


Co-Hydroprocessing of Fossil Middle Distillate and Bio-Derived Durene-Rich Heavy Ends under Hydrotreating Conditions



Abstract
:1. Introduction
1.1. Processing of Feedstocks Rich in Heavy Mono-Ring Aromatics
1.2. Modeling of Hydroprocessing
2. Materials and Methods
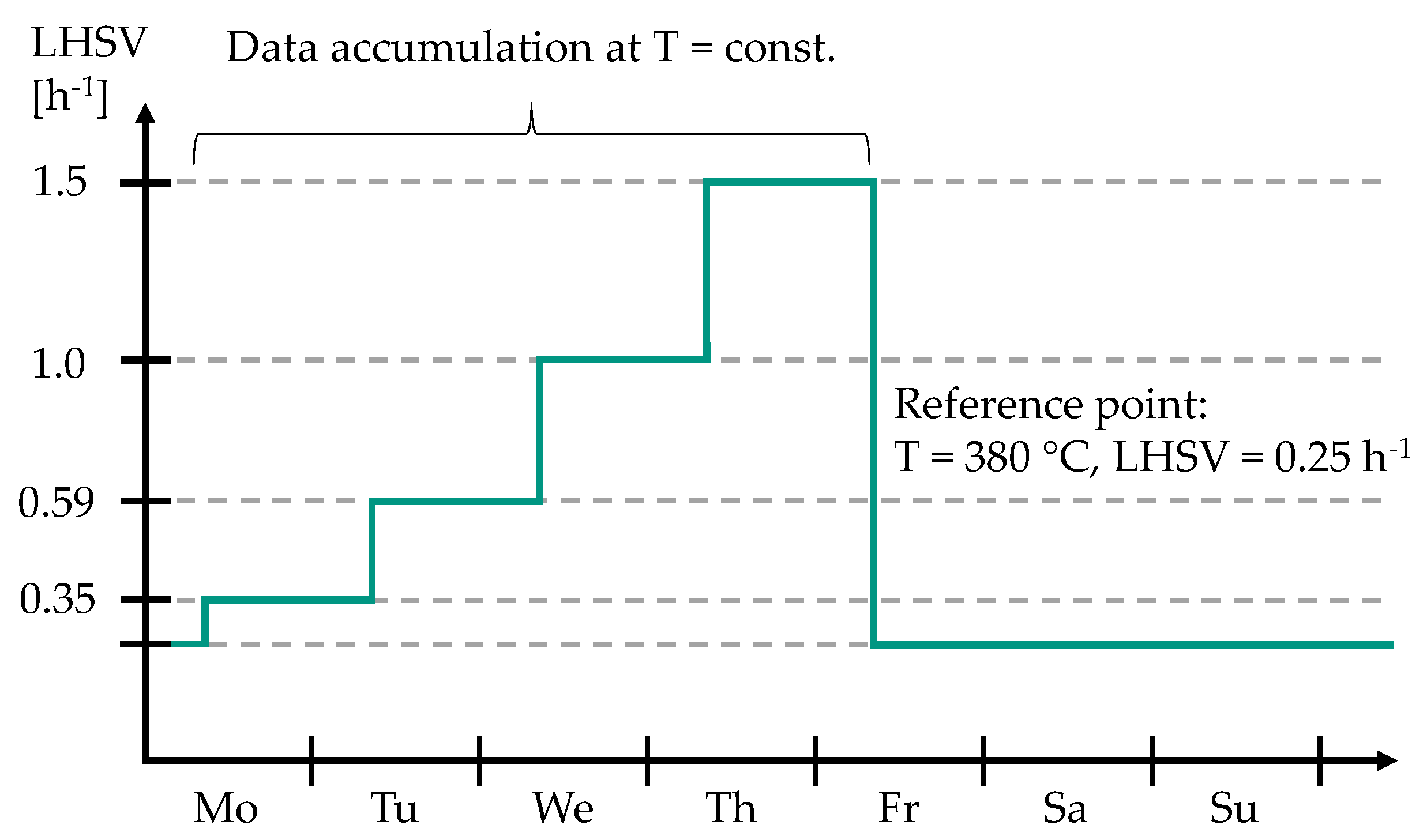
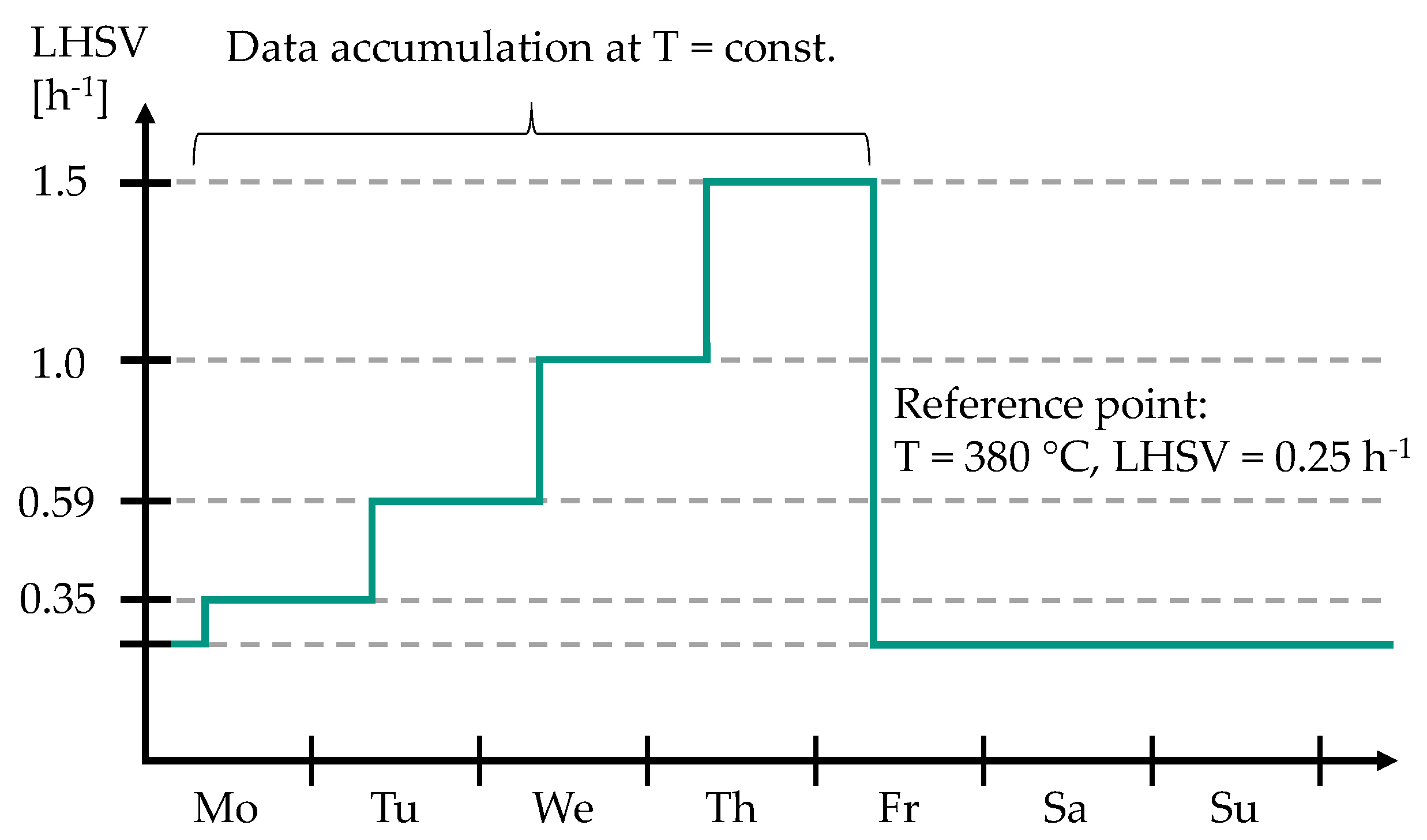
2.1. Experimental Setup and Procedure
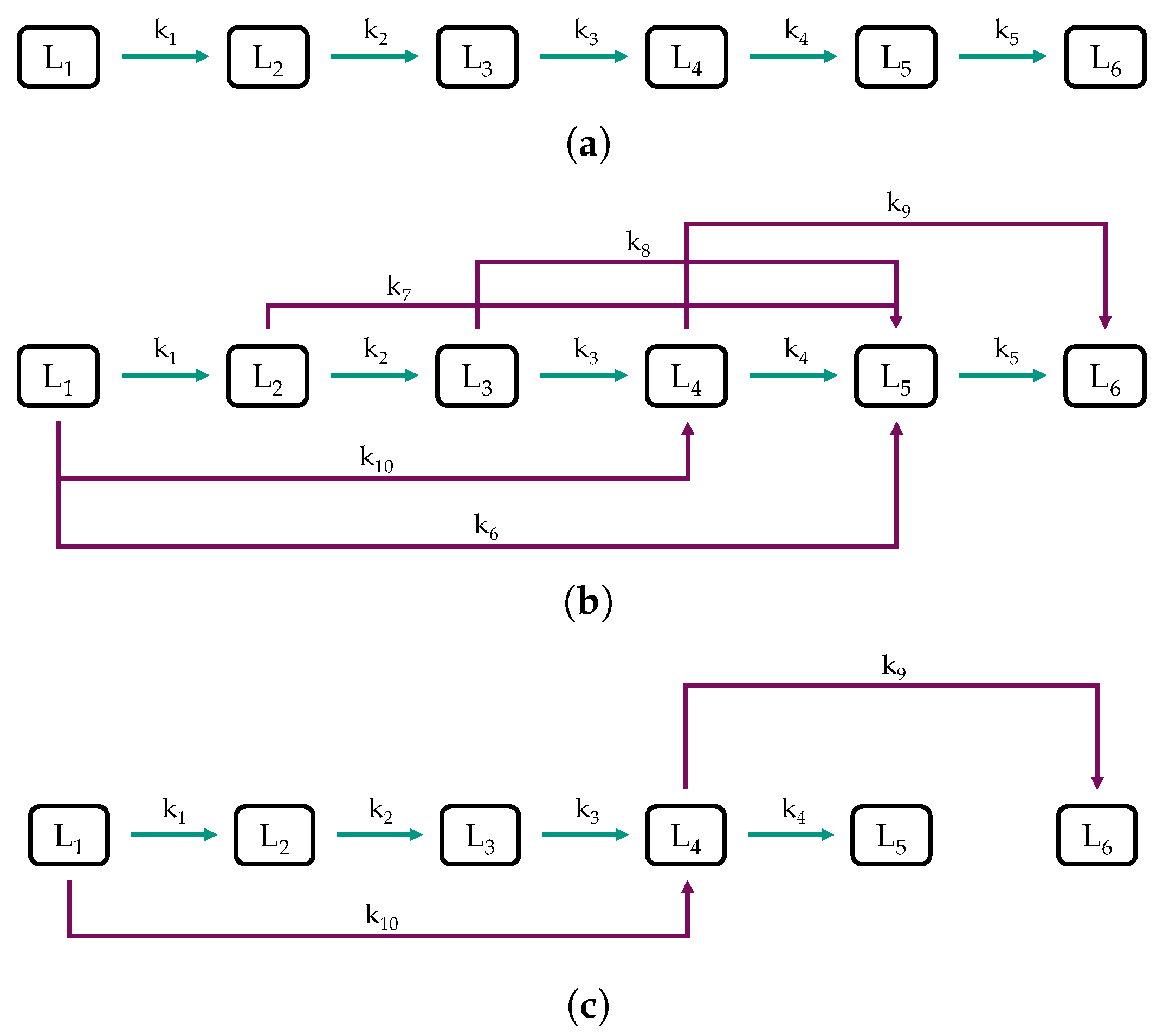
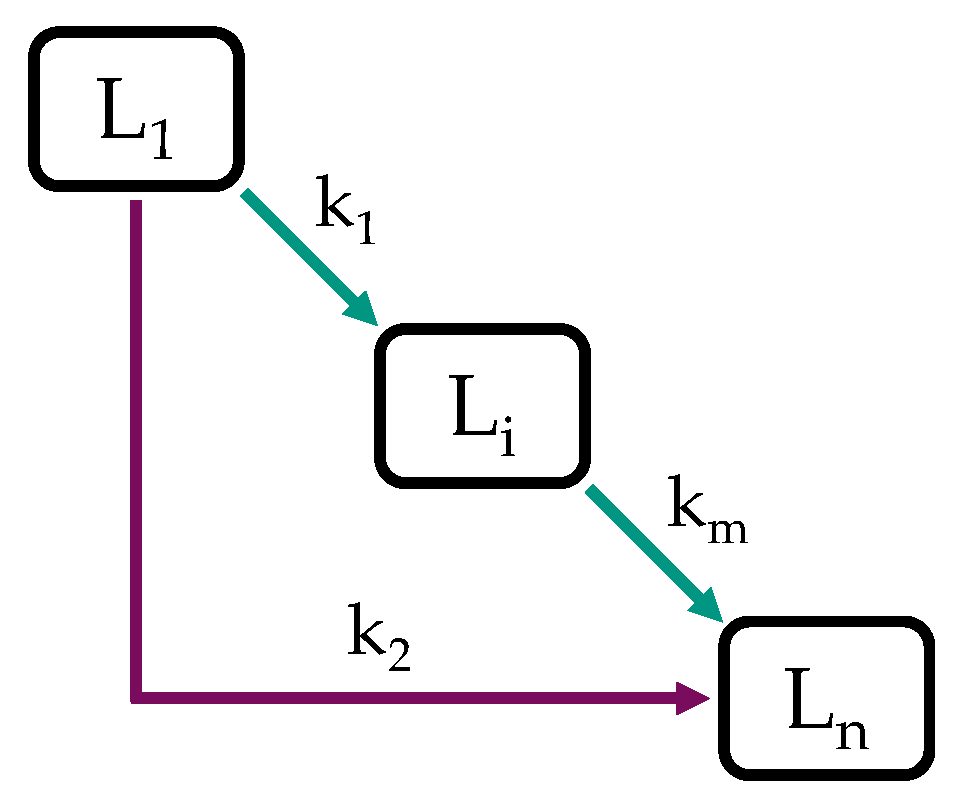
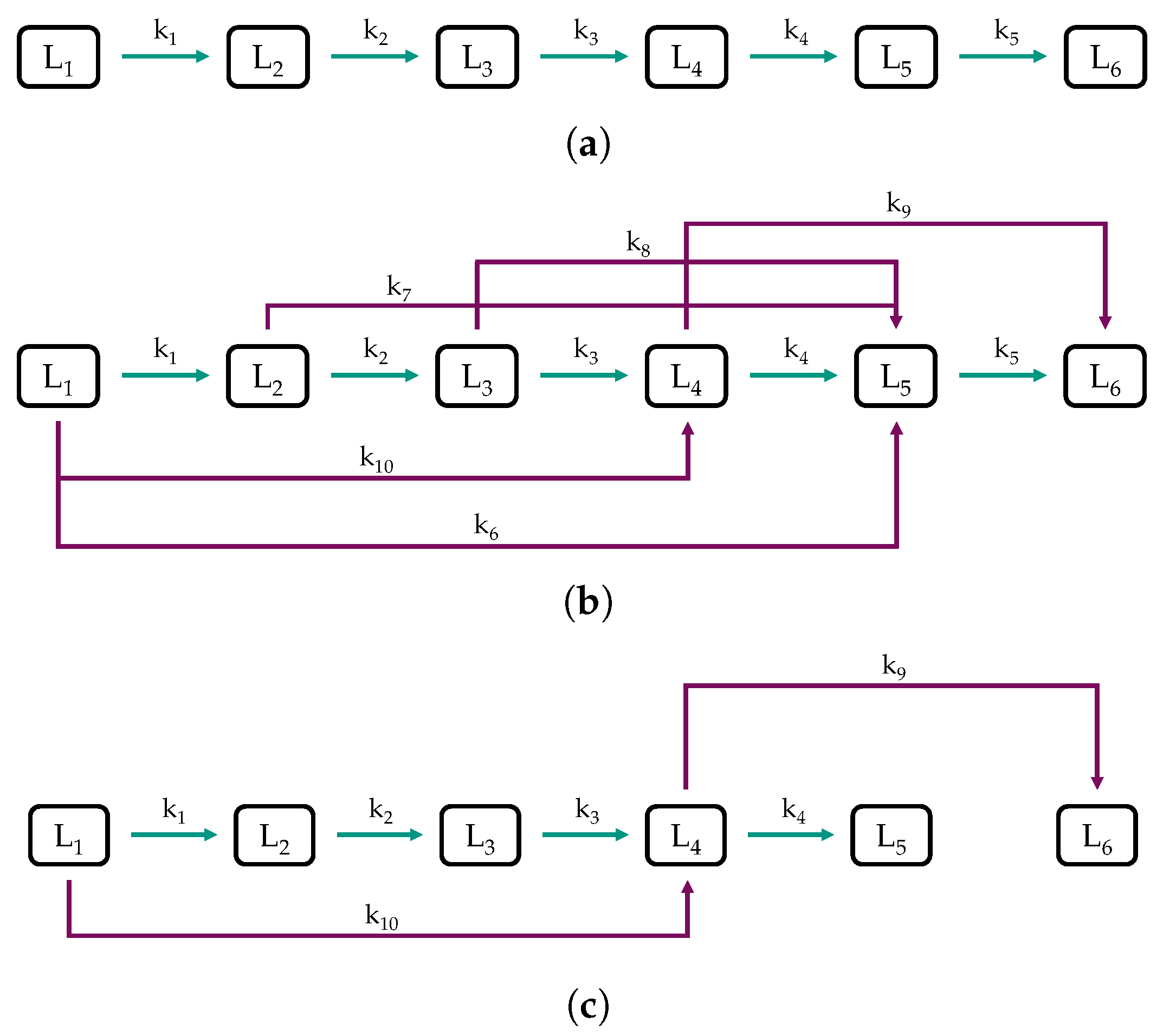
2.2. Model Structure and Procedure
3. Results and Discussion
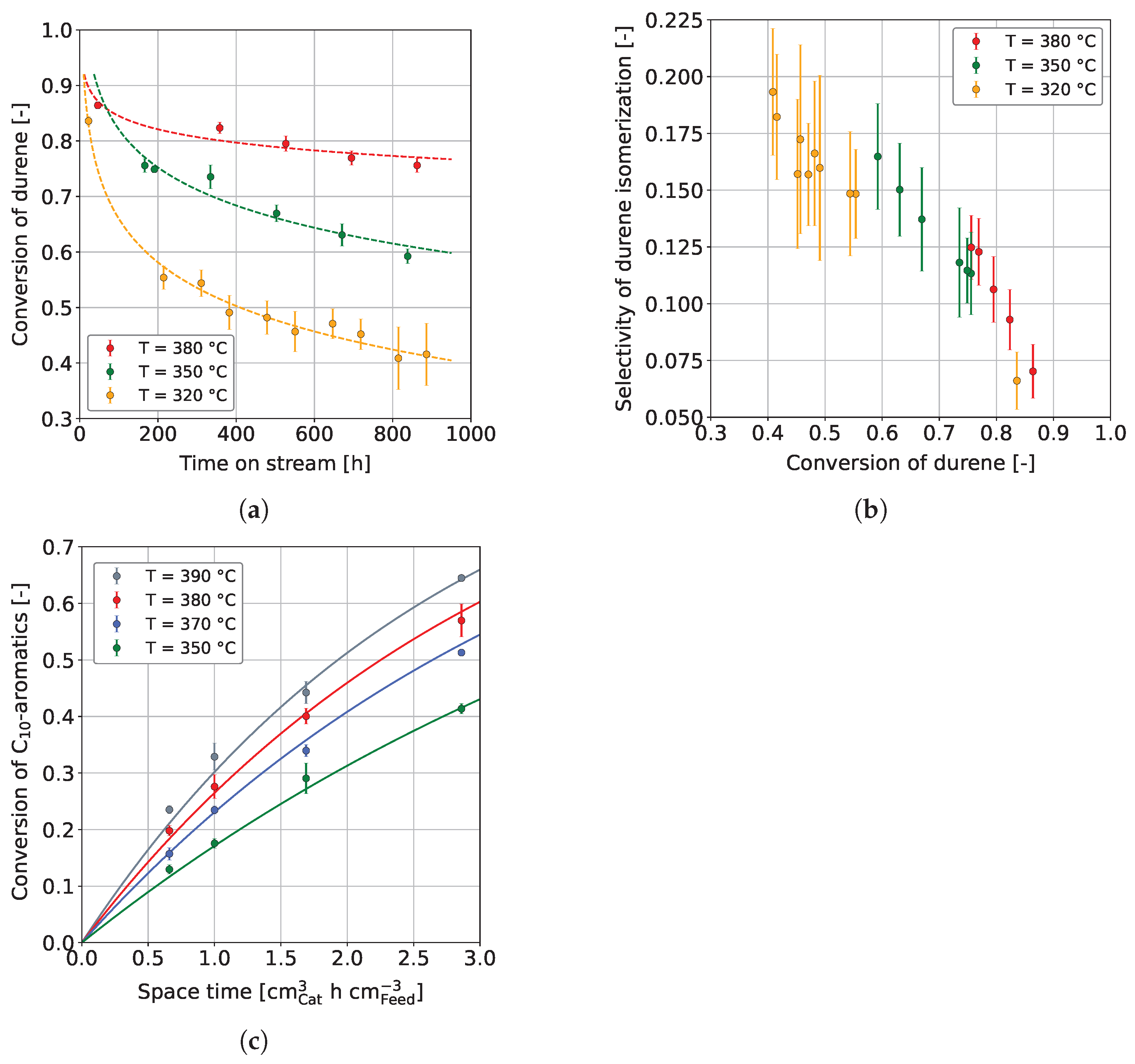
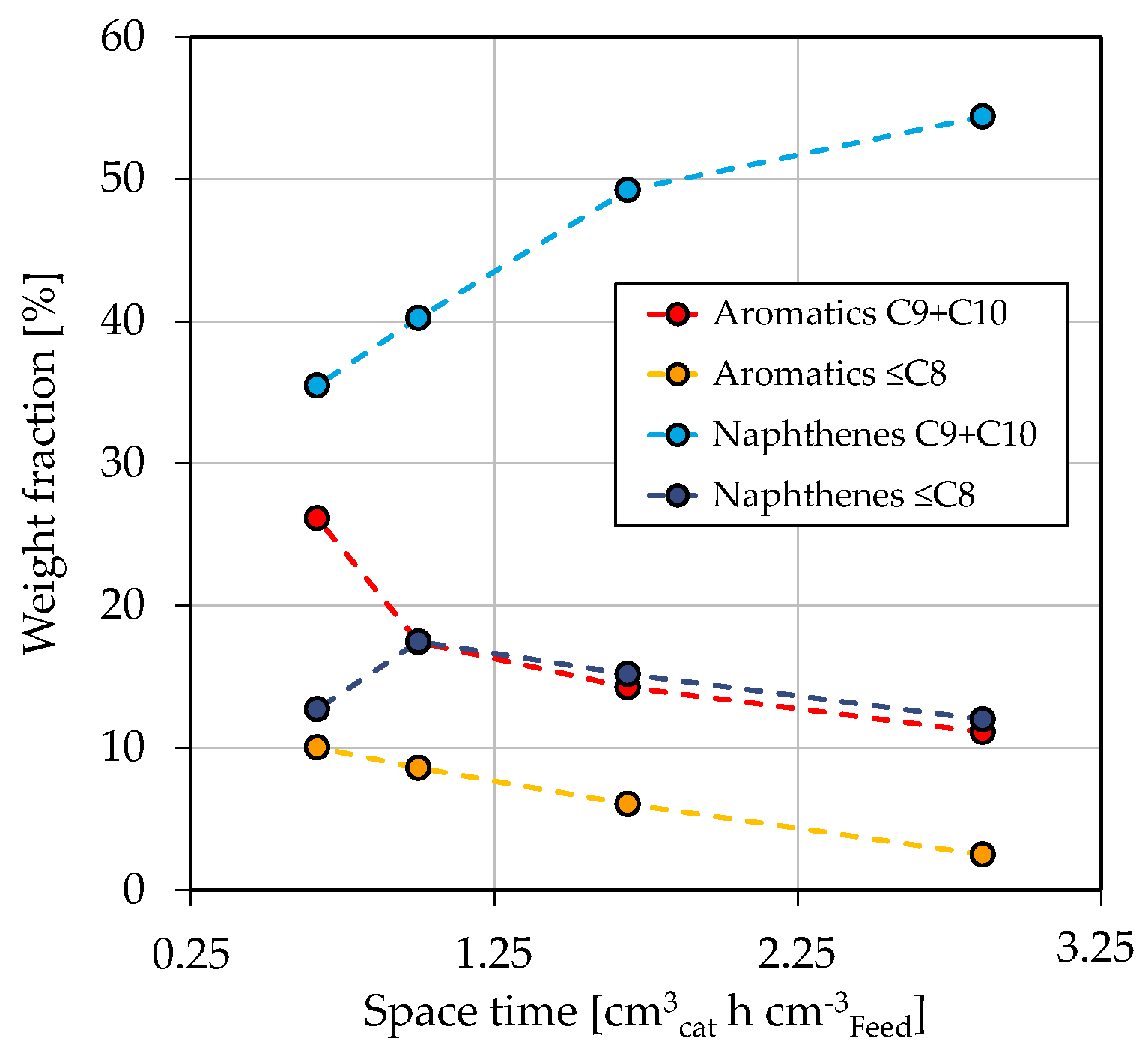
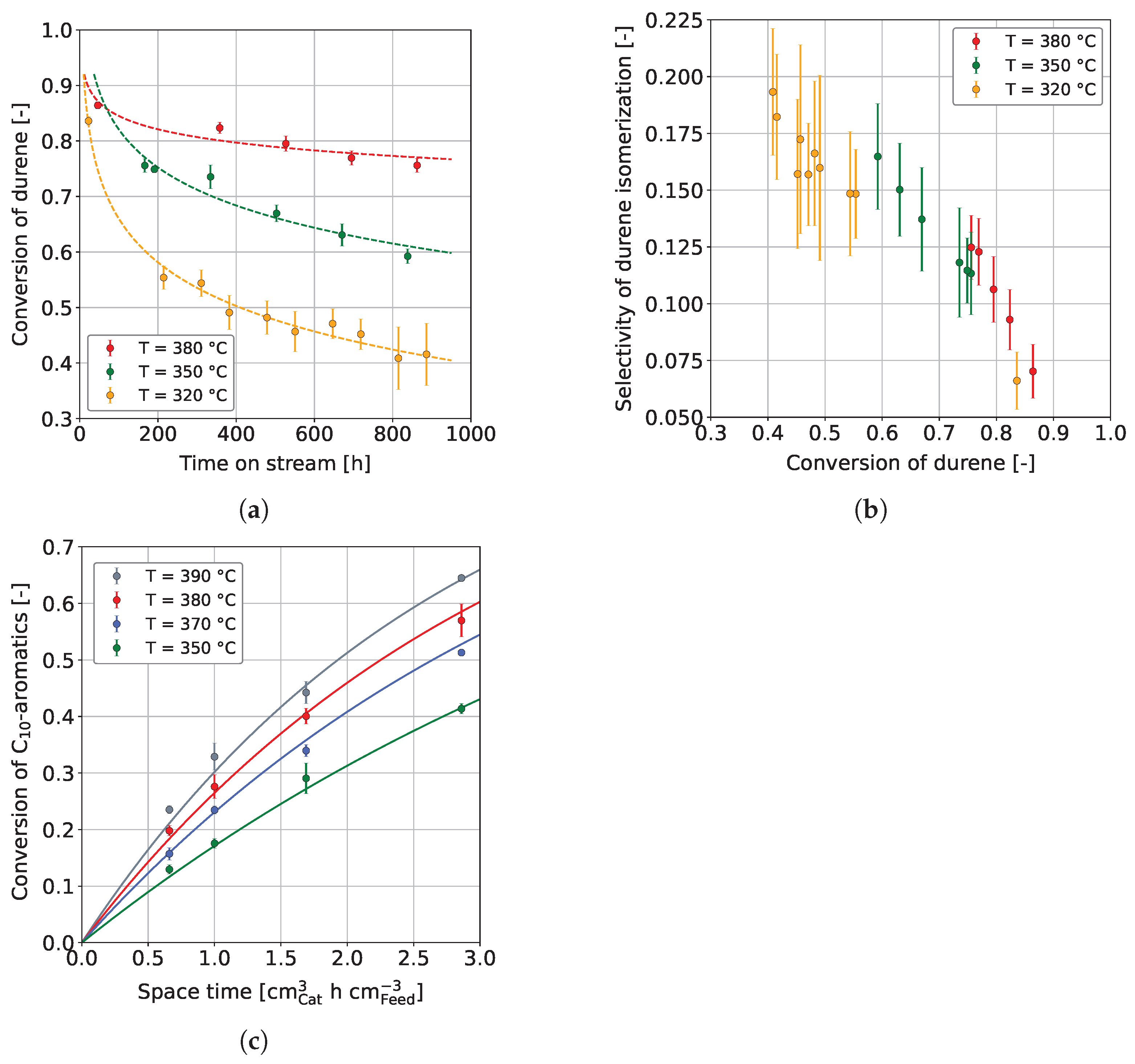
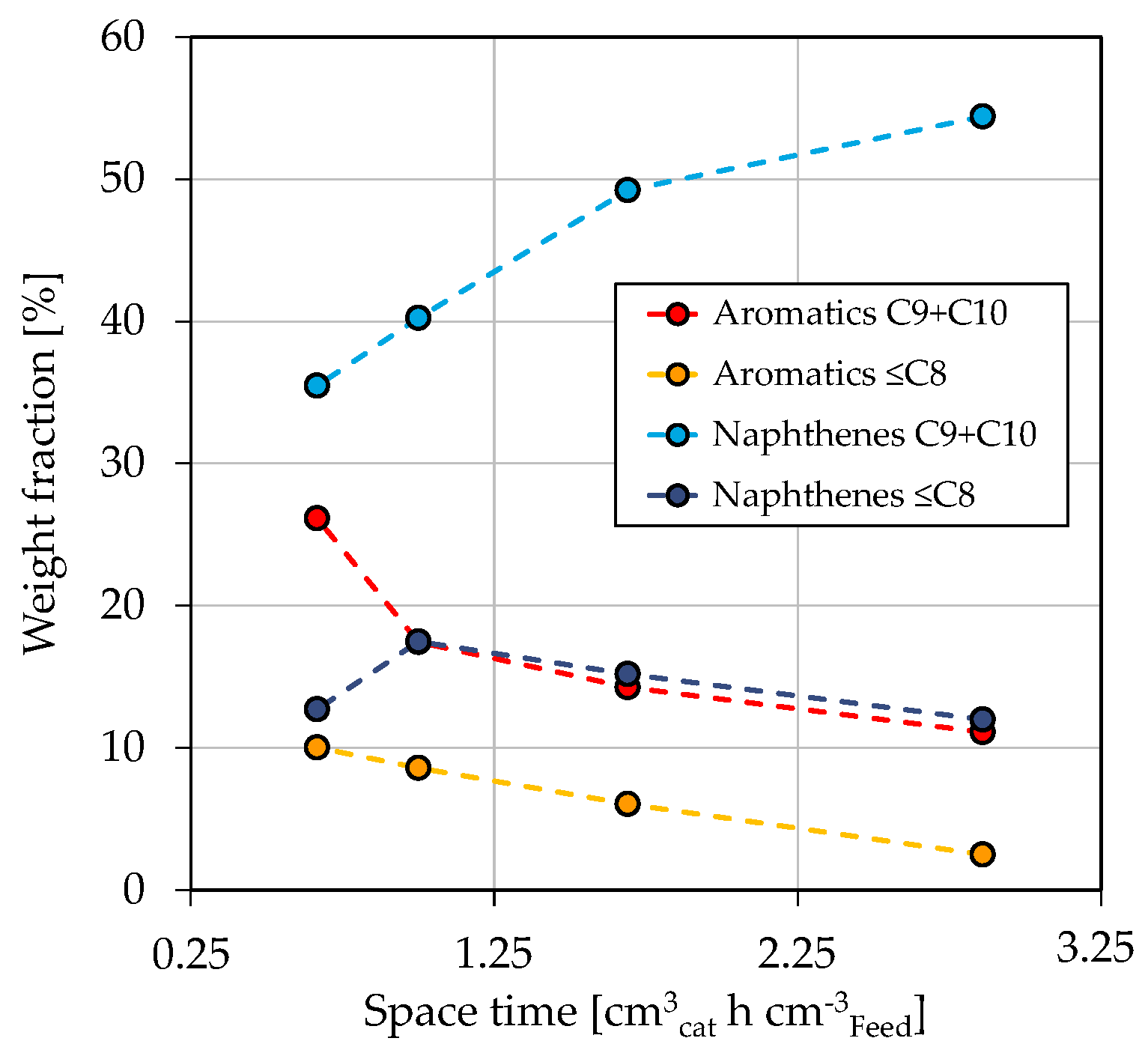
3.1. Co-Hydroprocessing of Middle Distillate and Heavy Mono-Ring Aromatics
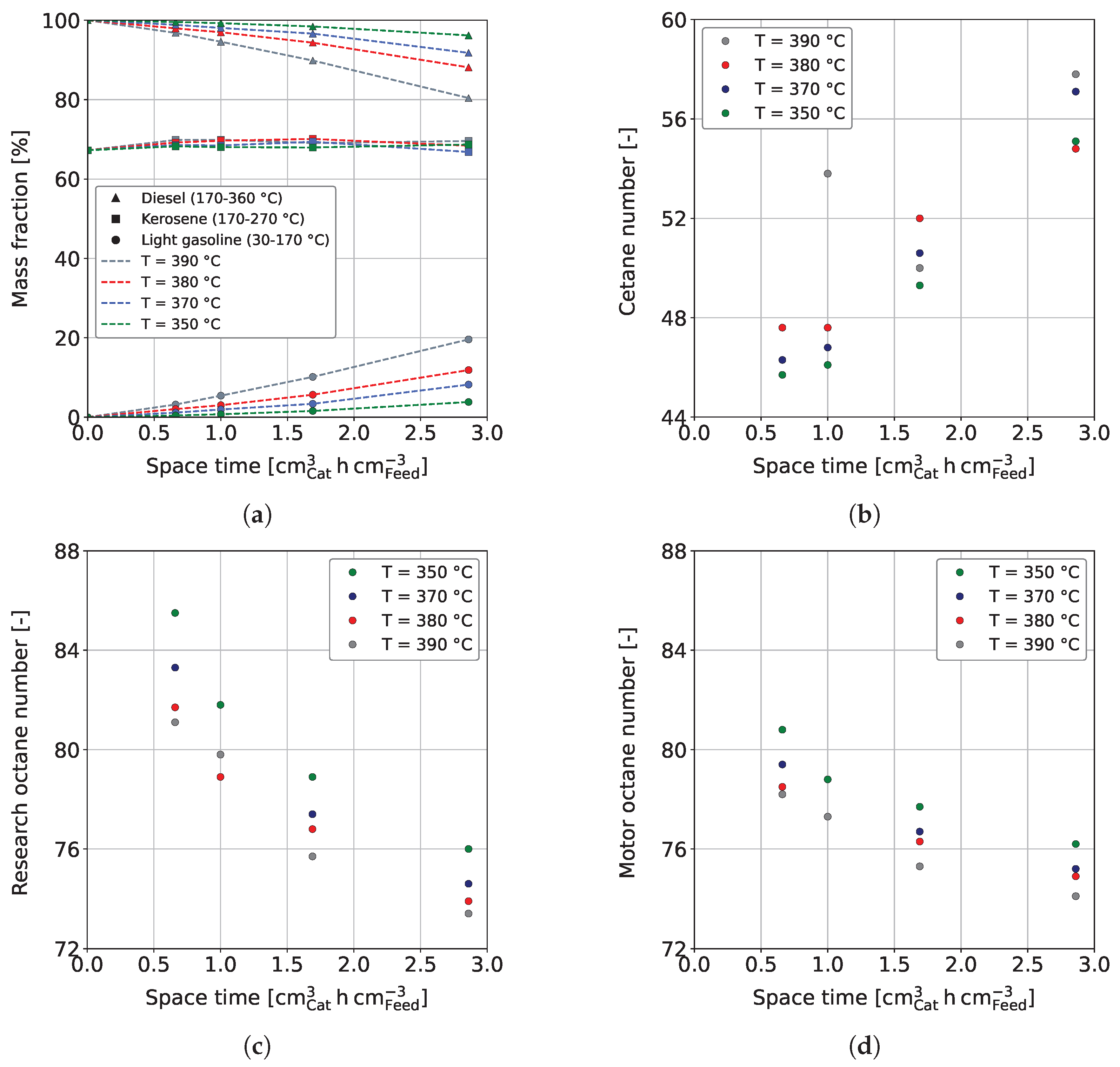
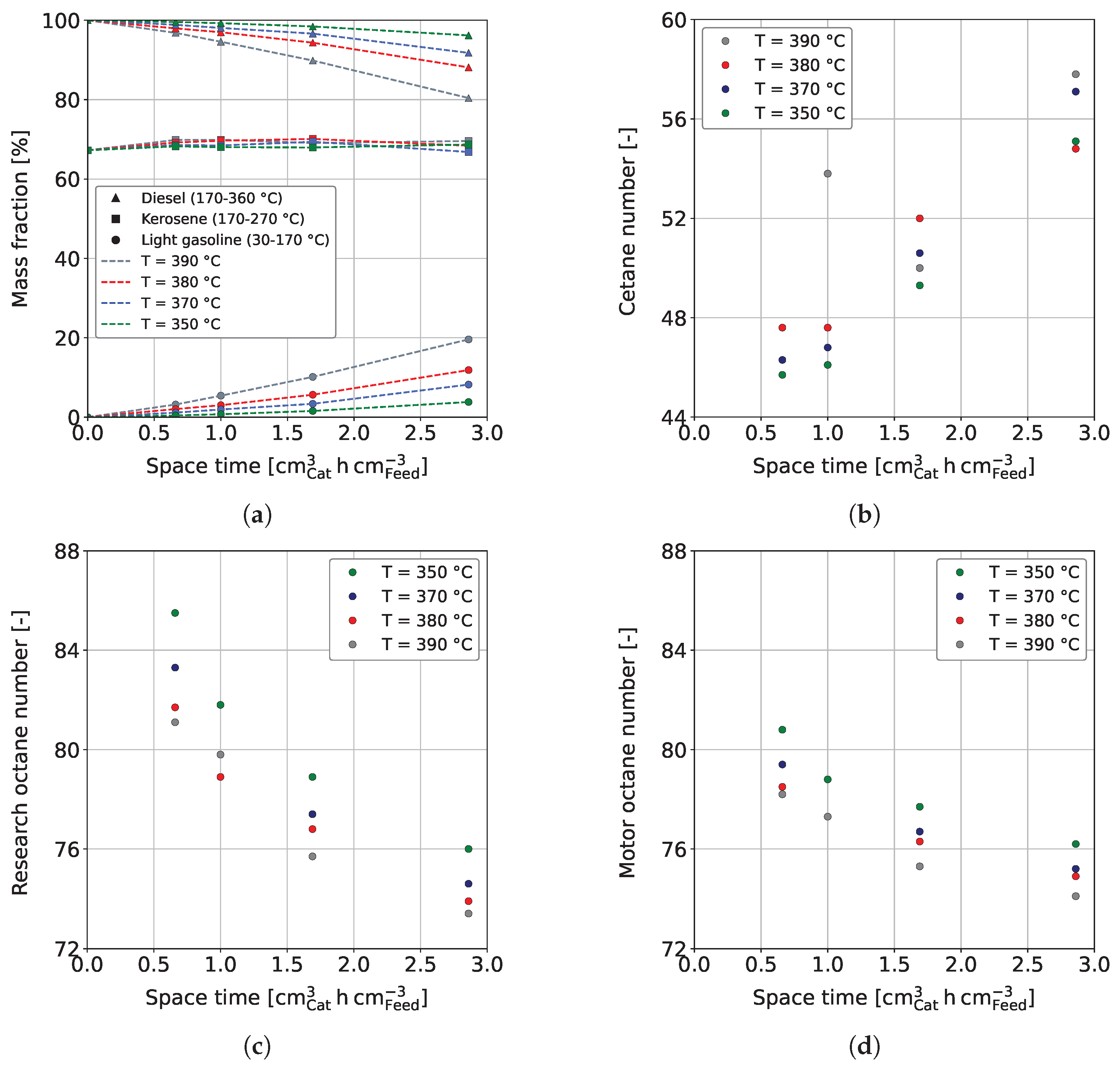
3.2. Fuel Yields and Qualities
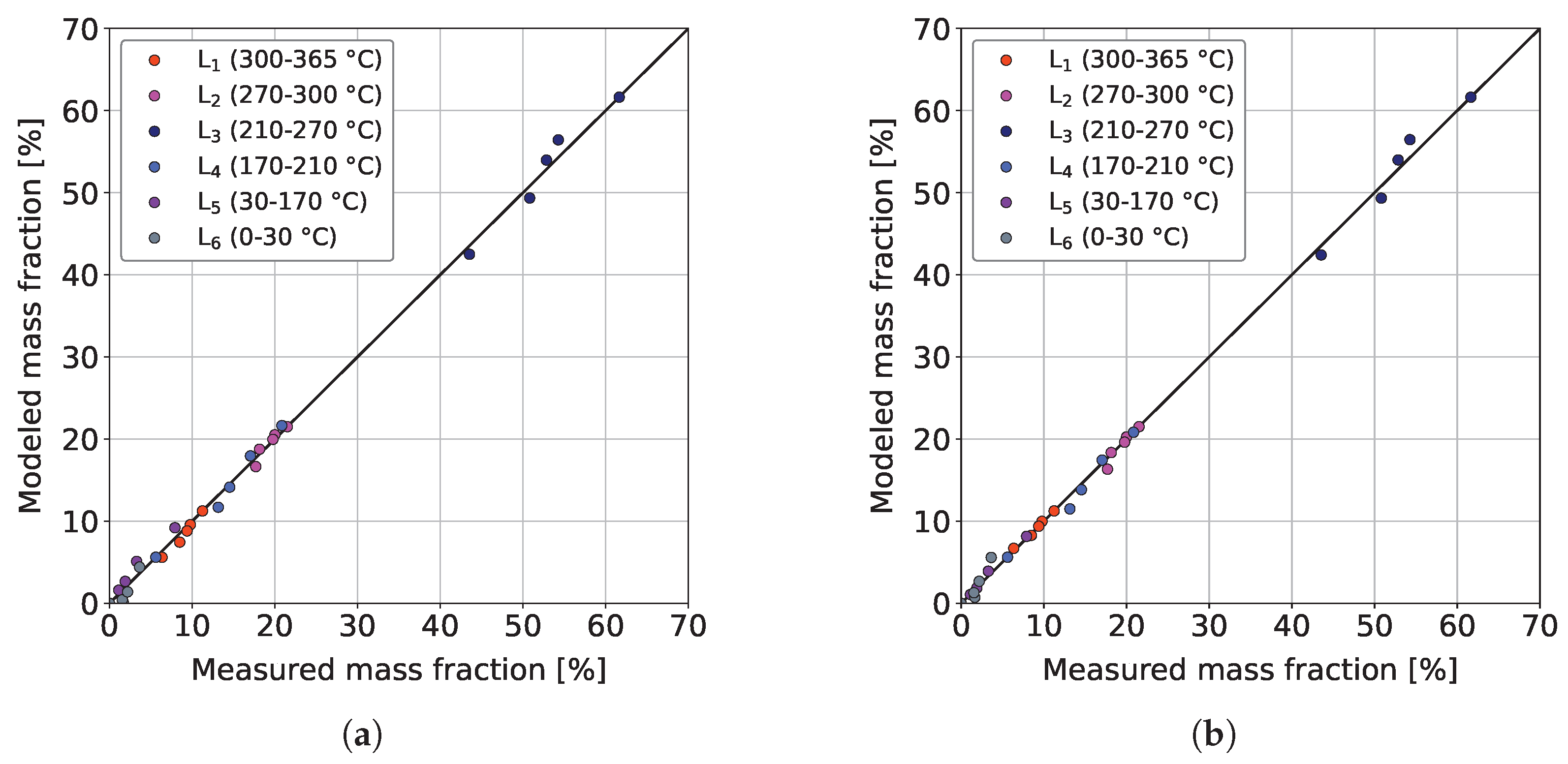
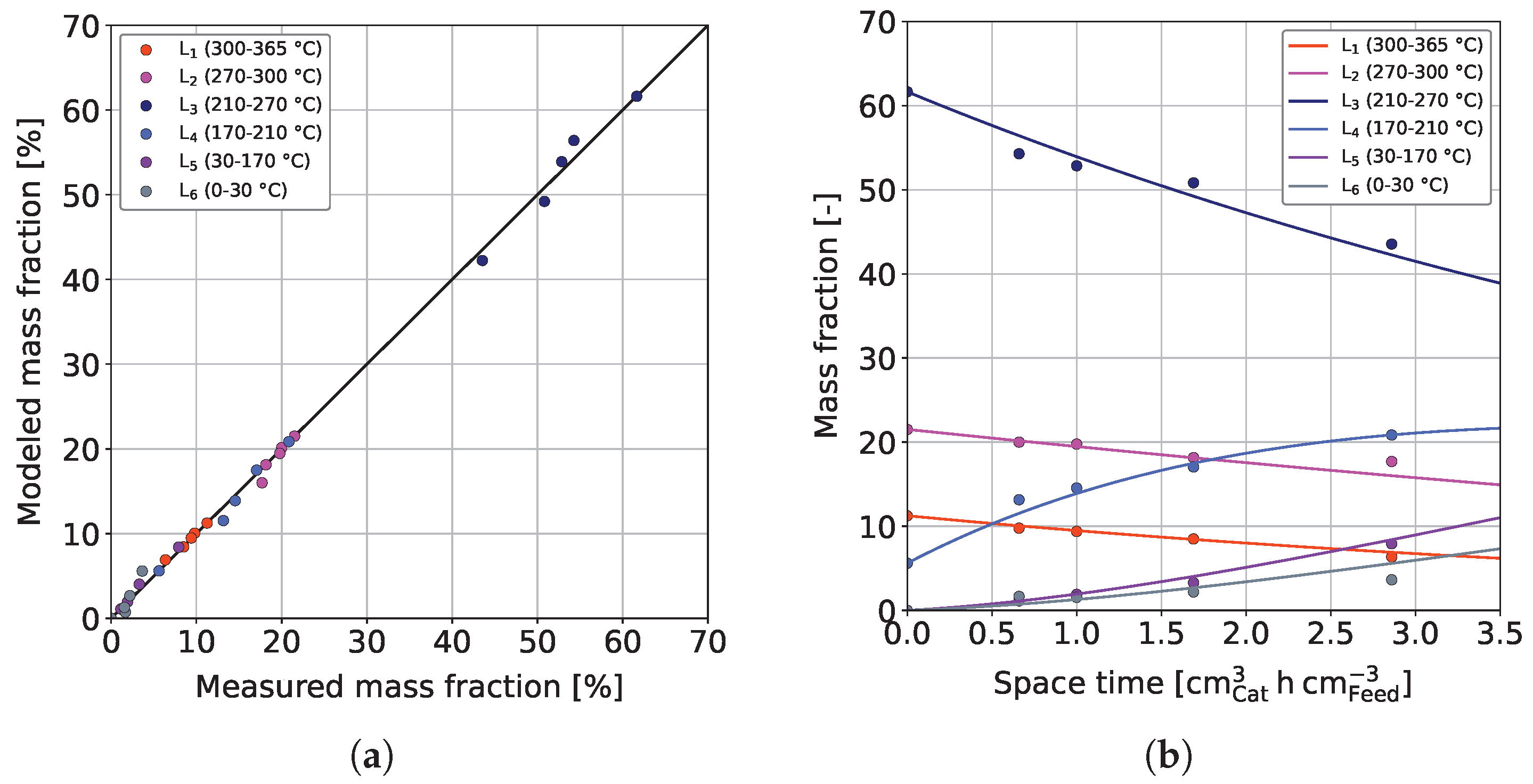
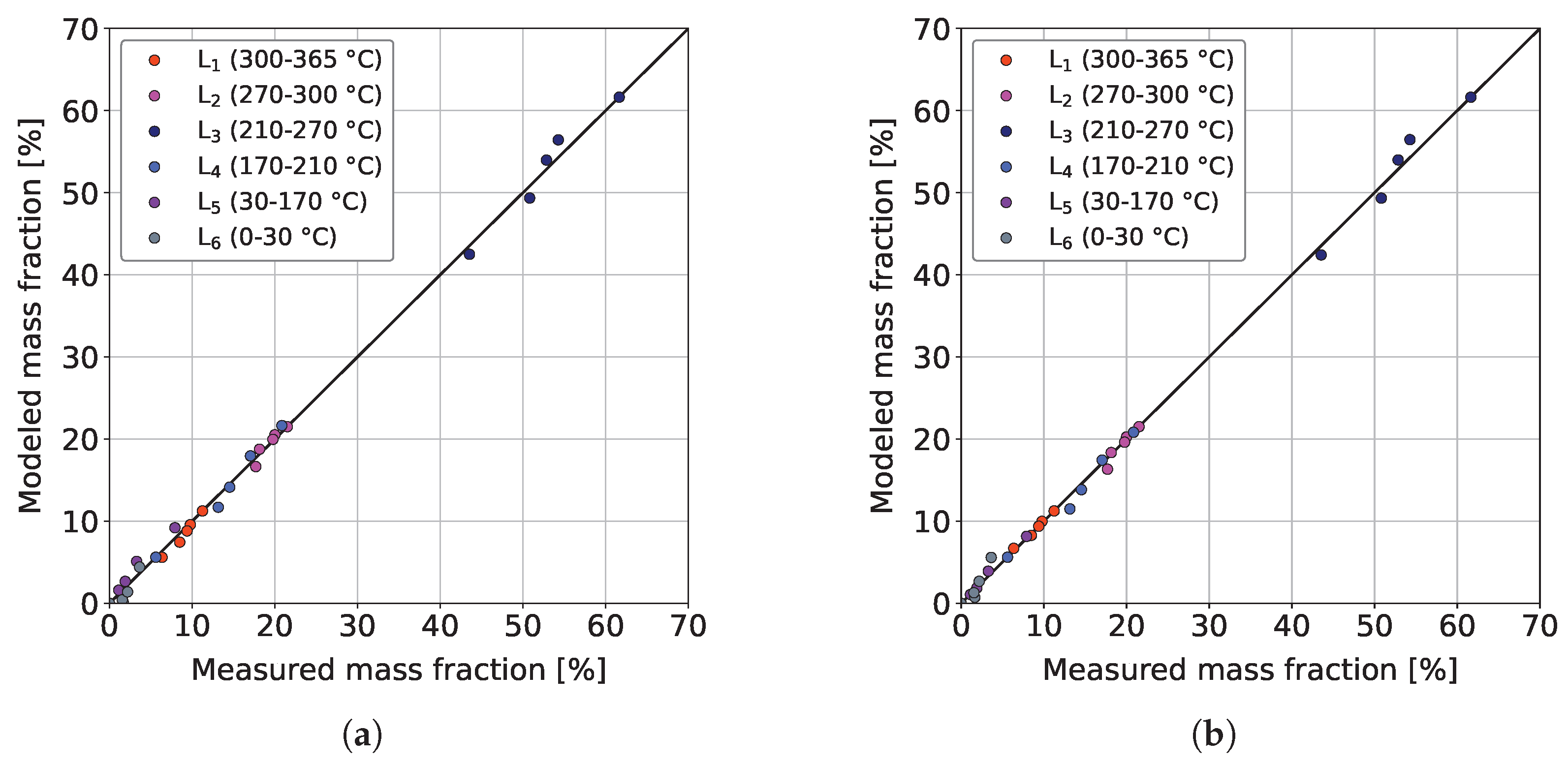
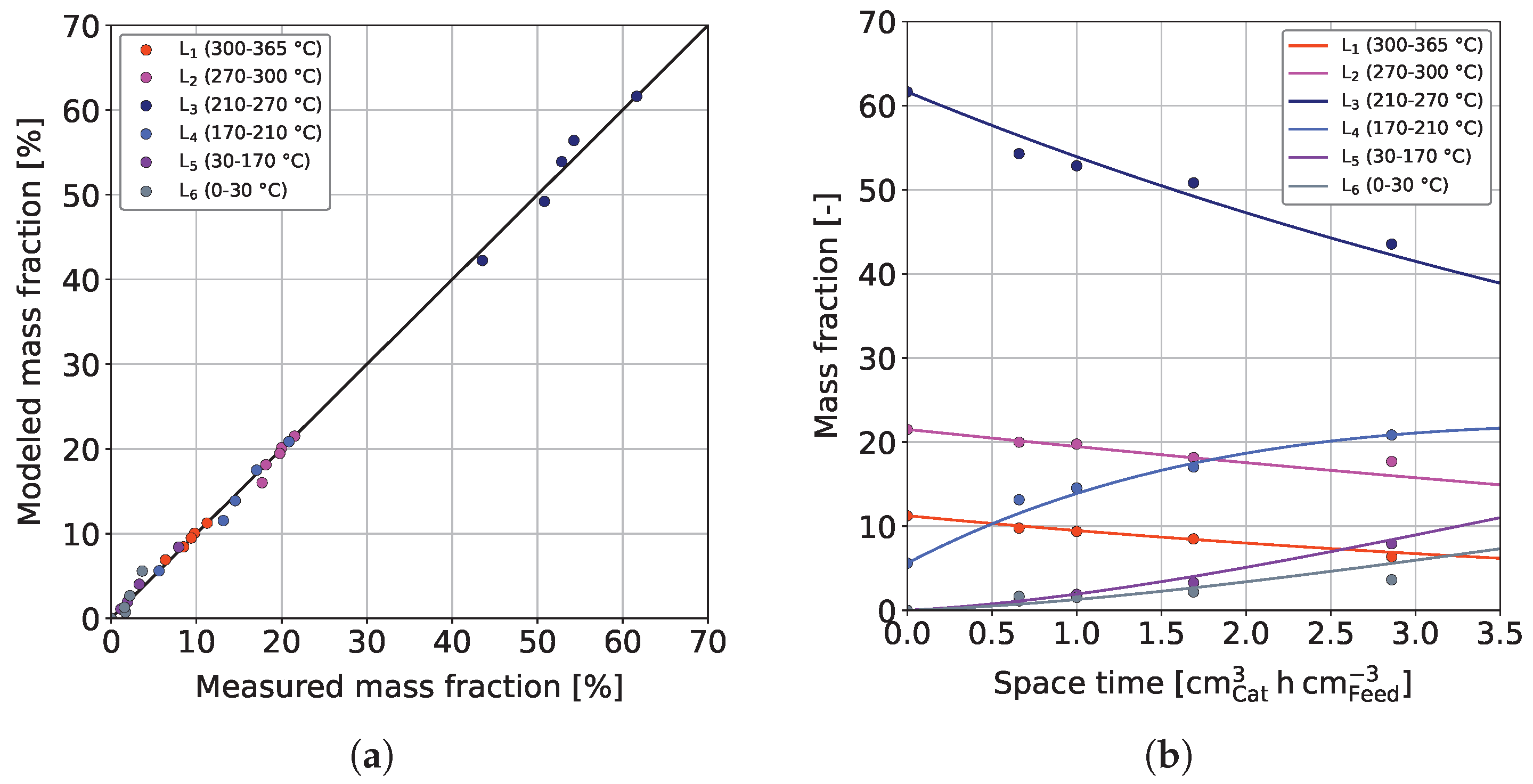
3.3. Parameter Estimation and Model Validation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Our World in Data. Global Direct Primary Energy Consumption. Available online: https://ourworldindata.org/grapher/global-primary-energy (accessed on 6 February 2023).
- European Commission. Communication from the Commission of the European Parliament, the European Council, the Council, the European Economic and Social Committee and the Committee of the Regions: The European Green Deal; European Commission: Brussels, Belgium, 2019.
- The European Parliament and the Council of the European Union. Regulation (EU) 2021/1119 of the European Parliament and of the Council of 30 June 2021 Establishing the Framework for Achieving Climate Neutrality and Amending Regulations (EC) No 401/2009 and (EU) 2018/1999 (’European Climate Law’): Regulation 2021/1119; The European Parliament and the Council of the European Union: Strasbourg, France, 2021. [Google Scholar]
- European Commission. Communication from the Commission to the European Parliament, the Council, the European Economic and Social Committee and the Committee of the Regions: ’Fit for 55’: Delivering the EU’s 2030 Climat Target on the Way to Climate Neutrality; European Commission: Brussels, Belgium, 2021.
- Abnett, K. EU Approves Effective Ban on New Fossil Fuel Cars from 2035. Available online: https://www.reuters.com/markets/europe/eu-approves-effective-ban-new-fossil-fuel-cars-2035-2022-10-27/ (accessed on 25 November 2022).
- Federal Ministry for the Environment, Nature Conservation, Nuclear Safety and Consumer Protection. PtL Roadmap: Sustainable Aviation Fuel from Renewable Energy Sources for Aviation in Germany; Federal Ministry for the Environment, Nature Conservation, Nuclear Safety and Consumer Protection: Berlin, Germany, 2021.
- International Energy Agency. Global EV Outlook 2023: Catching up with Climate Ambitions; International Energy Agency (IEA): Paris, France, 2023. [Google Scholar]
- European Automobile Manufacturers’ Association. Vehicles in Use Europe; European Automobile Manufacturers’ Association (ACEA): Brussels, Belgium, 2023. [Google Scholar]
- Alvarez-Majmutov, A.; Badoga, S.; Chen, J.; Monnier, J.; Zhang, Y. Co-Processing of Deoxygenated Pyrolysis Bio-Oil with Vacuum Gas Oil through Hydrocracking. Energy Fuels 2021, 35, 9983–9993. [Google Scholar] [CrossRef]
- DIN Deutsches Institut für Normung e.V. Automotive Fuels–Unleaded Petrol–Requirements and Test Methods, German version EN 228:2012+A1:2017; Beuthe Verlag GmbH: Berlin, Germany, 2017. [Google Scholar]
- DIN Deutsches Institut für Normung e.V. Automotive fuels—Diesel—Requirements and Test Methods, German version EN 590:2022; Beuthe Verlag GmbH: Berlin, Germany, 2022. [Google Scholar]
- Jeuland, N.; Montagne, X.; Gautrot, X. Potentiality of Ethanol as a Fuel for Dedicated Engine. Oil Gas Sci. Technol.-Rev. l’IFP 2004, 59, 559–570. [Google Scholar] [CrossRef]
- Munsin, R.; Laoonual, Y.; Jugjai, S.; Imai, Y. An experimental study on performance and emissions of a small SI engine generator set fuelled by hydrous ethanol with high water contents up to 40%. Fuel 2013, 106, 586–592. [Google Scholar] [CrossRef]
- Haseeb, A.; Fazal, M.A.; Jahirul, M.I.; Masjuki, H.H. Compatibility of automotive materials in biodiesel: A review. Fuel 2011, 90, 922–931. [Google Scholar] [CrossRef]
- Chang, C.D.; Silvestri, A.J. The Conversion of Methanol and Other O-Compounds to Hydrocarbons over Zeolite Catalysts. J. Catal. 1977, 47, 249–259. [Google Scholar] [CrossRef]
- Olsbye, U.; Svelle, S.; Bjørgen, M.; Beato, P.; Janssens, T.V.W.; Joensen, F.; Bordiga, S.; Lillerud, K.P. Conversion of Methanol to Hydrocarbons: How Zeolite Cavity and Pore Size Controls Product Selectivity. Angew. Chem. Int. Ed. 2012, 51, 5810–5831. [Google Scholar] [CrossRef]
- Chang, C.D. The New Zealand Gas-to-Gasoline plant: An engineering tour de force. Catal. Today 1992, 13, 103–111. [Google Scholar] [CrossRef]
- Topp-Jørgensen, J. Topsøe Integrated Gasoline Synthesis—The Tigas Process. Stud. Surf. Sci. Catal. 1988, 36, 293–305. [Google Scholar] [CrossRef]
- Fujimoto, K.; Asami, K.; Saima, H.; Shikada, T.; Tominaga, H.O. Two-Stage Reaction System for Synthesis Gas Conversion to Gasoline. Ind. Eng. Chem. Prod. Res. Dev. 1986, 25, 262–267. [Google Scholar] [CrossRef]
- Trippe, F.; Fröhling, M.; Schultmann, F.; Stahl, R.; Henrich, E. Techno-Economic Analysis of Fast Pyrolysis as a Process Step Within Biomass-to-Liquid Fuel Production. Waste Biomass Valorization 2010, 1, 415–430. [Google Scholar] [CrossRef]
- Pfitzer, C.; Dahmen, N.; Tröger, N.; Weirich, F.; Sauer, J.; Günther, A.; Müller-Hagedorn, M. Fast Pyrolysis of Wheat Straw in the Bioliq® Pilot Plant. Energy Fuels 2016, 30, 8047–8054. [Google Scholar] [CrossRef]
- Dahmen, N.; Henrich, E.; Dinjus, E.; Weirich, F. The bioliq® bioslurry gasification process for the production of biosynfuels, organic chemicals, and energy. Energy Sustain. Soc. 2012, 2, 3. [Google Scholar] [CrossRef]
- Stiefel, M.; Ahmad, R.; Arnold, U.; Döring, M. Direct synthesis of dimethyl ether from carbon-monoxide-rich synthesis gas: Influence of dehydration catalysts and operating conditions. Fuel Process. Technol. 2011, 92, 1466–1474. [Google Scholar] [CrossRef]
- Zimmermann, M.C.; Otto, T.N.; Wodarz, S.; Zevaco, T.A.; Pitter, S. Mesoporous H–ZSM–5 for the Conversion of Dimethyl Ether to Hydrocarbons. Chem. Ing. Tech. 2019, 91, 1302–1313. [Google Scholar] [CrossRef]
- Amelang, S. Pilot Project in Germany’s Largest Refinery to Produce Synthetic Fuels. 2021. Available online: https://www.cleanenergywire.org/news/pilot-project-germanys-largest-refinery-produce-synthetic-fuels (accessed on 8 March 2023).
- Liederman, D.; Yurchak, S.; Kuo, J.C.W.; Lee, W. Mobil Methanol-to-Gasoline Process. J. Energy 1982, 6, 340–341. [Google Scholar] [CrossRef]
- Petit, A.; Montagne, X. Effects of the Gasoline Composition on Exhaust Emissions of Regulated and Speciated Pollutants. SAE Tech. Pap. 1993, 932681, 1–11. [Google Scholar] [CrossRef]
- Calcote, H.F.; Manos, D.M. Effect of Molecular Structure on Incipient Soot Formation. Combust. Flame 1983, 49, 289–304. [Google Scholar] [CrossRef]
- Lim, S.S.; Vos, T.; Flaxman, A.D.; Danaei, G.; Shibuya, K.; Adair-Rohani, H.; AlMazroa, M.A.; Amann, M.; Anderson, H.R.; Andrews, K.G.; et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2224–2260. [Google Scholar] [CrossRef]
- Chang, C.D. Hydrocarbons from Methanol. Catal. Rev. Sci. Eng. 1983, 25, 1–118. [Google Scholar] [CrossRef]
- Bisht, D.; Petri, J. Considerations for Upgrading Light Cycle Oil with Hydroprocessing Technologies. Indian Chem. Eng. 2014, 56, 321–335. [Google Scholar] [CrossRef]
- Mokrani, T.; Scurrell, M. Gas Conversion to Liquid Fuels and Chemicals: The Methanol Route–Catalysis and Processes Development. Catal. Rev. 2009, 51, 1–145. [Google Scholar] [CrossRef]
- Vivas, A.H.; Joensen, F. Process and Catalyst for Upgrading Gasoline. U.S. Patent US10150714B2, 11 December 2018. [Google Scholar]
- Graf, D.; Neuner, P.; Rauch, R. Hydroprocessing and Blending of a Biomass-Based DTG-Gasoline. Energy Eng. 2022, 119, 2169–2192. [Google Scholar] [CrossRef]
- van Dyk, S.; Su, J.; Ebadian, M.; Saddler, J. Production of lower carbon-intensity fuels by co-processing biogenic feedstocks: Potential and challenges for refineries. Fuel 2022, 324, 124636. [Google Scholar] [CrossRef]
- van Dyk, S.; Su, J.; Mcmillan, J.D.; Saddler, J. Potential synergies of drop–in biofuel production with further co–processing at oil refineries. Biofuels Bioprod. Biorefin. 2018, 13, 760–775. [Google Scholar] [CrossRef]
- Lappas, A.A.; Bezergianni, S.; Vasalos, I.A. Production of biofuels via co-processing in conventional refining processes. Catal. Today 2009, 145, 55–62. [Google Scholar] [CrossRef]
- Marker, T.L. Opportunities for Biorenewables in Oil Refineries; U.S. Department of Energy: Washington, DC, USA, 2005. [CrossRef]
- Le, W.; Wang, Y.; Zheng, L.; Shi, M.; Li, J. Design and optimization of bio-oil co-processing with vacuum gas oil in a refinery. Energy Convers. Manag. 2019, 195, 620–629. [Google Scholar] [CrossRef]
- Tsai, T.C.; Liu, S.B.; Wang, I. Disproportionation and transalkylation of alkylbenzenes over zeolite catalysts. Appl. Catal. A Gen. 1999, 181, 355–398. [Google Scholar] [CrossRef]
- Vogt, E.T.C.; Weckhuysen, B.M. Fluid catalytic cracking: Recent developments on the grand old lady of zeolite catalysis. Chem. Soc. Rev. 2015, 44, 7342–7370. [Google Scholar] [CrossRef]
- Bezergianni, S.; Dimitriadis, A.; Kikhtyanin, O.; Kubička, D. Refinery co-processing of renewable feeds. Prog. Energy Combust. Sci. 2018, 68, 29–64. [Google Scholar] [CrossRef]
- Al-Sabawi, M.; Chen, J.; Ng, S. Fluid Catalytic Cracking of Biomass-Derived Oils and Their Blends with Petroleum Feedstocks: A Review. Energy Fuels 2012, 26, 5355–5372. [Google Scholar] [CrossRef]
- Pinho, A.d.R.; de Almeida, M.B.; Mendes, F.L.; Ximenes, V.L.; Casavechia, L.C. Co-processing raw bio-oil and gasoil in an FCC Unit. Fuel Process. Technol. 2015, 131, 159–166. [Google Scholar] [CrossRef]
- Thegarid, N.; Fogassy, G.; Schuurman, Y.; Mirodatos, C.; Stefanidis, S.; Iliopoulou, E.F.; Kalogiannis, K.; Lappas, A.A. Second-generation biofuels by co-processing catalytic pyrolysis oil in FCC units. Appl. Catal. B Environ. 2014, 145, 161–166. [Google Scholar] [CrossRef]
- Jones, D.S.J.S.; Pujadó, P.R. Handbook of Petroleum Processing; Springer: Dordrecht, The Netherlands, 2006. [Google Scholar] [CrossRef]
- van Dyk, S.; Su, J.; Mcmillan, J.D.; Saddler, J. ’Drop-In’ Biofuels: The Key Role That Co-Processing Will Play in Its Production; IEA Bioenergy: Paris, France, 2019. [Google Scholar]
- Zacher, A.H.; Olarte, M.V.; Santosa, D.M.; Elliott, D.C.; Jones, S.B. A review and perspective of recent bio-oil hydrotreating research. Green Chem. 2014, 16, 491–515. [Google Scholar] [CrossRef]
- Furimsky, E. Selection of catalysts and reactors for hydroprocessing. Appl. Catal. A Gen. 1998, 171, 177–206. [Google Scholar] [CrossRef]
- Ancheyta-Juárez, J.; Aguilar-Rodríguez, E.; Salazar-Sotelo, D.; Betancourt-Rivera, G.; Leiva-Nuncio, M. Hydrotreating of straight run gas oil–light cycle oil blends. Appl. Catal. A Gen. 1999, 180, 195–205. [Google Scholar] [CrossRef]
- Nakakita, K.; Ban, H.; Takasu, S.; Hotta, Y.; Inagaki, K.; Weissman, W.; Farrell, J.T. Effect of Hydrocarbon Molecular Structure in Diesel Fuel on In-Cylinder Soot Formation and Exhaust Emissions. SAE Int. J. Fuels Lubr. 2003, 112, 1763–1775. [Google Scholar] [CrossRef]
- Sági, D.; Baladincz, P.; Varga, Z.; Hancsók, J. Co-processing of FCC light cycle oil and waste animal fats with straight run gas oil fraction. J. Clean. Prod. 2016, 111, 34–41. [Google Scholar] [CrossRef]
- Jiang, Y.; Phillips, S.D.; Singh, A.; Jones, S.B.; Gaspar, D.J. Potential economic values of low-vapor-pressure gasoline-range bio-blendstocks: Property estimation and blending optimization. Fuel 2021, 297, 120759. [Google Scholar] [CrossRef]
- Laxminarasimhan, C.S.; Verma, R.P.; Ramachandran, P.A. Continuous lumping model for simulation of hydrocracking. AIChE J. 1996, 42, 2645–2653. [Google Scholar] [CrossRef]
- Weitkamp, J. Catalytic Hydrocracking—Mechanisms and Versatility of the Process. ChemCatChem 2012, 4, 292–306. [Google Scholar] [CrossRef]
- Sullivan, R.F.; Egan, C.J.; Langlois, G.E.; Sieg, R.P. A New Reaction That Occurs in the Hydrocracking of Certain Aromatic Hydrocarbons. J. Am. Chem. Soc. 1961, 83, 1156–1160. [Google Scholar] [CrossRef]
- Ancheyta, J.; Sánchez, S.; Rodríguez, M.A. Kinetic modeling of hydrocracking of heavy oil fractions: A review. Catal. Today 2005, 109, 76–92. [Google Scholar] [CrossRef]
- Palos, R.; Gutiérrez, A.; Hita, I.; Castaño, P.; Thybaut, J.W.; Arandes, J.M.; Bilbao, J. Kinetic Modeling of Hydrotreating for Enhanced Upgrading of Light Cycle Oil. Ind. Eng. Chem. Res. 2019, 58, 13064–13075. [Google Scholar] [CrossRef]
- Hita, I.; Aguayo, A.T.; Olazar, M.; Azkoiti, M.J.; Bilbao, J.; Arandes, J.M.; Castaño, P. Kinetic Modeling of the Hydrotreating and Hydrocracking Stages for Upgrading Scrap Tires Pyrolysis Oil (STPO) toward High-Quality Fuels. Energy Fuels 2015, 29, 7542–7553. [Google Scholar] [CrossRef]
- Soto-Azuara, L.A.; Ramírez-López, R.; del Carmen Monterrubio-Badillo, M.; Elizalde, I. Mathematical modeling of the hydrocracking kinetics of a heavy oil fraction using the discrete lumping approach: The effect of the variation of the lump number. React. Kinet. Mech. Catal. 2022, 135, 655–667. [Google Scholar] [CrossRef]
- Ayasse, A.R.; Nagaishi, H.; Chan, E.W.; Gray, M.R. Lumped kinetics of hydrocracking of bitumen. Fuel 1997, 76, 1025–1033. [Google Scholar] [CrossRef]
- Jarullah, A.T.; Mujtaba, I.M.; Wood, A.S. Enhancement of Productivity of Distillate Fractions by Crude Oil Hydrotreatment: Development of Kinetic Model for the Hydrotreating Process. Comput. Aided Chem. Eng. 2011, 29, 261–265. [Google Scholar] [CrossRef]
- Alhumaidan, F.; Lababidi, H.M.S.; Al-Adwani, H. Hydrocracking of atmospheric residue feedstock in hydrotreating processes. Kuwait J. Sci. Eng. 2010, 37, 129–159. [Google Scholar]
- Sánchez, S.; Rodríguez, M.A.; Ancheyta, J. Kinetic Model for Moderate Hydrocracking of Heavy Oils. Ind. Eng. Chem. Res. 2005, 44, 9409–9413. [Google Scholar] [CrossRef]
- Martínez, J.; Ancheyta, J. Kinetic model for hydrocracking of heavy oil in a CSTR involving short term catalyst deactivation. Fuel 2012, 100, 193–199. [Google Scholar] [CrossRef]
- Sadighi, S.; Ahmad, A.; Rashidzadeh, M. 4-Lump kinetic model for vacuum gas oil hydrocracker involving hydrogen consumption. Korean J. Chem. Eng. 2010, 27, 1099–1108. [Google Scholar] [CrossRef]
- Ramírez, S.; Martínez, J.; Ancheyta, J. Kinetics of thermal hydrocracking of heavy oils under moderate hydroprocessing reaction conditions. Fuel 2013, 110, 83–88. [Google Scholar] [CrossRef]
- Becker, P.J.; Celse, B.; Guillaume, D.; Dulot, H.; Costa, V. Hydrotreatment modeling for a variety of VGO feedstocks: A continuous lumping approach. Fuel 2015, 139, 133–143. [Google Scholar] [CrossRef]
- Martens, G.G.; Thybaut, J.W.; Marin, G.B. Single-Event Rate Parameters for the Hydrocracking of Cycloalkanes on Pt/US-Y Zeolites. Ind. Eng. Chem. Res. 2001, 40, 1832–1844. [Google Scholar] [CrossRef]
- Texier, S. Activation of alumina-supported hydrotreating catalysts by organosulfides: Comparison with H2S and effect of different solvents. J. Catal. 2004, 223, 404–418. [Google Scholar] [CrossRef]
- ASTM D6730-01; Standard Test Method for Determination of Individual Components in Spark Ignition Engine Fuels by 100-Metre Capillary (with Precolumn) High-Resolution Gas Chromatography. ASTM International: West Conshohocken, PA, USA, 2001. [CrossRef]
- ASTM D7345; Standard Test Method for Distillation of Petroleum Products and Liquid Fuels at Atmospheric Pressure (Micro Distillation Method). ASTM International: West Conshohocken, PA, USA, 2017. [CrossRef]
- Qader, S.A.; Hill, G.R. Hydrocracking of Gas Oil. Ind. Eng. Chem. Process. Des. Dev. 1969, 8, 98–105. [Google Scholar] [CrossRef]
- Cooper, B.H.; Donnis, B.B. Aromatic saturation of distillates: An overview. Appl. Catal. A Gen. 1996, 137, 203–223. [Google Scholar] [CrossRef]
- Alcázar, L.A.; Ancheyta, J. Sensitivity analysis based methodology to estimate the best set of parameters for heterogeneous kinetic models. Chem. Eng. J. 2007, 128, 85–93. [Google Scholar] [CrossRef]
- Newville, M.; Stensitzki, T.; Allen, D.B.; Antonino, I. LMFIT: Non-Linear Least-Square Minimization and Curve-Fitting for Python 2014. Available online: https://zenodo.org/record/11813 (accessed on 31 July 2023).
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef]
- Vivas-Báez, J.C.; Servia, A.; Pirngruber, G.D.; Dubreuil, A.C.; Pérez-Martínez, D.J. Insights in the phenomena involved in deactivation of industrial hydrocracking catalysts through an accelerated deactivation protocol. Fuel 2021, 303, 120681. [Google Scholar] [CrossRef]
- Meurer, A.; Kern, J. Fischer–Tropsch Synthesis as the Key for Decentralized Sustainable Kerosene Production. Energies 2021, 14, 1836. [Google Scholar] [CrossRef]
- Kubic, W.L. A Group Contribution Method for Estimating Cetane and Octane Numbers; Los Alamos National Laboratory: Los Alamos, NM, USA, 2016. Available online: https://www.osti.gov/biblio/1291241/ (accessed on 8 March 2023).
- Hawkins, D.M. The Problem of Overfitting. J. Chem. Inf. Comput. Sci. 2004, 44, 1–12. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Middle Distillate (MD) | bioliq® Heavy Gasoline (HG) | Feedstock | |
|---|---|---|---|
| [-] | 60.3 | N/A | N/A |
| [kg m] | 823.0 | 890.0 | 827.6 |
| [mg kg] | <5 | 0 | 0 |
| [°C] | −14 | N/A | N/A |
| [wt.%] | 5.7 | 97.0 | approx. 19.4 |
| IBP [°C] | 179.7 | 176.7 | 175.0 |
| T25 [°C] | 247.7 | 185.7 | 230.6 |
| T50 [°C] | 263.2 | 190.0 | 257.5 |
| T75 [°C] | 277.6 | 198.2 | 275.1 |
| FBP [°C] | 365.2 | 289.8 | 365.2 |
| Lump | Boiling Range [°C] | Approx. Composition | L @ = 0 [wt.%] |
|---|---|---|---|
| 1 | 300–365 | Mainly Par. | 11.25 |
| 2 | 270–300 | Aro., Par. | 21.51 |
| 3 | 210–270 | Aro., Par. | 61.64 |
| 4 | 170–210 | Aro., Par. and Naph. | 5.61 |
| 5 | 30–170 | Aro., Par. and Naph. | 0 |
| 6 | 0–30 | Par. | 0 |
| [cm h cm] | [kJ mol] | ||||||
|---|---|---|---|---|---|---|---|
| Sequential | Combined | Reduced | Sequential | Combined | Reduced | ||
| 0.206 | 0.119 | 0.123 | 116.3 | 0 | 0 | ||
| 0.156 | 0.135 | 0.142 | 142.4 | 79.9 | 89.8 | ||
| 0.186 | 0.181 | 0.185 | 54.1 | 19.1 | 22.9 | ||
| 0.264 | 0.160 | 0.161 | 97.0 | 109.9 | 122.4 | ||
| 0.360 | 6.09 × 10 | - | 0 | 24.9 | - | ||
| - | 3.20 × 10 | - | - | 0 | - | ||
| - | 9.25 × 10 | - | - | 1845.5 | - | ||
| - | 9.65 × 10 | - | - | 15.8 | - | ||
| - | 0.126 | 0.126 | - | 12.1 | 11.0 | ||
| - | 0.036 | 0.025 | - | 382.6 | 435.4 | ||
| Parameters | Sequential Model | Reduced Model | Combined Model |
|---|---|---|---|
| 10 | 12 | 20 | |
| 6 | 6 | 6 | |
| 16 | 16 | 16 | |
| 86 | 84 | 76 | |
| [-] | 0.9971 | 0.9976 | 0.9978 |
| [kg kg] | 27.16 | 22.24 | 20.51 |
| F | - | 9.29 () | 0.8 () |
| - | 3.11 | 2.06 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Graf, D.; Waßmuth, J.; Rauch, R. Co-Hydroprocessing of Fossil Middle Distillate and Bio-Derived Durene-Rich Heavy Ends under Hydrotreating Conditions. Reactions 2023, 4, 531-551. https://doi.org/10.3390/reactions4030032
Graf D, Waßmuth J, Rauch R. Co-Hydroprocessing of Fossil Middle Distillate and Bio-Derived Durene-Rich Heavy Ends under Hydrotreating Conditions. Reactions. 2023; 4(3):531-551. https://doi.org/10.3390/reactions4030032
Chicago/Turabian StyleGraf, David, Johannes Waßmuth, and Reinhard Rauch. 2023. "Co-Hydroprocessing of Fossil Middle Distillate and Bio-Derived Durene-Rich Heavy Ends under Hydrotreating Conditions" Reactions 4, no. 3: 531-551. https://doi.org/10.3390/reactions4030032
APA StyleGraf, D., Waßmuth, J., & Rauch, R. (2023). Co-Hydroprocessing of Fossil Middle Distillate and Bio-Derived Durene-Rich Heavy Ends under Hydrotreating Conditions. Reactions, 4(3), 531-551. https://doi.org/10.3390/reactions4030032