2(OnPr)2O] (C2)
The reaction of 3-methylsalicylic acid (1.22 g, 8.27 mmol) with zirconium n-propoxide (1.82 g, 3.90 mmol) in 30 mL n-propanol was performed under the same experimental conditions as in the previous reaction. The reaction yielded an orange solid product. EA (C30H34O12Zr2, (3-Me-SA-OH)2(3-Me-SA)Zr2(OnPr)2O, Mw = 769.03 g/mol): Calc. C 46.85, H 4.46%. Found: C 45.58, H 3.64%. TGA: % loss= 69.51%. 1H NMR (CDCl3) δ/ppm: 0.84 (t, 6H, J = 7.4 Hz, CH3, OnPr), 1.42 (sext, 4H, J = 7.4 Hz, CH2, OnPr), 2.28 (s, CH3, 3-Me-SA), 3.35 (t, 4H, J = 6.6 Hz, OCH2, OnPr), 6.81 (t, 2H, J = 7.65 Hz, H-5, ph), 7.36 (d, 2H, J = 7.22 Hz, H-4, ph), 7.75 (d, 2H, J = 7.94 Hz, H-6, ph), 10.5 (s, 2H, 2OH). 13C NMR (CDCl3) δ/ppm: 10.35 (CH3, OnPr), 15.8 (CH3, 3-Me-SA), 25.3 (CH2, OnPr), 62.4 (OCH2, OnPr), 113.9 (C-1, ph), 118.6 (C-5, ph), 126.5 (C-3, ph), 128.5 (C-6, ph), 136.8 (C-4, ph), 163.4 (C-2, ph), 174.0 (COO). FTIR (cm−1): 3290 (OH), 3018 (Csp2-H), 2957 (Csp3-H), 2916, 2875, 1601 (COO, asym), 1537, 1460, 1429 (CH3 bending), 1394 (COO, sym), 1239 (Csp2-O), 1191, 1157, 1081, 1005 (Csp3-O), 874, 754, 658.
2.4. Preparation of [(4-Me-SA-OH)2(4-Me-SA)Zr2(OnPr)2O] (C3)
The reaction of 4-methylsalicylic acid (1.22 g, 8.10 mmol) with zirconium n-propoxide (1.86 g, 3.98 mmol) in 30 mL n-propanol was performed under the same experimental conditions as in the previous reaction. The reaction yielded an orange solid product. EA (C30H34O12Zr2, (4-Me-SA-OH)2(4-Me-SA)Zr2(OnPr)2O, Mw = 769.03 g/mol): Calc. C 46.85, H 4.46%. Found: C 45.13, H 3.63%. MS, Calc.: 766.01 Da for C30H34O12Zr2, Found: 764.54 Da for C30H34O12Zr2H +. 1H NMR (CDCl3) δ/ppm: 0.84 (t, 6H, J = 7.4 Hz, CH3, OnPr), 1.49 (sext, 4H, J = 7.4 Hz, CH2, OnPr), 2.48 (s, 6H, CH3, 3-Me-SA), 3.60 (t, 4H, J = 6.6 Hz, OCH2, OnPr), 6.90 (t, 2H, J = 7.65 Hz, H-5, ph), 7.28 (d, 2H, J = 7.22 Hz, H-4, ph), 7.75 (d, 2H, J = 7.94 Hz, H-6, ph), 10.5 (s, 2H, 2OH). 13C NMR (CDCl3) δ/ppm: 10.35 (CH3, OnPr), 21.8 (CH3, 4-Me-SA), 25.5 (CH2, OnPr), 66.8 (OCH2, OnPr), 110.9 (C-1, ph), 117.6 (C-5, ph), 120.5 (C-3, ph), 130.9 (C-6, ph), 146.8 (C-4, ph), 161.1 (C-2, ph), 173.0 (COO). FTIR (cm−1): 3262 (OH), 3018 (Csp2-H), 2961 (Csp3-H), 2922, 2876, 1614 (COO, asym), 1577, 1494, 1437 (CH3 bending and COO, sym), 1382, 1249 (Csp2-O), 1167, 1111, 1038, 1010 (Csp3-O), 957, 866, 781, 754, 704, 619, 527.
2.5. Preparation of [(3,5-But2-SA-OH)2(3,5-But2-SA)Zr2(OnPr)2O] (C4)
The reaction of 3,5-di-tertbutylsalicylic acid (0.5 g, 2.1 mmol) with zirconium n-propoxide (0.45 g, 0.96 mmol) in 30 mL n-propanol was performed under the same experimental conditions as in the previous reaction. The reaction yielded a white solid product. EA (C51H76O12Zr2, (3,5-But2-SA-OH)2(3,5-But2-SA)Zr2(OnPr)2O, Mw = 1063.60 g/mol): Calc. C 57.59, H 7.20%. Found: C 56.65, H 7.53%. Mass Spectra (MS), Calc.: 1060.34 Da for C51H76O12Zr2, Found: 1060.33 Da for C51H76O12Zr2. 1H NMR (CDCl3) δ/ppm: 0.85 (t, 6H, J = 7.4 Hz, CH3, OnPr), 1.42 (sext, 4H, J = 7.4 Hz, CH2, OnPr), 1.48–1.60 (a few singlets, 36H, tertbutyl), 3.64 (t, 4H, J = 6.6 Hz, OCH2, OnPr), 6.90 (t, 2H, J = 7.65 Hz, H-5, ph), 7.28 (d, 2H, J = 7.22 Hz, H-4, ph), 7.40 (d, 2H, J = 9.07 Hz, ph), 11.0 (s, 2H, 2OH). 13C NMR (CDCl3) δ/ppm: 10.4 (CH3, OnPr), 25.6 (CH2, OnPr), 25.8–31.3 (3,5-But2), 64.8 (OCH2, OnPr), 84.0, 86.1, 108.5, 123.4, 130.7, 140.1, 148.7, 165.6 (C-2, ph), 176.0 (COO). FTIR (cm−1): 3203 (OH), 2956 (Csp3-H), 2908, 2871, 1613 (COO, asym), 1534, 1445 (CH3 bending), 1389 (COO, sym), 1361, 1281, 1243 (Csp2-O), 1199, 1148, 1024, 1045, 1006 (Csp3-O), 959, 896, 851, 809, 752, 723, 677, 641, 541.
2.6. Preparation of [(SA-OH)2(SA)Zr2(OnBu)2O] (C5)
Salicylic acid (0.70 g, 5.07 mmol) was added to the solution of zirconium n-butoxide (1.20 g, 2.51 mmol) in 30 mL n-butanol. The reaction mixture was stirred at RT for 3 h. Then, the volatiles were removed from the solution by vacuum evaporator at 50 °C for 3 h. The resulting white solid product was then washed three times with hexane and dried in an evaporator under reduced pressure. EA (C29H32O12Zr2, (SA-OH)2(SA)Zr2(OnBu)2O, Mw = 755.01 g/mol): Calc. C 46.13, H 4.27%. Found: C 45.18, H 3.57%. 1H NMR (CDCl3) δ/ppm: 0.93 (t, 6H, J = 7.4 Hz, CH3, OnBu), 1.25–1.60 (m, 8H, J = 7.4 Hz, CH2, OnBu), 3.70 (t, 4H, J = 6.6 Hz, OCH2, OnBu), 6.91 (brd, 2H, H-3, ph), 6.99 (t, 2H, J = 5.6 Hz, H-5, ph), 7.57 (brd, 2H, H-4, ph), 7.99 (d, 2H, J = 6.32 Hz, H-6, ph), 10.5 (s, 2H, 2OH). 13C NMR (CDCl3) δ/ppm: 13.8 (CH3, OnBu), 18.8 (CH2, OnBu), 34.70 (CH2, OnBu), 62.9 (OCH2, OnBu), 111.7 (C-1, ph), 117.6 (C-3, ph), 119.3 (C-5, ph), 130.8 (C-6, ph), 136.4 (C-4, ph), 162.1 (C-2, ph), 173.28 (COO). FTIR (cm−1): 3238 (OH), 3057 (Csp2-H), 2958 (Csp3-H), 1580 (COO, asym), 1523, 1463, 1399 (COO, sym), 1241 (Csp2-O), 1147, 1101, 1031 (Csp3-O), 954, 888, 864, 806, 755.
2.7. Preparation of [(3-Me-SA-OH)2(3-Me-SA)Zr2(OnBu)2O] (C6)
The reaction of 3-methylsalicylic acid (0.81 g, 5.49 mmol) with zirconium n-butoxide (1.22 g, 2.55 mmol) in 30 mL n-butanol was performed under the same experimental conditions as in the previous reaction. The reaction yielded a white solid product. EA (C32H38O12Zr2, (3-Me-SA-OH)2(3-Me-SA)Zr2(OnBu)2O, Mw = 797.09 g/mol): Calc. C 48.22, H 4.81%. Found: C 46.50, H 4.15%. MS, Calc.: 833.01, 835.01, 837.01 for C32H38O12Zr2 + K, Found: 834.65 for C32H38O12Zr2 + K. 1H NMR (CDCl3) δ/ppm: 0.91 (t, 6H, J = 7.4 Hz, CH3, OnBu), 1.32–1.54 (m, 8H, CH2, OnBu), 2.28 (s, CH3, 3-Me-SA), 3.60 (t, 4H, J = 6.6 Hz, OCH2, OnBu), 6.81 (t, 2H, J = 7.65 Hz, H-5, ph), 7.36 (d, 2H, J = 7.22 Hz, H-4, ph), 7.82 (d, 2H, J = 7.94 Hz, H-6, ph), 10.7 (s, 2H, 2OH). 13C NMR (CDCl3) δ/ppm: 13.6 (CH3, OnBu), 15.8 (CH3, 3-Me-SA), 18.44 (CH2, OnBu), 34.3 (CH2, OnBu), 63.2 (OCH2, OnBu), 110.6 (C-1, ph), 118.6 (C-5, ph), 126.7 (C-3, ph), 128.5 (C-6, ph), 137.3 (C-4, ph), 163.4 (C-2, ph), 174.3 (COO). FTIR (cm−1): 2955 (Csp3-H), 2918, 2868, 1661 (COO, asym), 1328, 1238 (Csp2-O), 1155, 1081, 1005 (Csp3-O), 872, 752, 656, 526.
2.8. Preparation of [(4-Me-SA-OH)2(4-Me-SA)Zr2(OnBu)2O] (C7)
The reaction of 4-methylsalicylic acid (0.78 g, 5.18 mmol) with zirconium n-butoxide (1.21 g, 2.52 mmol) in 30 mL n-propanol was performed under the same experimental conditions as in the previous reaction. The reaction yielded a white solid product. EA (C32H38O12Zr2, (4-Me-SA-OH)2(4-Me-SA)Zr2(OnBu)2O, Mw = 797.09 g/mol): Calc. C 48.22, H 4.81%. Found: C 46.49, H 4.12%. MS: Calc. C32H38O12Zr2 + K=833.01, 835.01, 837.01, Found: 834.65. 1H NMR (CDCl3) δ/ppm: 0.83 (t, 6H, J = 7.4 Hz, CH3, OnBu), 1.03–1.37 (m, 8H, CH2, OnBu), 2.36 (s, CH3, 3-Me-SA), 3.49 (t, 4H, J = 6.6 Hz, OCH2, OnBu), 6.75 (t, 2H, J = 7.65 Hz, H-5, ph), 7.46 (d, 2H, J = 7.22 Hz, H-4, ph), 7.71 (d, 2H, J = 7.94 Hz, H-6, ph), 10.5 (s, 2H, 2OH). 13C NMR (CDCl3) δ/ppm: 12.3 (CH3, OnBu), 19.7 (CH2, OnBu), 21.8 (CH3, 3-Me-SA), 35.7 (CH2, OnBu), 63.0 (OCH2, OnBu), 111.4 (C-1, ph), 117.6 (C-5, ph), 130.5 (C-3, ph), 131.6 (C-6, ph), 146.4 (C-4, ph), 161.3 (C-2, ph), 173.0 (COO). FTIR (cm−1): 3246 (OH), 2957 (Csp3-H), 2927, 2869, 1577 (COO, asym), 1496, 1438, 1381 (COO, asym), 1250 (Csp2-O), 1166, 1108, 1067, 1031 (Csp3-O), 953, 856, 828, 780, 754, 647, 618, 500.
2.9. Preparation of [(3,5-But2-SA-OH)2(3,5-But2-SA)Zr2(OnBu)2O] (C8)
The reaction of 3,5-di-tertbutylsalicylic acid (0.5 g, 2.1 mmol) with zirconium n-butoxide (0.46 g, 0.96 mmol) in 30 mL n-butanol was performed under the same experimental conditions as in the previous reaction. The reaction yielded a yellow solid product. EA (C53H80O12Zr2, (3,5-But2-SA-OH)2(3,5-But2-SA)Zr2(OnBu)2O, Mw = 1091.66 g/mol): Calc. C 58.31, H 7.39%. Found: C 58.26, H 7.56%. TGA: % loss = 83.37%. 1H NMR (CDCl3) δ/ppm: 0.91 (t, 6H, J = 7.4 Hz, CH3, OnBu), 1.05–1.39 (m, 8H, CH2, OnBu), 1.42–1.59 (a few singlets, 36H, tertbutyl), 3.60 (t, 4H, J = 6.6 Hz, terminal-OCH2, OnBu), 7.84–7.28 (d, 6H, J = 9.07 Hz, ph), 11.2 (s, 2H, 2OH). 13C NMR (CDCl3) δ/ppm: 10.1 (CH3, OnBu), 19.7 (CH2, OnBu), 34.3 (CH2, OnBu), 29.5–35.1 (3,5-But2), 62.8 (OCH2, OnBu), 84.0, 86.1, 111.0, 124.5, 131.2, 137.3, 140.6, 159.6 (C-2, ph), 176.1 (COO). FTIR (cm−1): 3176 (OH), 2955 (Csp3-H), 2909, 2870, 1653 (COO, asym), 1613, 1551, 1441 (CH3 bending), 1389 (COO, sym), 1361, 1280, 1241 (Csp2-O), 1198, 1150, 1021, 1067, 1026 (Csp3-O), 940, 851, 807, 721, 670, 640, 541, 514, 478, 431.
2.10. ROP of Ɛ-Caprolactone with the Catalyst [(4-Me-SA-OH)2(4-Me-SA)Zr2(OnBu)2O]
Catalyst C7 (20 mg) was mixed with Ɛ-caprolactone (1.2 mL) in a small flask under N2 gas. The solvent-free or bulk mixture was stirred in a hot plate stirrer at 90–110 °C for 8–24 h as shown in Table 1 and Table 2. 1H NMR (CDCl3, ppm), δ: 4.06 (t, J = 6.64 Hz, εCH2-O), 2.31 (t, J = 7.46 Hz, αCH2-C=O), 1.65 (m, J = 7.34 Hz, β,δCH2), 1.40 (m, J = 7.25 Hz, γCH2). 13C NMR (CDCl3), δ/ppm: 176 (C=O), 68.2 (εCH2O), 34.8 (αCH2), 30.1 (γCH2), 29.2 (δCH2), 23.7 (βCH2), [O=C-αCH2βCH2γCH2δCH2εCH2O-]. The ROP reactions of Ɛ-caprolactone with all new catalysts (C1–C6, C8) were performed as described above and under the same experimental conditions.

Table 1.
Results for Ɛ-CL polymers (at 90 °C) obtained from GPC measurements.
Table 2.
Results for Ɛ-CL polymers (at 110 °C) obtained from GPC measurements.
3. Results and Discussion
Reactions of Zr(OnPr)4 and Zr(OnBu)4 with salicylate derivatives in 1:2 mole ratios in propanol/butanol at RT produced the products of [(SA-OH)2(SA)Zr2(OnPr)2O], [(3-Me-SA-OH)2(3-Me-SA)Zr2(OnPr)2O], [(4-Me-SA-OH)2(4-Me-SA-OH)Zr2(OnPr)2O], [(3,5-But2-SA-OH)2(3,5-But2-SA)Zr2(OnPr)2O], [(SA-OH)2(SA)Zr2(OnBu)2O], [(3-Me-SA-OH)2(3-Me-SA)Zr2(OnBu)2O], [(4-Me-SA-OH)2(4-Me-SA)Zr2(OnBu)2O], and [(3,5-But2-SA-OH)2(3,5-But2-SA)Zr2(OnBu)2O]. These complexes were coded from C1 to C8, respectively. The formulations of the C1–C8 complexes were based on 1H, 13C NMR, FTIR, mass (MS), and TGA measurements as well as the results of elemental analysis. When salicylic acid derivatives were added to the starting zirconium alkoxide solutions, some of the propoxy or butoxy groups in the starting zirconium-based precursors were replaced by salicylate groups and some of them were condensed to form oxo groups depending on the reaction time and environment. The presence and number of oxo groups were determined by mass measurements, elemental and thermogravimetric analysis. 1H NMR spectra of salicylate zirconium compounds confirmed the expected peaks, peak areas, and peak multiplicities for the organic groups as suggested in the formulation. For instance, 1H NMR spectrum of [(SA-OH)2(SA)Zr2(OnPr)2O] (C1) demonstrated triplets at 0.91 ppm for CH3 (J = 7.4 Hz) protons, sextet at 1.61 ppm for CH2 protons, triplet at 3.35 ppm for OCH2 of OnPr, doublets or triplets at 6.32–7.90 ppm for salicylate protons, and around at 10.5 ppm for non-bonded OH proton (Figure S1). The fact that salicylic acid, which gives a carboxyl (COOH) peak at ~11 ppm before coordinating to zirconium, does not give a peak at ~11 ppm after coordinating specifies that the salicylate is fully bonded to zirconium via the carboxyl group. However, the appearance of two proton peaks belonging to OH peaks at around 10.5 indicated that only one unit of salicylate was connected to both carboxyl and OH. The other two were bonded only through COO groups. Substituted phenyl protons were also present in the 1H NMR spectrum as given above.
The 13C NMR spectra of the zirconium compounds showed some shifts compared to those of the free salicylate derivatives. These shifts were more predominant at the carboxyl carbons (COO) and at the carbons containing the OH group (C-OH) bound to zirconium. For example, the 13C NMR spectrum of [(3-Me-SA-OH)2(3-Me-SA)Zr2(OnBu)2O] gave peaks at 173.3 ppm for C=O and 162.1 ppm for C-O (3-Me-C5H3-C-OH) as seen in Figure S2. These peak values were different from the free ligand values. The chemical shift values (δ) and coupling constants (J) reported in the experimental part are in agreement with the published literature values for similar carboxylate or salicylate compounds [19].
As shown in Scheme 1, the binding of carboxylate groups as chelate and bidentate was inferred from the FTIR measurements of the C1–C8 compounds. FTIR spectra of free salicylic acid and its derivatives show characteristic intense bands at ~1670 and 1440 cm−1 corresponding to asymmetric and symmetric stretching vibrations of carboxyl groups.
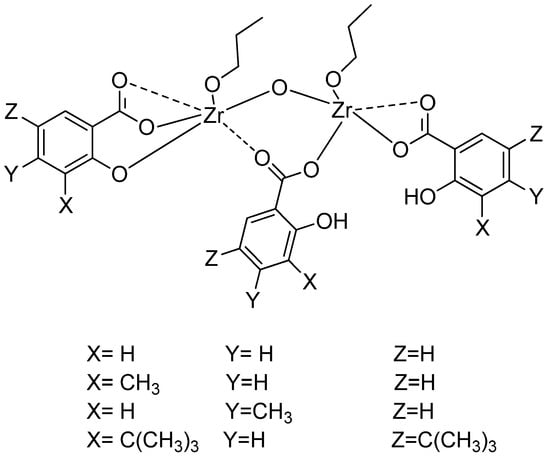
Scheme 1.
The open structures of compounds C1–C4.
After the reaction of salicylate derivatives with zirconium alkoxides, the asymmetric COO vibration band at ~1670 cm−1 appears in the ~1570–1620 cm−1 region, i.e., at lower wave number or energy (Figure 1). For instance, the coordinated carboxylate bands for C3 appeared at ~1614 cm−1 for νCOOasym and ~1437 cm−1 for νCOOsym. The Δυasym-sym value (177 cm−1) was smaller than 220 cm−1, indicating that zirconium atoms and the carboxylate group of salicylate were bonded in the bidentate and chelate coordinate mode, i.e., not mono dentate [20]. The IR spectra of catalysts C1–C8 also showed a broad peak at ~3175–3300 cm−1 because of the O-H stretching, indicating the presence of hydroxide group on the salicylate ligand which was not linked to the zirconium atom. All these values are in agreement with those given for a number of transition metal–carboxylate complexes in the literature [21].
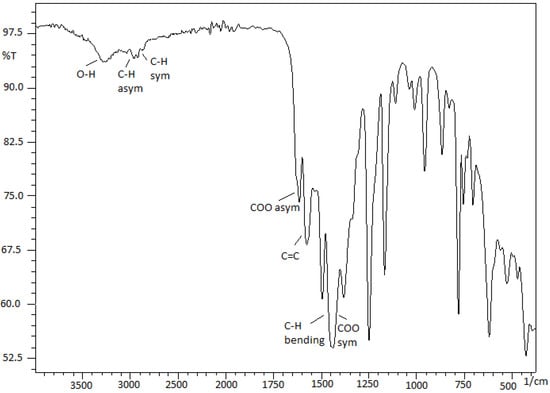
Figure 1.
FTIR spectrum of C3.
The masses of salicylate or substituted salicylate zirconium complexes analyzed under positive ionization conditions were determined using high-resolution mass spectrometry (HRMS). The m/z spectrum of [(SA-OH)2(SA)Zr2(OnPr)2O] complex gave molecular ion at m/z 713.42 and 714.42 Da which was 12 Da lower than the predicted protonated ion, (SA-OH)2(SA)Zr2(OnPr)2O + H]+ or [C27H28O12Zr2 + H]+ (Figure 2). At this stage, the formula to be proposed for this complex is the dimeric compound of [C27H28O12Zr294 + H]+. This proposed dimeric formula was also supported by the percentages of carbon and hydrogen found by elemental analysis and the ratios of hydrogen areas found by 1H NMR results. However, the reaction of zirconium propoxide with salicylate under different experimental conditions was formulated as Zr10O6(OH)4(OOC-C6H4OH)8(OOC-C6H4O)8·6PrOH by U. Schubert and co-workers [22]. Different formulations of salicylate zirconium complexes depend on several experimental parameters like carboxylate/zirconium alkoxy ratio, reaction times, hydrolysis, crystallization conditions, solvent volatilization conditions and solvents used. The parameters of the experiments influence which types of clusters are formed. Changing this parameter allows the synthesis of cluster-type compounds of different sizes and shapes.
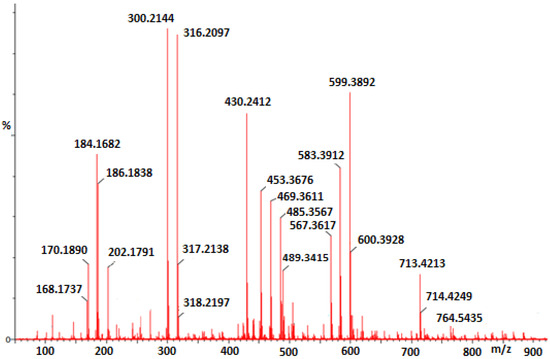
Figure 2.
Mass spectrum of C1.
Based on mass spectroscopy measurements it was suggested to be a dimer, but the difference of about 12–16 Da in the measurement results is perhaps evidence that these compounds may be tetramers or even hexamers. A similar result was observed in other mass measurements (Figure S3), so it is an indication that it has broken over the oxygen bridge. Since it is very difficult to prepare single crystals, the simplest unit was estimated from mass measurements.
Thermogravimetric measurements of the compounds were also taken and interpreted to support or confirm the proposed C1–C8 formulas by elemental analysis, 1H-NMR, and mass measurements. For instance, the thermal decomposition graph of compound-2 (C2), TGA of which was taken under artificial air flow, showed that it was stable up to 250 °C, with only a 10% weight loss up to this temperature. This may be a result of the removal of moisture and solvent impurities. Thermogravimetric analysis (TGA) of compound-2 showed that the weight loss was considerable up to 580 °C, with a small amount from 580 °C to 700 °C. Another way of putting it is that the complete degradation of compound-2 to ZrO2 continued until about 700 °C (calc. ZrO2 weight was 32.05%). As seen in Figure 3, the total weight loss up to 790 °C was found to be 69.51% by TGA (weight loss calculated from the proposed formula was 67.95%).
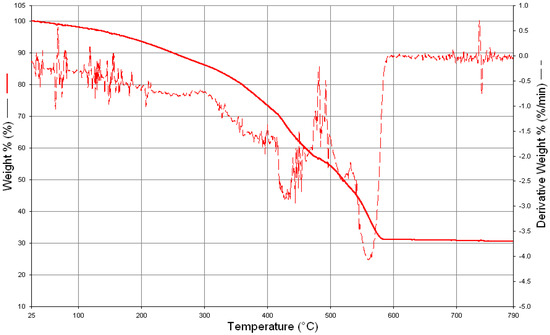
Figure 3.
TGA of [(3-Me-SA-OH)2(3-Me-SA)Zr2(OnPr)2O] compound (C2).
The difference between the calculated and measured weight loss based on the proposed formula (69.51–67.95 = 1.56%) also supported the proposed formula of compound-2. As in the TGA measurements, the elemental analysis results of the complexes deviate slightly from the theoretical values (0.5–1.5% for carbon). This is due to the fact that the starting materials are very reactive and a very small amount of them are converted into oxide (ZrO2) in the complexation process, which is not desirable. Due to the low solubility of both complexes and ZrO2, 100% purification may not be achieved. This caused small deviations in the elemental analysis measurements.
All new compounds were characterized and formulated similar to the above zirconium compounds. After all these characterizations, the structures of compounds (C1–C4) can be drawn as seen in Scheme 1. The open structure of C5–C8 is the same as C1–C4, except that butoxide groups are present instead of propoxide groups.
A brief comparison between the compounds reported in this paper and those in our previous study [19] shows that in this study, both the propoxide and butoxide compounds of zirconium were complexed with a series of ligands containing electron-donating groups (Me and tBu) on salicylate and active catalysts were synthesized. In our previous study, only the reaction of zirconium propoxide with salicylate ligands containing electron-withdrawing groups (such as Cl−, NO2−, and OH−) was investigated. As expected, a slight difference in the structure, i.e., formulation, of the compounds was noted, which is due to the reactivity of the ligands and the slightly different experimental conditions. In this study, elemental analysis, 1H-NMR and mass measurements showed that there is an oxo group and that this oxo group may be between two zirconium as in the literature studies [11,22]. As mentioned above, we can estimate the probability of an oxo between two zirconium atoms using 1H-NMR data. The fact that the salicylate groups attached to zirconium have the same chemical shift values in the 1H-NMR spectra indicates that the structure is symmetric or that the organic groups are equally bonded to the zirconium atoms. In other words, it shows that the chemical environment of zirconium is the same. Therefore, this indicates that the oxo group may be between two Zr atoms, not on a single atom as in the literature [11,22].
These zirconium compounds (C1–C8) were utilized as catalysts to initiate the ring-opening polymerization (ROP) of Ɛ-CL under different experimental conditions. In ROP reactions using single-ended metal alkoxide compounds as catalysts, alkoxide groups are the active groups initiating the polymerization. In methy/tbutyl-salicylate zirconium compounds, the polymerization of Ɛ-caprolactone by n-propoxide or n-butoxide group was also initiated. The chemical and physical properties of PCL polymers obtained from ROP of Ɛ-CL were determined by 1H, 13C NMR, FTIR, and GPC measurements. In the 1H NMR spectra of polycaprolactone (PCL), peaks were assigned as follows: δ 4.07 ppm (Hε), δ 2.31 ppm (Hα) δ 1.39 (Hγ) and 1.66 ppm (Hβ+δ), characterizing the polymer chain (Figure 4). (Pure Ɛ-caprolactone: 1H NMR (CDCl3) δ: 4.23 (m, ε-CH2), 2.64 (m, α-CH2), 1.86 (m, δ-CH2), 1.77 (m, β-CH2 + γ-CH2)). In the 13C NMR spectrum of PCL, peaks were assigned as follows: 173.79 (C=O), 64.38 (εCH2O), 34.34 (αCH2), 28.57 (δCH2), 25.75 (βCH2), 24.80 (γCH2). [O=C-αCH2βCH2γCH2δCH2εCH2O-]. These values are consistent with those in the literatures [23,24].
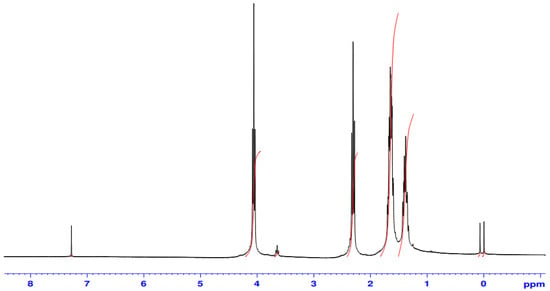
Figure 4.
1H NMR spectrum of PCL polymerized by C7.
The improvement in “single-site” metal alkoxide catalysts has been an important target to understand the mechanism of ring-opening polymerization (ROP) and to produce polymers with controllable molecular weights and polydispersities [25,26]. Single-site metal alkoxide compounds containing different auxiliary ligands were synthesized and used as efficient initiators with high catalytic activities for the controlled polymerization of Ɛ-CL [26,27]. Considering the catalytic advantages of single-site metal alkoxy complexes, new catalysts have been synthesized between salicylate ligands containing electron-donating groups and zirconium alkoxides. Since C1–C8 compounds have an active alkoxide group (one alkoxide for every Zr atom), these complexes were utilized as catalysts for activity towards ROP of Ɛ-CL. As shown in Scheme 2, when Ɛ-CL is added to zirconium catalysts at room temperature and after stirring for a while, the 1HNMR spectrum shows that the Ɛ-caprolactone and the alkoxide group bind together to the zirconium atom. However, polymerization does not occur at this temperature. When the amount of Ɛ-caprolactone is increased and the temperature is raised, the reaction proceeds as described in the mechanism below.
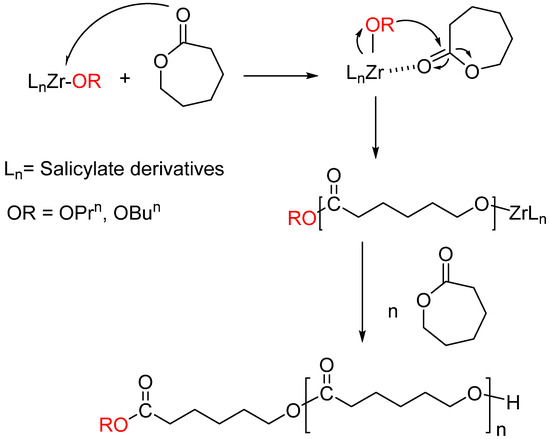
Scheme 2.
Polymerization mechanism between Ɛ-caprolactone and zirconium catalysts (C1–C8).
The reaction mechanism starting from Ɛ-caprolactone and continuing until PCL formation is shown in Scheme 2. Zirconium complexes bearing salicylate ligands (C1–C8) performed ROP of Ɛ-caprolactone through a coordination–insertion mechanism. Interpretation of the results obtained from spectroscopic measurements shows that Ɛ-CL first attacks the zirconium atom, and then the nucleophile OnPr− or OnBu− ion attacks the O-C=O carbon atom in Ɛ-CL, and the reaction proceeds in this way [8].
GPC was used to determine the percentage conversion of Ɛ-CL monomers to PCL, whether the polymer formed was viable, whether it contained different phases, the molecular weight (Mw) and molecular weight distribution index (PDI: Mw/Mn) of PCL. The PCL with different average Mw or number average Mn were obtained at 90–110 °C for different hours stirring (Table 1 and Table 2). For PCL prepared by stirring with catalyst C7 at 110 °C for 24 h, the main peak appeared at 18,905 Da for weight average molecular weight (Mw) and at 14,617 Da for number average molecular weight (Mn). The ratio of the Mw/Mn was 1.29 (Figure S4). As can be seen from Table 1 and Table 2, in addition to temperatures and times, the molecular weights of PCL also changed due to the change in the position and composition of the electron-donating groups on the salicylate derivatives connected to zirconium. In conclusion, both the electron-donating power and resonance effects of functional organic groups (Me, tBu, OH, and COO) on the salicylate parts were important for the activity of the alkoxy group attached to the central zirconium atom. Due to the electron-donating groups, electrons increase in both the salicylate ligand and zirconium center. As the electron-donating tendency of the substituent (Me and tBu) on the salicylate ligand increases, the Lewis acidity of the zirconium center will decrease. Hence, Ɛ-CL monomers will be somewhat slower to readily attack the zirconium center to initiate ring-opening polymerization of Ɛ-CL compared to those containing electron-withdrawing groups. But the easiest and most controlled way to synthesize a ~30,000 Da PCL is to work with electron server groups. The steric effect may be the reason why the catalysts (C4 and C8) containing tert-butyl group are weaker in polymerization. Due to the large steric effect, the Ɛ-CL monomer has difficulty in binding to the zirconium center. When the substituted groups on the salicylate and other experimental parameters are the same, if there is a difference in activity in the catalyst reaction, it is due to the difference in alkoxy groups attached to the zirconium. For example, the C2 catalyst formed a PCL of 9150 Da in 24 h at 110 °C, while the C6 catalyst formed a PCL of 5110 Da under the same conditions. The only reason for this difference was the different alkoxy groups attached to zirconium. Zirconium compounds (C1–C8) containing salicylate ligands with electron-donating groups on them were effective catalysts for the synthesis of PCL from Ɛ-caprolactone by ring-opening polymerization with average size compared with known metal alkoxy catalysts. PCLs with an average size of 10,000–30,000 Da are used in important areas such as drug release because they dissolve more easily compared to large-molecule PCLs [28]. Zirconium compounds of salicylate ligands containing electron-donating groups formed polymers with lower molecular weight and lower poly dispersity indices than Zirconium compounds of salicylate ligands containing electron-withdrawing groups in Ɛ-caprolactone polymerization [19].
DSC analysis was performed to determine the melting point and degree of crystallinity of PCL prepared with C7 (Figure 5). As seen in Figure 5, PCL had a melting temperature at 61.14 °C, an enthalpy of melting at 88.68 j/g and a glass transition temperature at −63.14 °C.
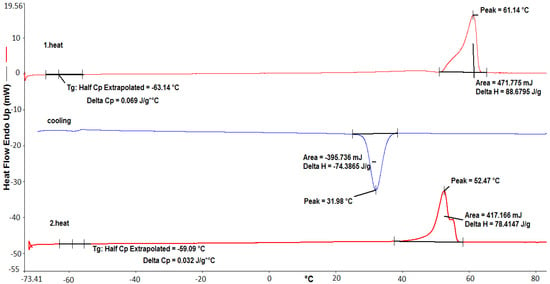
Figure 5.
DSC heating curves of PCL prepared with C7.
In the DSC curve, the initial temperature of crystallization was evident at approximately 37 °C. The crystallization exotherm peak with a maximum appeared at 31.98 °C. The TGA measurement of C8, which exhibits similar behavior, is given in Figure S5. These DSC data are in agreement with published papers on the physical properties of polycaprolactone [29,30].
4. Conclusions
This study demonstrated that salicylate derivatives containing an electron-donating group are suitable ligands for zirconium propoxide/butoxide to form stable complexes containing one alkoxy group per zirconium atom. Eight new zirconium compounds (C1–C8) were synthesized and their structures were elucidated by TGA measurements, elemental analysis, mass measurements, 1H-NMR, 13C-NMR, and FTIR spectroscopy. These zirconium compounds bearing substituted salicylate ligands (C1–C8) were utilized as catalysts for ROP of Ɛ-caprolactone at 90 and 110 °C. The C1–C8 catalysts produced PCL with narrow molecular weight distributions (Mw/Mn = 1.0–1.3), showing the properties of single-site metal alkoxide catalysts. Furthermore, the position of the substituents on the salicylate ligand bound to the zirconium atom was found to be efficient in the polymerization reaction. The structure of PCL was elucidated by NMR spectroscopy, GPC, and DSC. Briefly, these complexes are efficient catalysts for the formation of medium-sized PCL (Mw = ~30,000 Da) polymers by ROP of Ɛ-caprolactone compared to known acid catalysts.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/reactions5030029/s1, Figure S1. 1H NMR spectrum of C1; Figure S2. 13C NMR spectrum of C6; Figure S3. MS spectrum of C7; Figure S4. Gel permeation chromatogram of PCL synthesized with the catalyst C7, (24 h, 110 °C); Figure S5. TGA spectrum of C8.
Author Contributions
G.U.: data curation, investigation, drawing graphics, formal analysis, writing—original draft. A.K.: conceptualization, methodology, resources, supervision, project administration, funding acquisition, writing—review editing, visualization. All authors have read and agreed to the published version of the manuscript.
Funding
This research received funding from Kocaeli University (Project No: 2021/2719).
Data Availability Statement
All data generated or analyzed during this study are included in this published article.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Kayan, G.Ö.; Kayan, A. Polycaprolactone Composites/Blends and Their Applications Especially in Water Treatment. ChemEngineering 2023, 7, 104. [Google Scholar] [CrossRef]
- Pawar, R.; Pathan, A.; Nagaraj, S.; Kapare, H.; Giram, P.; Wavhale, R. Polycaprolactone and its derivatives for drug delivery. Polym. Adv. Technol. 2023, 34, 3296–3316. [Google Scholar] [CrossRef]
- Mandal, P.; Shunmugam, R. Polycaprolactone: A biodegradable polymer with its application in the field of self-assembly study. J. Macromol. Sci. A 2020, 58, 111–129. [Google Scholar] [CrossRef]
- Liu, Z.H.; Li, Y.; Zhang, C.J.; Zhang, Y.Y.; Cao, X.H.; Zhang, X.H. Synthesis of high-molecular-weight poly (ε-caprolactone) via heterogeneous zinc-cobalt (III) double metal cyanide complex. Giant 2020, 3, 100030. [Google Scholar] [CrossRef]
- El Yousfi, R.; Brahmi, M.; Dalli, M.; Achalhi, N.; Azougagh, O.; Tahani, A.; El Idrissi, A. Recent advances in nanoparticle development for drug delivery: A comprehensive review of polycaprolactone-based multi-arm architectures. Polymers 2023, 15, 1835. [Google Scholar] [CrossRef] [PubMed]
- Dias, J.R.; Sousa, A.; Augusto, A.; Bártolo, P.J.; Granja, P.L. Electrospun polycaprolactone (PCL) degradation: An in vitro and in vivo study. Polymers 2022, 14, 3397. [Google Scholar] [CrossRef] [PubMed]
- Yalcin, G.; Yildiz, U.; Kayan, A. Preparation of Al, Ti, Zr-perfluoroheptanoate compounds and their use in ring opening polymerization. Appl. Catal. A Gen. 2012, 423, 205–210. [Google Scholar] [CrossRef]
- Kayan, A. Recent studies on single site metal alkoxide complexes as catalysts for ring opening polymerization of cyclic compounds. Catal. Surv. Asia 2020, 24, 87–103. [Google Scholar] [CrossRef]
- Jansen, J.H.; Powell, A.B.; Specht, S.E.; Gerislioglu, S.; Hermans, I. Understanding the structure and reactivity of mixed titanium (IV) alkoxide and tin (II)/(IV) carboxylates as esterification catalysts. ACS Sustain. Chem. Eng. 2022, 10, 2484–2493. [Google Scholar] [CrossRef]
- Brown, S.E.; Mantaloufa, I.; Andrews, R.T.; Barnes, T.J.; Lees, M.R.; De Proft, F.; Pike, S.D. Photoactivation of titanium-oxo cluster [Ti6O6(OR)6(O2CtBu)6]: Mechanism, photoactivated structures, and onward reactivity with O2 to a peroxide complex. Chem. Sci. 2023, 14, 675–683. [Google Scholar] [CrossRef]
- Schubert, U. En route from metal alkoxides to metal oxides: Metal oxo/alkoxo clusters. J. Sol-Gel Sci. Technol. 2023, 105, 587–595. [Google Scholar] [CrossRef]
- Kayan, G.Ö.; Kayan, A. Inorganic–organic hybrid materials of zirconium and aluminum and their usage in the removal of methylene blue. J. Inorg. Organomet. Polym. Mater. 2021, 31, 3613–3623. [Google Scholar] [CrossRef]
- Du, Q.; Rao, R.; Bi, F.; Yang, Y.; Zhang, W.; Yang, Y.; Zhang, X. Preparation of modified zirconium-based metal-organic frameworks (Zr-MOFs) supported metals and recent application in environment: A review and perspectives. Surf. Interfaces 2022, 28, 101647. [Google Scholar] [CrossRef]
- Zhang, H.; Xiong, P.; Li, G.; Liao, C.; Jiang, G. Applications of multifunctional zirconium-based metal-organic frameworks in analytical chemistry: Overview and perspectives. TrAC Trends Anal. Chem. 2020, 131, 116015. [Google Scholar] [CrossRef]
- Peprah, F.; Tarantola, G.E.; Plaman, A.S.; Vu, E.L.; Huynh, A.B.; Durr, C.B. Synthesis and catalytic activity of single-site group V alkoxide complexes for the ring-opening polymerization of ε-caprolactone. Dalton Trans. 2024, 53, 7073–7080. [Google Scholar] [CrossRef]
- Su, R.R.; Ganta, P.K.; Cheng, C.A.; Hu, Y.T.; Chang, Y.C.; Chang, C.J.; Wu, K.H. Ring-opening polymerization of ε-caprolactone and L-lactide using ethyl salicylate-bearing zinc complexes as catalysts. Mol. Catal. 2023, 537, 112965. [Google Scholar] [CrossRef]
- Buchard, A.; Davidson, M.G.; Gobius du Sart, G.; Jones, M.D.; Kociok-Köhn, G.; McCormick, S.N.; Mckeown, P. Coordination of ε-Caprolactone to a Cationic Niobium (V) Alkoxide Complex: Fundamental Insight into Ring-Opening Polymerization via Coordination–Insertion. Inorg. Chem. 2023, 62, 15688–15699. [Google Scholar] [CrossRef]
- Mankaev, B.N.; Karlov, S.S. Metal complexes in the synthesis of biodegradable polymers: Achievements and prospects. Materials 2023, 16, 6682. [Google Scholar] [CrossRef]
- Mert, O.; Kayan, A. Synthesis and characterization of substituted salicylate zirconium compounds and their catalytic activity over ε-caprolactone. J. Incl. Phenom. Macrocycl. Chem. 2014, 80, 409–416. [Google Scholar] [CrossRef]
- Stefanov, B.I. Optically Transparent TiO2 and ZnO Photocatalytic Thin Films via Salicylate-Based Sol Formulations. Coatings 2023, 13, 1568. [Google Scholar] [CrossRef]
- Mao, Y.; Chen, G.H.; Yi, X.; Kang, Y.; Zhang, J.; Zhang, L. Preparation and visible-light response of salicylate-stabilized heterobimetallic Pb–Ti–Oxo clusters initiated via auxiliary quaternary ammonium salts and a solvent effect. Inorg. Chem. 2022, 61, 5017–5024. [Google Scholar] [CrossRef] [PubMed]
- Kickelbick, G.; Schubert, U. Hydroxy carboxylate substituted oxozirconium clusters. J. Chem. Soc. Dalton Trans. 1999, 8, 1301–1306. [Google Scholar] [CrossRef]
- Tuancharoensri, N.; Ross, G.M.; Kongprayoon, A.; Mahasaranon, S.; Pratumshat, S.; Viyoch, J.; Ross, S. In situ compatibilized blends of PLA/PCL/CAB melt-blown films with high elongation: Investigation of miscibility, morphology, crystallinity and modelling. Polymers 2023, 15, 303. [Google Scholar] [CrossRef] [PubMed]
- Limwanich, W.; Meepowpan, P.; Dumklang, M.; Funfuenha, W.; Rithchumpon, P.; Punyodom, W. Non-isothermal kinetics of the organocatalytic ring-opening polymerization of ε-caprolactone with metal-free α-hydroxy acids: Eco-friendly and facile synthesis process. Thermochim. Acta 2024, 736, 179734. [Google Scholar] [CrossRef]
- Rosa, R.P.; Ferreira, F.V.; Lona, L.M. Modeling of Ring Opening Polymerization: A short review with insights on how to develop the method of moments. Chem. Eng. Sci. 2021, 246, 116934. [Google Scholar] [CrossRef]
- Yildiz, B.C.; Kayan, A. Ti (IV)-silyliminophenolate catalysts for ϵ-caprolactone and L-Lactide polymerization. Sustain. Chem. Pharm. 2021, 21, 100416. [Google Scholar] [CrossRef]
- Limwanich, W.; Rakbamrung, N.; Meepowpan, P.; Funfuenha, W.; Kongsuk, J.; Punyodom, W. Solvent-free ring-opening polymerization of ε-caprolactone initiated by Mg (II), Sn (II), Zn (II), Al (III), and Sn (IV) derivatives: A comparative study. React. Kinet. Mech. Catal. 2023, 136, 381–395. [Google Scholar] [CrossRef]
- Malikmammadov, E.; Tanir, T.E.; Kiziltay, A.; Hasirci, V.; Hasirci, N. PCL and PCL-based materials in biomedical applications. J. Biomater. Sci. Polym. Ed. 2018, 29, 863–893. [Google Scholar] [CrossRef]
- Steinman, N.Y.; Bentolila, N.Y.; Domb, A.J. Effect of Molecular Weight on Gelling and Viscoelastic Properties of Poly (caprolactone)–b-Poly (ethylene glycol)–b-Poly (caprolactone)(PCL–PEG–PCL) Hydrogels. Polymers 2020, 12, 2372. [Google Scholar] [CrossRef]
- Mahović Poljaček, S.; Priselac, D.; Tomašegović, T.; Elesini, U.S.; Leskovšek, M.; Leskovac, M. Effect of the Addition of Nano-Silica and Poly (ε-caprolactone) on the Mechanical and Thermal Properties of Poly (lactic acid) Blends and Possible Application in Embossing Process. Polymers 2022, 14, 4861. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).