1. Introduction
The threat of climate change as well as geopolitical tensions are gradually reducing or even prohibiting the use of fossil fuels. Hydrogen, produced from renewable energy sources, is widely studied as a replacement [
1]. Among the hydrogen storage technologies, the most mature are compression or liquefaction and one of the most studied is physisorption. However, they have drawbacks regarding safety, due to high compression pressures and the risk of leakage and evaporation of flammable hydrogen [
2,
3,
4]. With regard to chemical storage of hydrogen in carrier molecules, this technology can involve a wide variety of chemical reactions and storage solutions. The most common ways involve power-to-X systems, by CO
2 and/or N
2 hydrogenation to fuels capable of being stored under reasonable conditions over extended periods [
1]. However, these systems often lack in reversibility because hydrogen is hardly retrieved [
1,
5]. Another solution consists in binding hydrogen to metals under a solid state [
6]. Anyway, the most promising solutions in terms of hydrogen gravimetric density are currently not reversible and the reversible ones are limited in terms of hydrogen content. Another way is the use of a liquid organic hydrogen carrier (LOHC), which involves reversible reactions, is easy to transport and stable over long periods [
1,
4]. Indeed, a LOHC consists in a couple of hydrogen-lean|hydrogen-rich molecules, which are respectively catalytically hydrogenated and dehydrogenated into the other molecule in order to store and release hydrogen. These organic liquids are fluids sharing similar properties with hydrocarbon fuels or common chemicals. Their stability at ambient temperature and atmospheric pressure in the liquid phase makes them compatible with the existing fuel infrastructures and thus LOHCs can be easily transported in tankers, trucks and/or pipelines [
1,
4,
7].
Multiple LOHC systems have already been identified. The main ones highlighted in the literature are aromatics|cycloalkanes, such as toluene|methylcyclohexane (TOL|MCH), dibenzyltoluene|perhydro-dibenzyltoluene (H0-DBT|H18-DBT), benzyltoluene|perhydro-benzyltoluene (H0-BT|H12-BT) and couples based on N-heterocycles, such as N-ethylcarbazole|perhydro-N-ethylcarbazole (H0-NEC|H12-NEC). The first cycloalkane-based LOHCs were developed because of their good hydrogen capacities (5.8–6.2%), their wide availabilities and the amount of knowledge on the processes of hydrogenation and dehydrogenation. However, reaction enthalpies, which dictate how much energy is lost during the storage process, are high for these LOHCs at around 65–69 kJ·mol
H2−1, although NEC LOHC has a slightly lower reaction enthalpy (52 kJ·mol
H2−1). Safety and toxicity profiles have also been improved from the early light aromatics LOHCs to NEC, BT and DBT LOHCs, due to their higher flash points and lesser carcinogenic properties. The liquid state criterion, which is a key point for the process designs, is hardly met for some LOHCs at some conditions. For example, H12-NEC is solid at ambient conditions. Moreover, DBT LOHCs have reaction intermediates with high viscosities. NEC has also reversibility issues with high degradation when subject to harsh dehydrogenation temperatures. Finally, the use of expensive noble metal catalysts and relatively severe reaction conditions hinder the economical or environmental viability of the aromatic-based LOHCs [
1,
3,
7,
8,
9,
10,
11,
12]. CEA has recently identified a bioavailable LOHC: γ-butyrolactone|1,4-butanediol (GBL|BDO) [
13]. The latter, described for the first time by Onoda et al. [
14], has low reaction enthalpies of 42 kJ·mol
H2−1 in liquid phase and 31 kJ·mol
H2−1 in gas phase, thus making it the most energy efficient. Its lower cycle operating temperature range (160–220 °C), compared to that of cycloalkane-based LOHCs _(300–350 °C), makes its process less energy consuming. BDO is partly produced from biomass, by hydrogenating bio-based succinic acid into GBL or by producing BDO directly from a fermentation of sugars over microorganisms [
14,
15,
16,
17], although most of it is currently produced from acetylene and formaldehyde [
15,
16,
17]. It has a H
2 capacity of 4.5 wt%, which is lower than that of the other LOHCs. The melting point of pure BDO at 20 °C could limit its use in colder weather or regions [
16], but as for the stoichiometric ratio in feed, it remains liquid at reaction operating temperature and pressure, so reaction product separation is easier. Although GBL and BDO have good safety and toxicity profiles, GBL is known as a drug precursor because of its psychotropic properties and chemical resemblance to γ-hydroxybutyric acid (GHB) [
18]. They are commonly used as solvents and monomers and their uses are allowed in industrial processes in most countries. See
Table S1 in Supplementary Materials for LOHC comparisons [
1,
16,
19,
20,
21,
22].
For all LOHCs, except for TOL|MCH, hydrogenation and dehydrogenation are performed in three-phase reactors. Although LOHCs are usually compared with respect to their mass H2 capacities, the volume H2 capacity is therefore also useful for the design of suitable reactors as it indicates the volume of hydrogen involved during the reactions. For all LOHCs, the ratio H2/LOHC is significant (613–856 LH2·L−1 LOHC) and could act as a barrier to liquid–solid transfer by forming dry zones on the surface of the catalyst pellets. Thus, the knowledge of the reaction kinetics in both the liquid and gas phases is necessary to accurately design a three-phase reactor. As the main quality of LOHCs is the perfect reversibility of their reactions, the formation of co-products in both phases and reactions must also be assessed. This study is therefore a part of a larger study on GBL|BDO. Here, the paper is focused on the kinetics of the hydrogenation in the gas phase. The kinetics of the hydrogenation in the liquid phase will be the subject of another article.
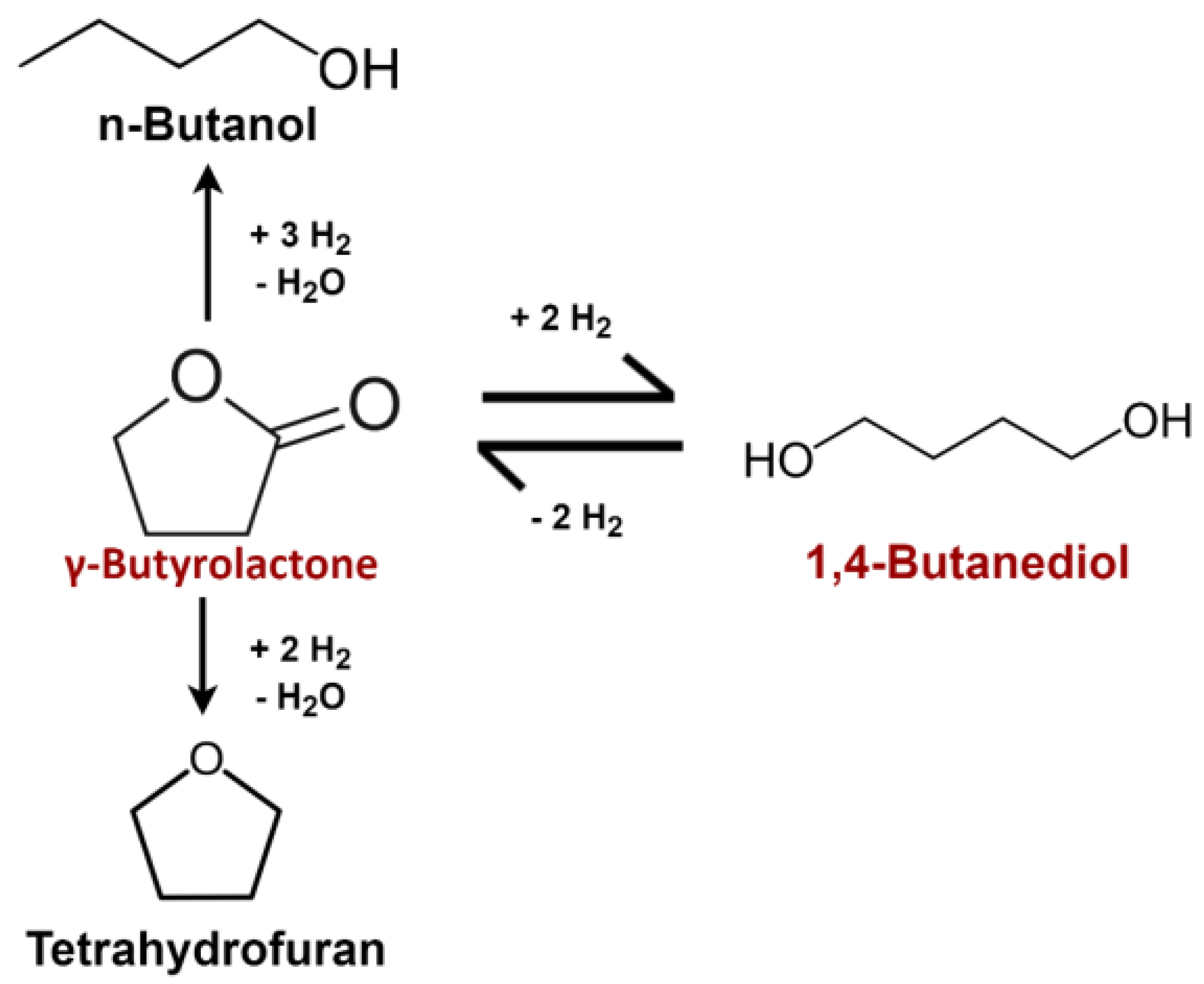
The reaction pathway describing the equilibrium between GBL and BDO has been debated within the literature. Most have studied the reaction on copper-based catalysts in the liquid phase with temperature and pressure ranging from 200 to 250 °C and from 30 to 90 bar [
14,
15,
23,
24], respectively. Two parallel reactions, resulting in a dehydration towards tetrahydrofuran (THF) and n-Butanol (BuOH), occur at high temperature and are catalyzed by acid sites present in the catalyst support [
23,
24,
25,
26,
27]; however, their mechanisms are unclear. Reaction intermediates, such as 2-Hydroxytetrahydrofuran, have been proposed as they could be involved in the formation of secondary products [
19,
24,
27]. Ichikawa et al. proposed a model taking into account its formation without measuring it [
27]. Chaudhari et al. proposed a mechanism where both GBL and BDO can lead up to the formation of THF and BuOH [
23]. However, no article seems to indicate that the latter are formed from intermediates, despites hydrogen imbalances between the reactants and the secondary products (except for the dehydration of BDO into THF). For instance, the experimental data in Schlander’s Ph.D. thesis [
25], performed in the gas phase, seem to indicate that GBL concentrations decrease faster than those of BDO as the secondary products build up in the mixture. As for Chaudhari et al., they contend THF is likely to be formed from the hydrogenation of GBL, rather than the dehydration of BDO [
23]; we have also used Schlander’s reaction pathway, shown in
Figure 1, for the kinetic modeling.
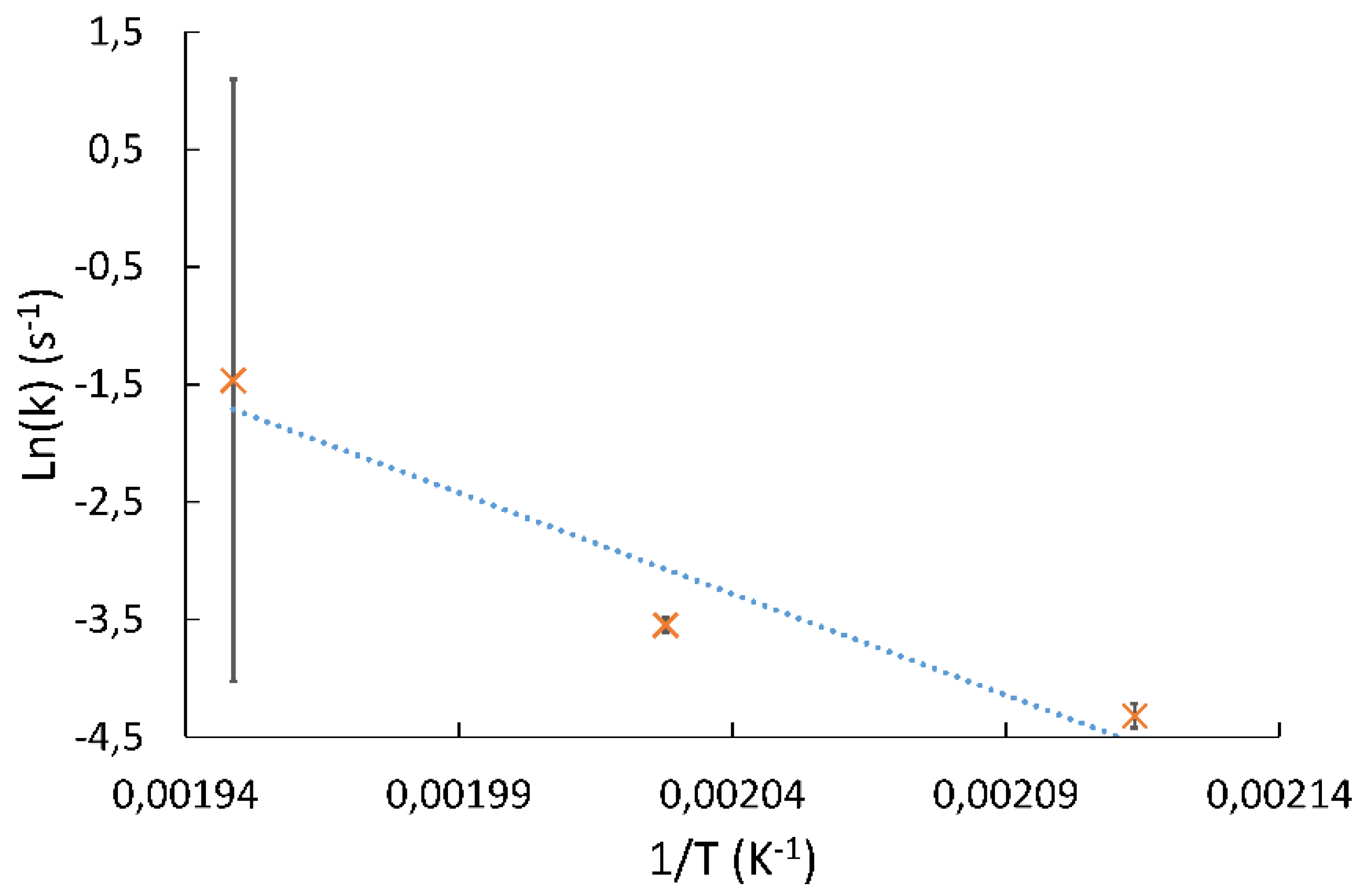
Kinetic models for the hydrogenation of GBL usually involve a simple power law model such as that proposed by Schlander [
15,
23,
25]. However, it (Model 1 in §2.2.1) proved unable to correctly predict the experimentally described pressure effect. Hermann et al. [
24] proposed a Langmuir–Hinshelwood type model because an intermediate is thought to be formed during the hydrogenation of GBL, but this approach was first ruled out because of the number of parameters needed to be estimated, giving the model too many degrees of liberty and thus making it hard to extrapolate. This is why we decided to revise the kinetic study of the GBL gas-phase hydrogenation. The aim was to build a kinetic model, able to predict the effect of temperature and pressure, to be included in a larger reactor model including all phases involved. As the data from Schlander’s works [
25,
26] were obtained under operating conditions that can mimic the composition of the gas phase in a trickle bed, the published data are used here to determine the appropriate kinetic law.
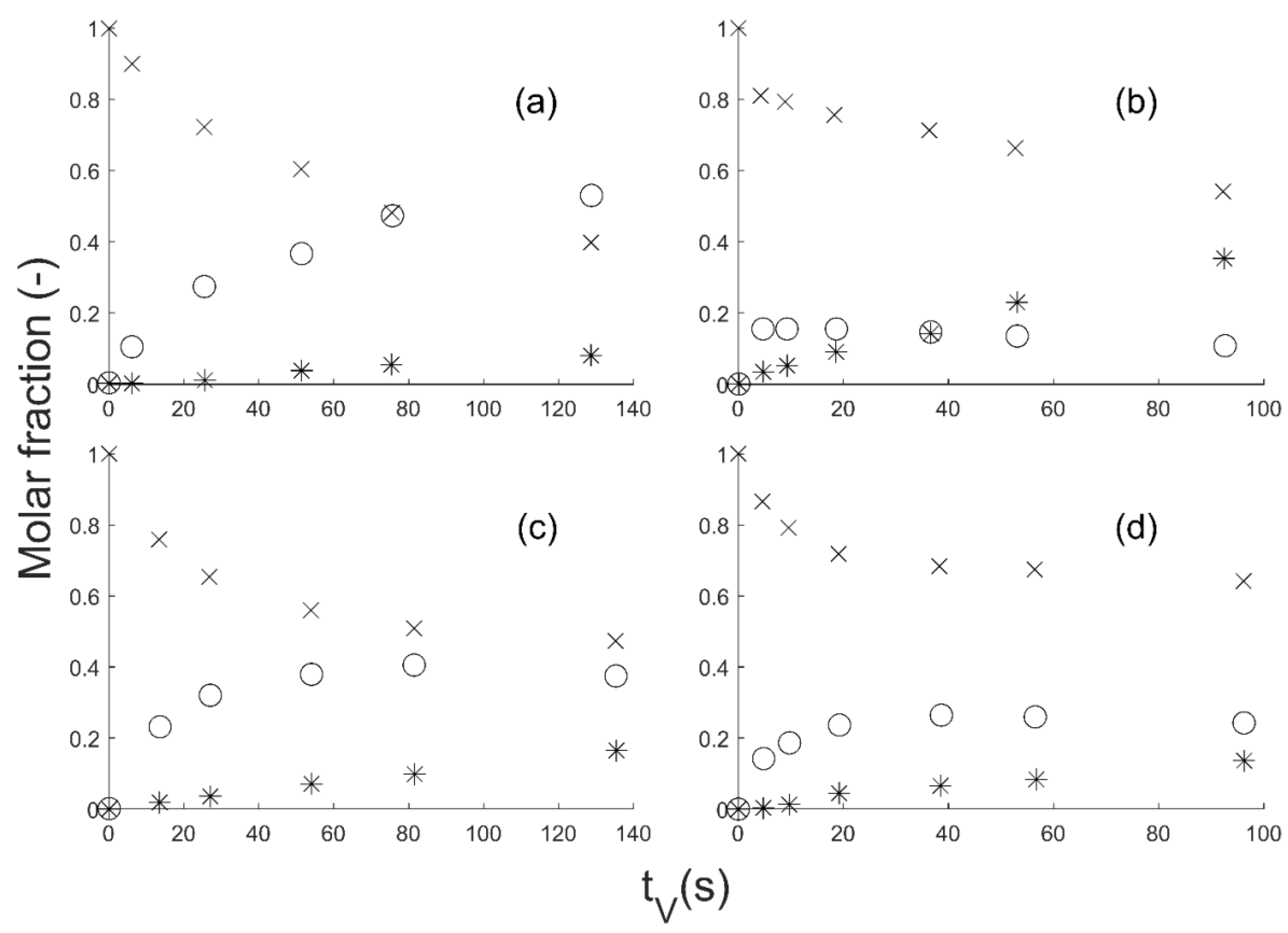
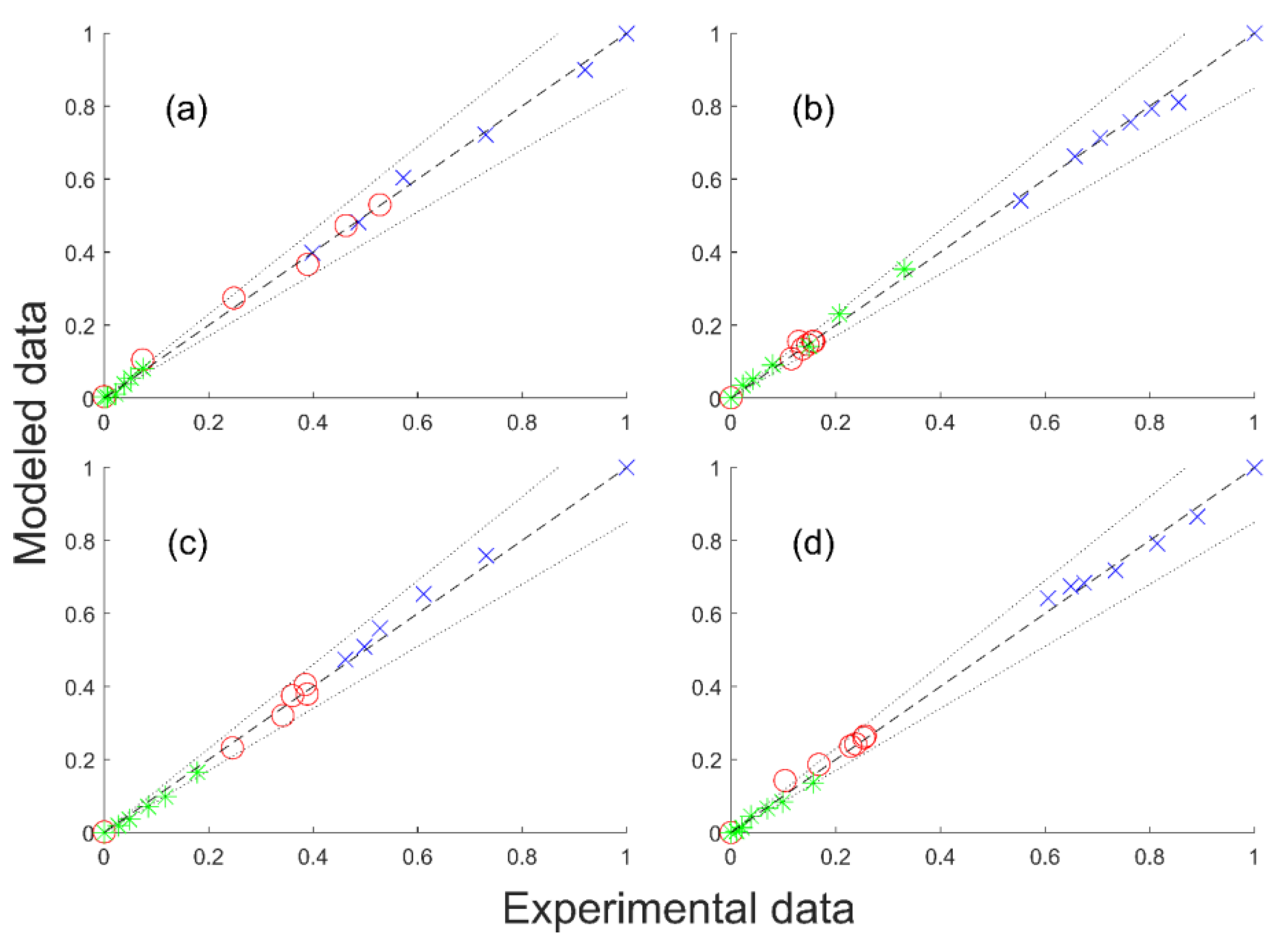
The GBL hydrogenation was done in gas phase on a CuZnO catalyst in an isothermal tubular reactor. The fixed bed was loaded on a reactor length of 80 mm with crushed catalyst and glass of 400 µm-diameter particles. The catalyst loading was about 8 g. The molar ratio H
2/GBL at the reactor entrance was about 90. Temperature and pressure ranges from 200 to 240 °C and from 25 to 35 bar, respectively, were screened. The experimental data presented in
Figure 2 are plotted as a function of the residence time t
V, which is the ratio between the reactor volume V
R and the total volume flow rate Q. The bimetallic catalyst was prepared by co-precipitation and contained 15 mol% Cu and 85 mol% ZnO [
28]. See
Section S2 in Supplementary Materials, and references [
25,
26,
28] for further details. Regarding the potential transfer limitation, Schlander showed that the apparent kinetics could not be explained either by internal or external mass transfer (
Section S3 in Supplementary Materials).
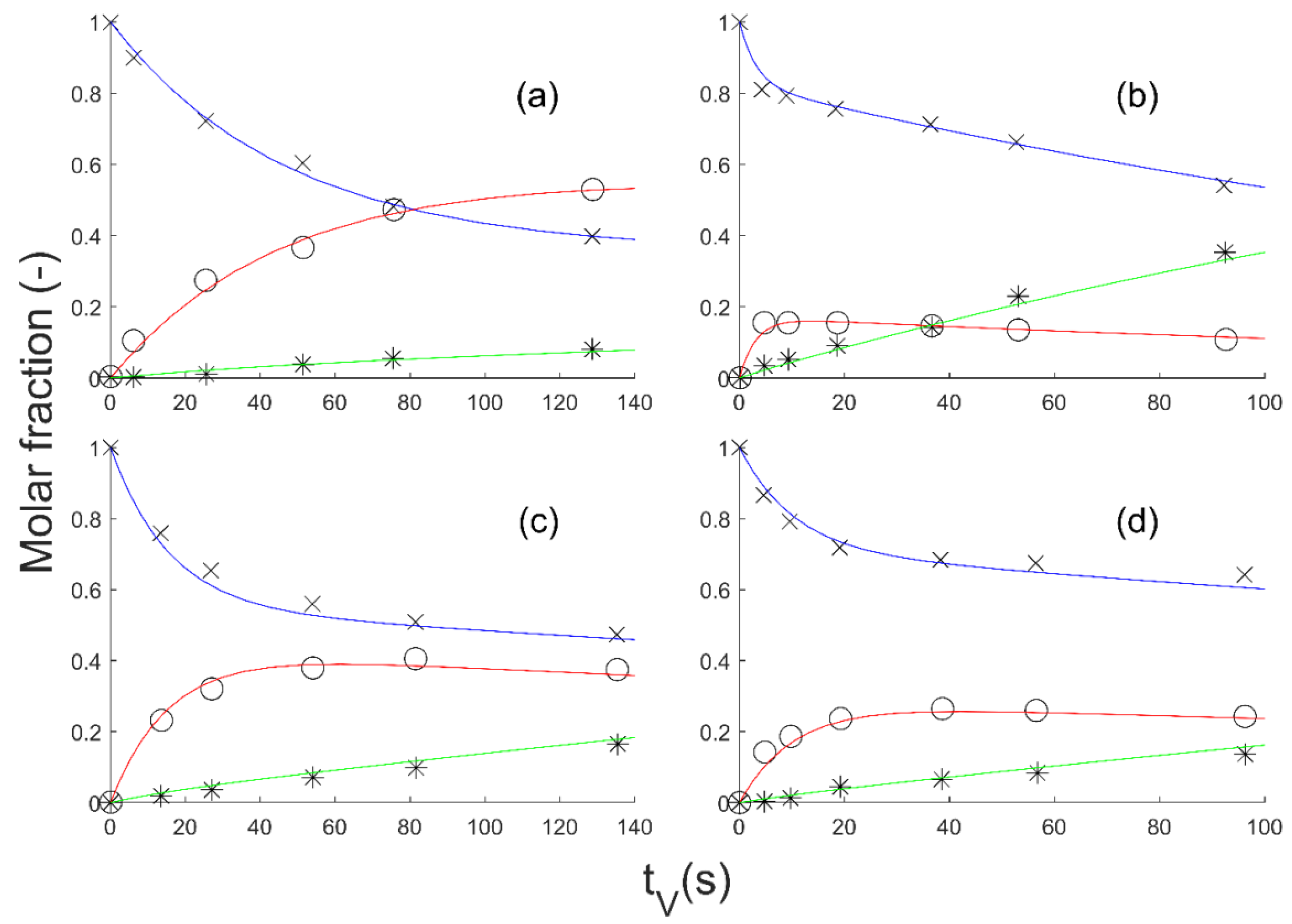
In this paper, based on these previous bibliographic results, a reactor model was first described and then several kinetic laws were tested and kinetic parameters estimated in order to predict the effect of temperature and pressure on the gas-phase hydrogenation of GBL into BDO. The kinetic laws were established in interaction with the thermodynamics to take into account the reversibility of the reactions.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}