3.3. Pure Water
Under an applied electric field with a reasonable potential difference, electrolysis of pure water results in consumption in protons (H+) and release of H2 at cathode and releasing O2 by consuming ‘hydroxyl’ (OH−) ions at anode. To complete the circuit, the OH− ion from cathode must be driven towards anode. At the anode, the OH− ion recombines with excess H+ into water molecules again. However, pure water has a low rate of dissociation as well as poor conductivity. Consequently, pure water goes through a very slow auto-ionization process. Hence, growing accumulation of H+ ions at the anode results in overpotential that slows down the electrolysis process, and the pH of the pure water is found to be basic near cathode and acidic near anode.
Addition of an electrolyte to pure water raises the conductivity of water. An aqueous electrolytic solution mostly dissociated into its corresponding anionic and cationic (let us say for NaCl, and Na+ and Cl−) counterpart, along with H+ and OH−.
Equilibrated aqueous solution:
With an applied electric field, the electrolytic anions and cations quickly remove any accumulation of surface charges around the electrodes, helping a continuous flow of electricity between the electrodes and through the solution, i.e., complete the circuit. Thus, the electrochemical reactions of the water at the electrodes compete with the electrolyte ions present at the solution, determining the excess or insufficient proton concentration around both electrodes. Generally, a combination of an electrolytic anion having a higher electrode potential (>1.23 V) and an electrolytic cation having negative potential (<0 V) is used as an electrolyte. Therefore, within a microchannel, adding an electrolyte kinetically controls the electrical circuit completion.
In the presence of an electrolyte, under an applied electric potential, the following electrochemical half-reactions complete around both electrodes are plausible.
Around the anode with 0.08 V potential, the oxidation of water with the release of O
2 results in the accumulation of H
+ and makes the solution more acidic, i.e., lowering the local pH around the anode. On the contrary, the reduction occurs near the neutral cathode (0 V) with the release of H
2, and the drop of proton concentration results in increasing the pH locally and making a basic environment around the cathode.
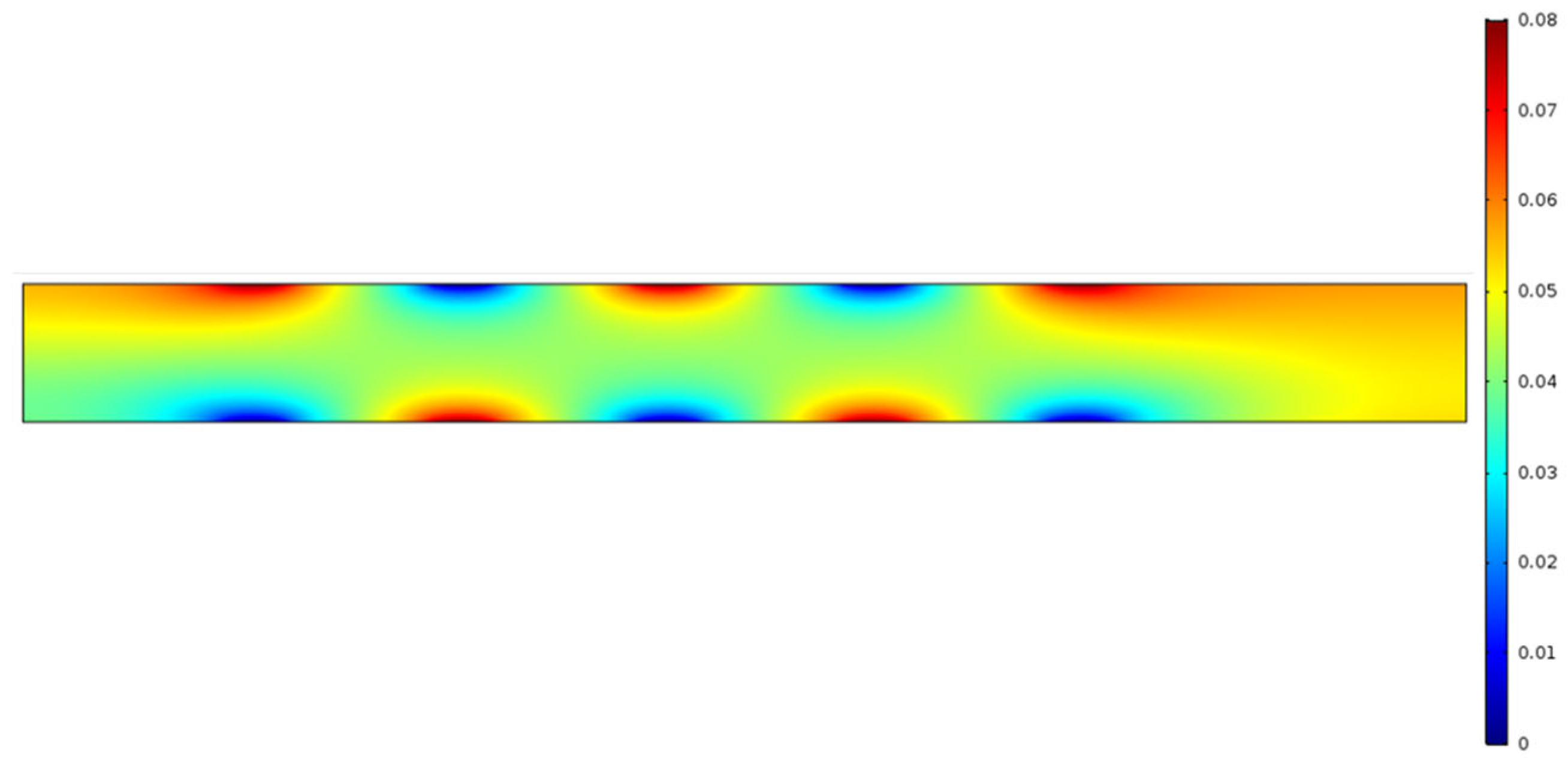
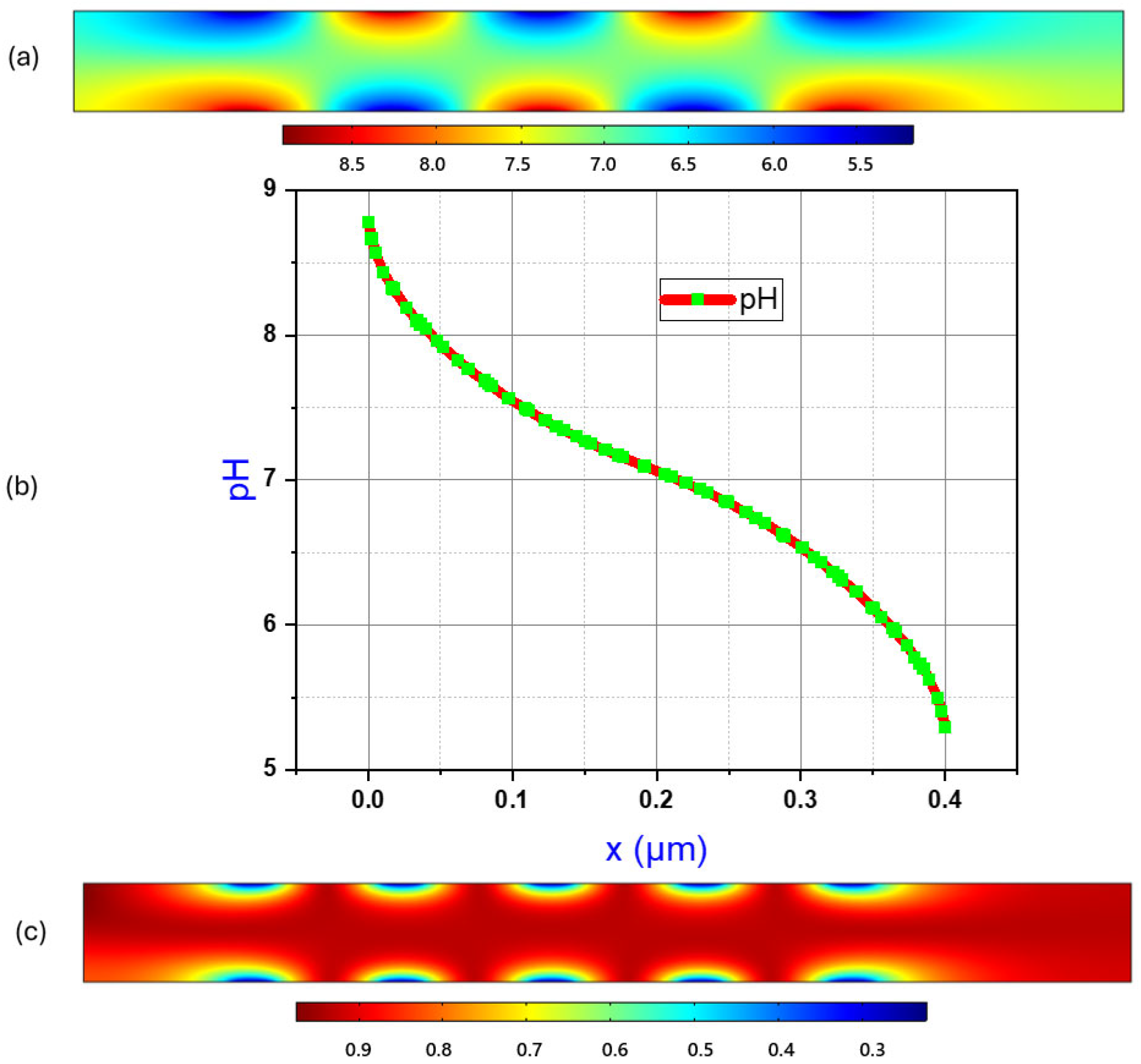
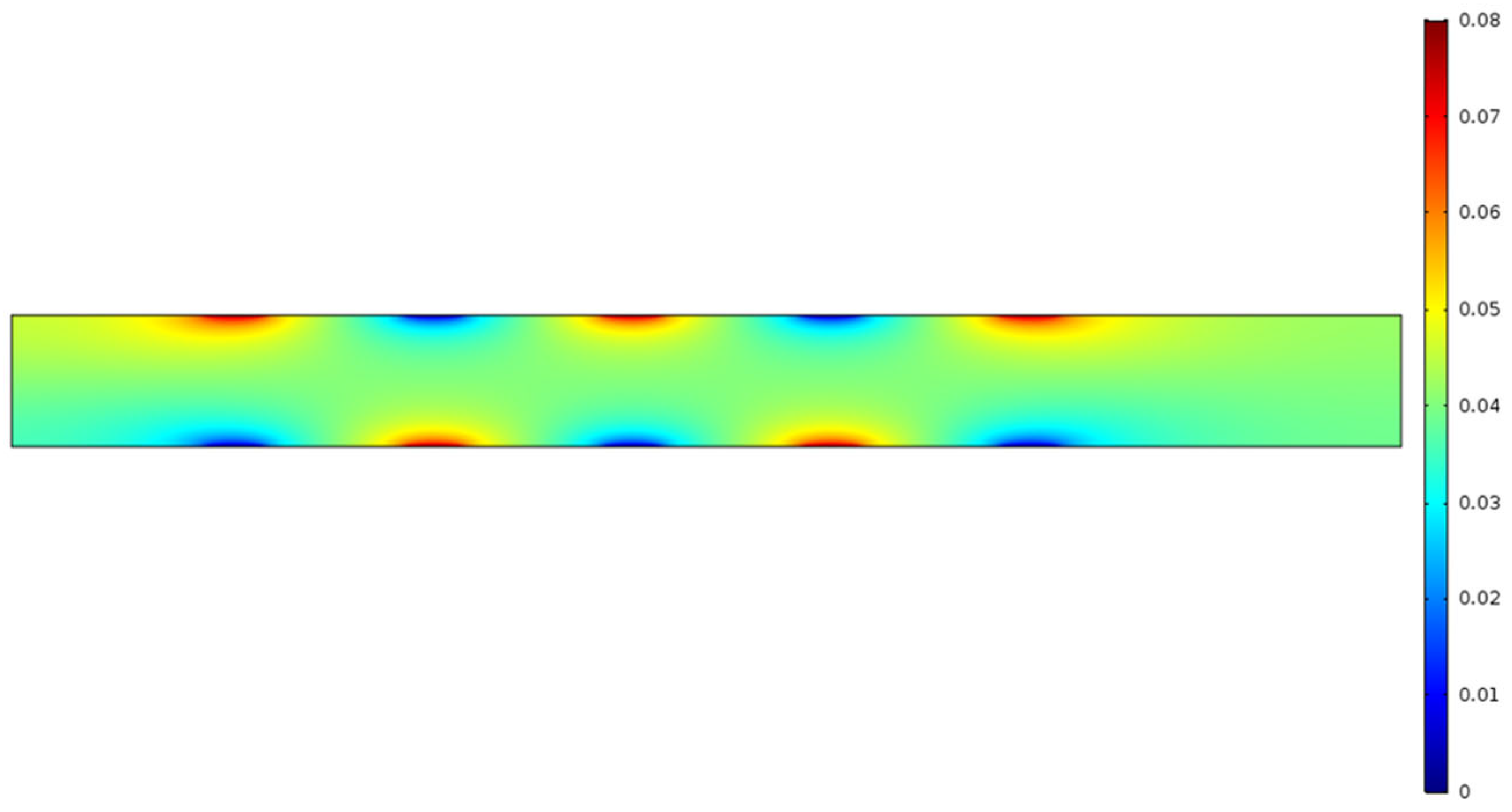
Figure 4a demonstrates the pH-gradient of the aqueous solution in the micro-channel in presence of an electrolyte, such as NaCl. The simulation clearly demonstrates that, under an applied electric field, for an aqueous electrolytic solution, the anode is acidic in nature (low pH; blue region), the cathode is basic in nature (high pH; red region), and there is a continuous gradient of pH that exists between each pair of electrodes, showing mostly neutral pH (green region) around the central part of the micro-channel.
Figure 4b supports the observation quantitatively.
The line graph in
Figure 4b plots measured pH values from the same simulation on the y axis with distance from the bottom of the channel plotted on the x axis. The coordinates (0.6 µm, 0.0 µm) and (0.6 µm, 0.4 µm) are used to plot a 2D cut line.
Figure 4b represents a cross section of the microchannel in this way as means to understand how pH gradients are created between anode and cathode across the channel. For both
Figure 4a,b, the highest pH reading is 8.78 and the lowest pH is 5.29. Recall that the microchannels in this experiment are a height of 0.4 μm.
3.6. Weak Acid and Base Surface Concentration Behavior
Next, we simulated a weak acid and weak base in the microchannel at the electrode array under the same conditions as previous simulations with 0.08 V of AC applied at the anode.
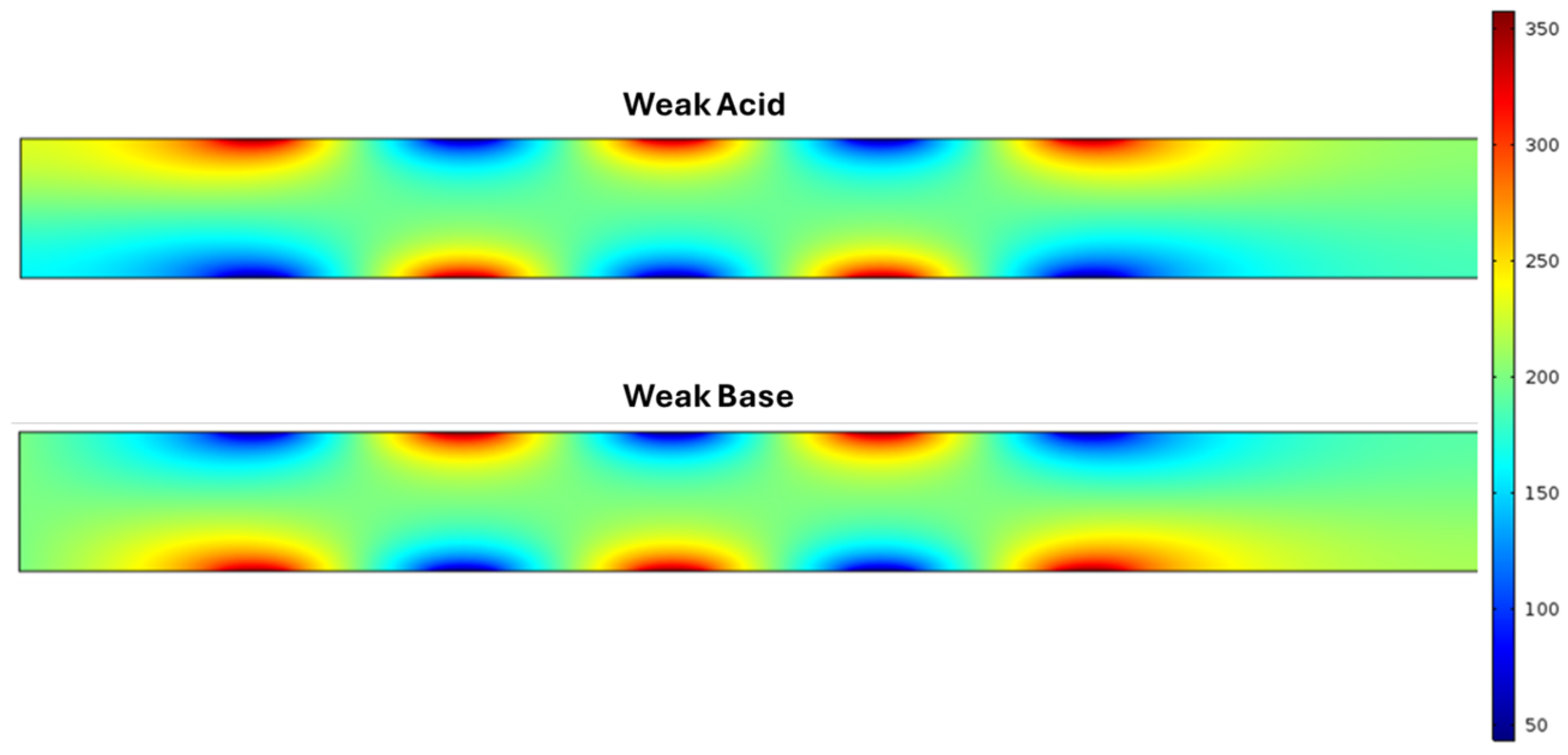
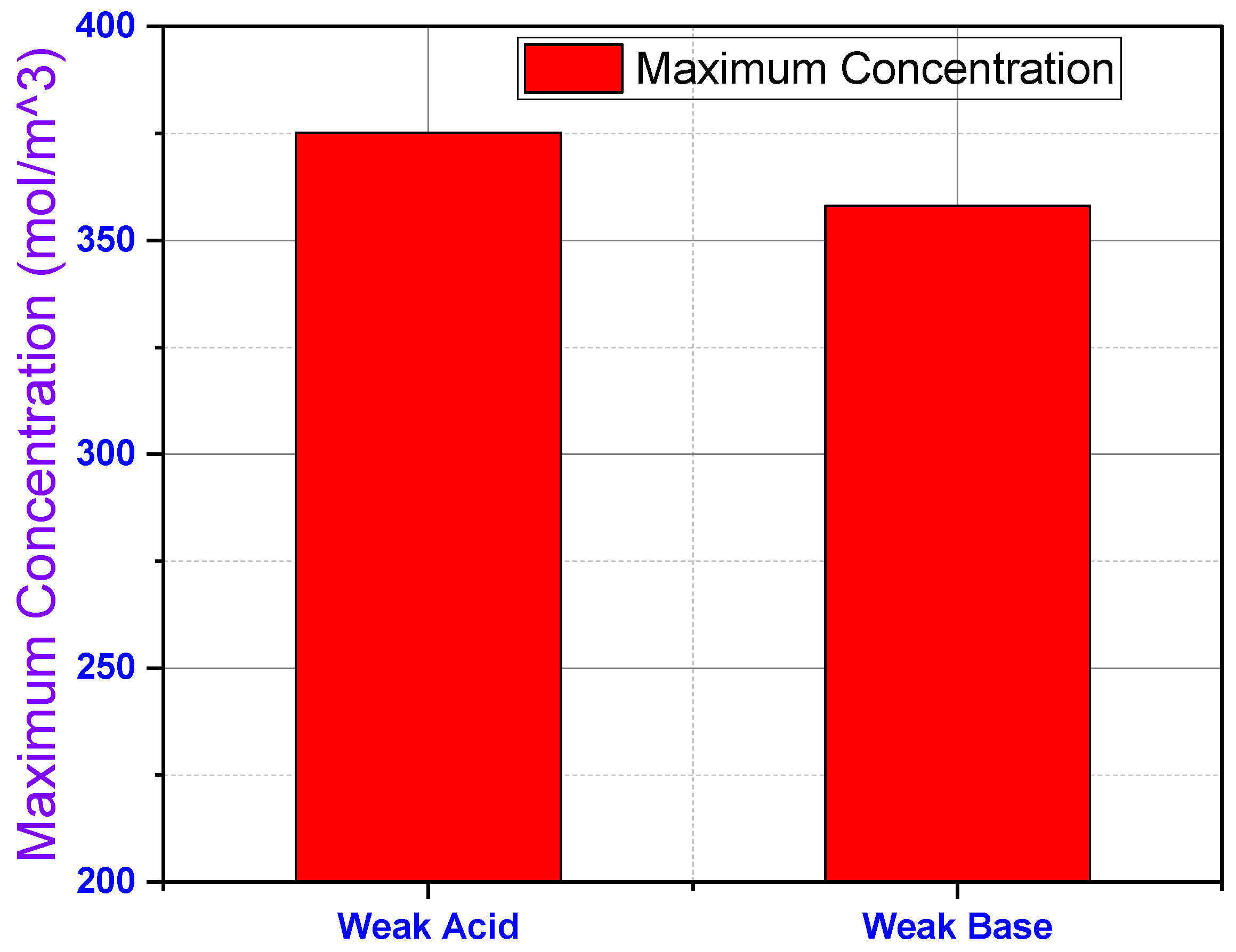
Figure 6 shows a surface graphic of Weak Acid (top). We recorded a maximum concentration of 375.24 mol/m
3 at the anode, and a notable region of low concentration is focused on the cathode. The regions of high and low concentration in these trials demonstrate the same semi-circular shape seen in previous trials at varying degrees. The surface graphic for Weak Base in
Figure 6 (bottom) shows an identical but inverse behavior at the anode and cathode sides of the electrode array. Here, the simulation of weak base under the same conditions results in the maximum concentration of 358.06 mol/m
3 occurring at the cathodes with the lowest concentration occurring at the anode. The bar graph in
Figure 7 shows the maximum concentrations of each simulation visualized in
Figure 6. These simulations demonstrate how the pH of a solution dictates the distribution of concentration in a fluid within a microchannel in the presence of AC.
From an electrochemical point of view, a most likely explanation of the behavior of weak acid and weak base in the microchannel is the following. Unlike the electrolytes, the weak acids and bases mostly remain undissociated in aqueous solution. While dissociated, the conjugate anion of a weak acid behaves as a strong base, and the conjugate cation of a weak base behaves as a strong acid. In the presence of electrolytic ions, the conjugated base and conjugated acid forms a ‘buffer solution’ with the corresponding weak acid and weak base, respectively. When the buffer solution is in the presence of a certain excess proton concentration, i.e., at a low pH, the conjugate anion of the dissociated weak acid immediately consumes H
+, driving the equilibrium towards undissociated weak acid. Similarly, an abundance of hydroxyl (OH
−) ions or lack of H
+ concentration at certain threshold, i.e., at a high pH, a weak base is forced to remain mostly undissociated. Following is the chemical equilibrium of a hypothetical weak acid (HA) and a weak base (BOH), respectively, in an aqueous solution of electrolyte.
Equations (13) and (14) represent the drive of the equilibrium of a weak acid and its conjugate salt form in presence of excess H+ and a weak base and its conjugate salt in presence of excess OH−. Consequently, under an electric field, the weak acid dissociation around the anode is less favored due to the acidic condition (see Equation (13); low pH). Most likely, the half reactions at both of electrodes in presence of a weakly acidic buffer solution are the following:
Equations (15)–(18) show that, at the cathode, the presence of an excess of OH− accelerates the dissociation of the weak acid (HA) to its conjugate base (A−), driving the electric flow in between the electrodes through the ion-transport of A− within the electrolytic solution. At anode, the excess of H+ is quickly removed by the A− ions and accumulates as HA around the anode, thus completing the circuit.
Conversely, due to the basic environment around the cathode (see Equation (16); high pH), the dissociation of a weak base is suppressed. The likely half reactions at the electrodes are the following in the presence of a weak base:
Equations (20)–(23) show that the low pH around the anode favors a weak base (BOH) to be dissociated into its strong conjugate acid (B
+), which transport the charges to cathode through the electrolytic solution and removes the accumulated OH
− at the electrode. Thus, the electric circuit becomes complete by the surface accumulation of the weak base around the cathode.
Figure 8 illustrates that the simulation agrees that the surface concentration of the weak acid is largely concentrated around the anode and the weak base around the cathode.
By definition, the pK
a (=−log
10K
a) is the measure of the strength of an acid or base based on their dissociation rate. A pK
a of an acid and the pH = −log
10C
H+ is related as following:
pK
a is the inherent property of a weak acid/base specifying their ability to donate or accept H
+. A low pK
a for an acid indicates high donation of protons, leading to a low pH medium. However, in an aqueous solution, as the dissociation strength of water (pK
w) is 14, the dissociation rate of a weak base (pK
b) in an aqueous solution is related to pH and pK
a in the following manner:
A low pK
b indicates strong dissociation of the corresponding base. The pK
a (=8) of the given aqueous solution of the weak acid and the pK
b (=14 − pK
a = 14 − 6 = 8) of the given aqueous solution of the weak base (given pK
a = 6) indicate that similar dissociation rates. In the presence of an electrolyte, the solution of both weak acid and base generates a mixture of buffer solution having a pH slightly less than 7.0. As a result, the accumulation of a weak acid at the anode is slightly higher than the accumulation of a weak base at the cathode, as evidently shown in
Figure 7.
A protein, in general, is comprised of various amino acids having multiple functional groups that can accept or donate protons based on the pH of the aqueous solution, if soluble in water. Hence, depending on the total pH of the solution, a protein can have either a net negative or a positive charge, behaving as a zwitterion. Having a net positive charge, a protein can act as an acid or having a net negative charge, a protein can act as a base. Therefore the protein solutions, may behave as a weak acid or weak base respectively, based on the dissociation strength of the proteins. However, at certain pKa range, the same protein solution may not have any negative or positive charge, which is otherwise known as ‘isoelectric point (pI)’. Hence, for a solution of multiple proteins, at a certain pH of the solution; if the pH > pI of a given protein in the solution, then the protein has a net negative charge in the solution; otherwise, if the pH < pI of the protein, then in the solution the protein remains positively charged. Therefore, under an applied electric field, the protein solution having pI less than the overall pH accumulates at the anode and solution having pI greater than the overall pH accumulates at the cathode within the microchannel. Utilizing this property of protein in the presence of a buffer, and under a certain potential difference between the electrodes, the simulations are run within the microchannel for a mixture of five hypothetical proteins, having various pI values, in the presence of a buffer solution.
3.7. Protein Molar Concentrations
A constant 0.08 V of AC was simulated at the electrode array for the protein solutions 1 through 5, having pI = 5.0, 6.0, 7.5, 8.5, and 9.5 respectively. Diffusivity for all proteins was set at 5 × 10
−10 m/s
2 with an inlet concentration of 5 mM. Our results show that the 5 protein solutions demonstrated notably different behavior in the distribution of molar concentration.
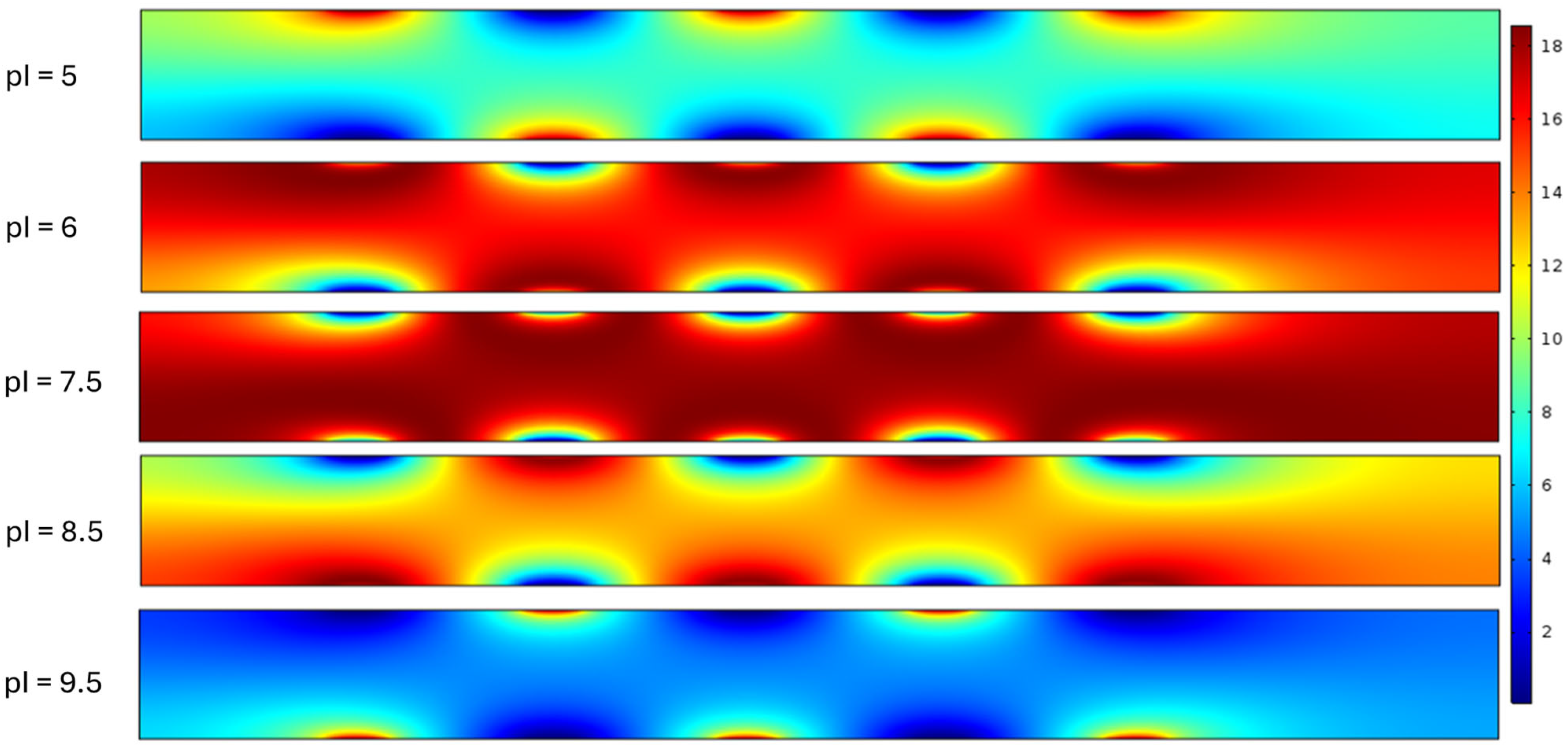
Figure 8 shows a compilation of graphical renderings describing recorded surface concentration in the microchannel at the array. Since numerical concentration results varied between experiments, color coding in this graphic is relative with extremes described between ‘high’ and ‘0’. Channel height is measured in μm from the bottom of the microchannel (0 μm) to the top of the microchannel (0.4 μm). All five proteins in the study have different pH.
In the protein 1 (pI = 5.0) solution we see the greatest molar concentration of 11.72 mol/m
3 at the site of positive charge. Concentration decreases moving vertically across the channel toward the cathode where concentration is effectively 0, or 0.209 mol/m
3. Referring to
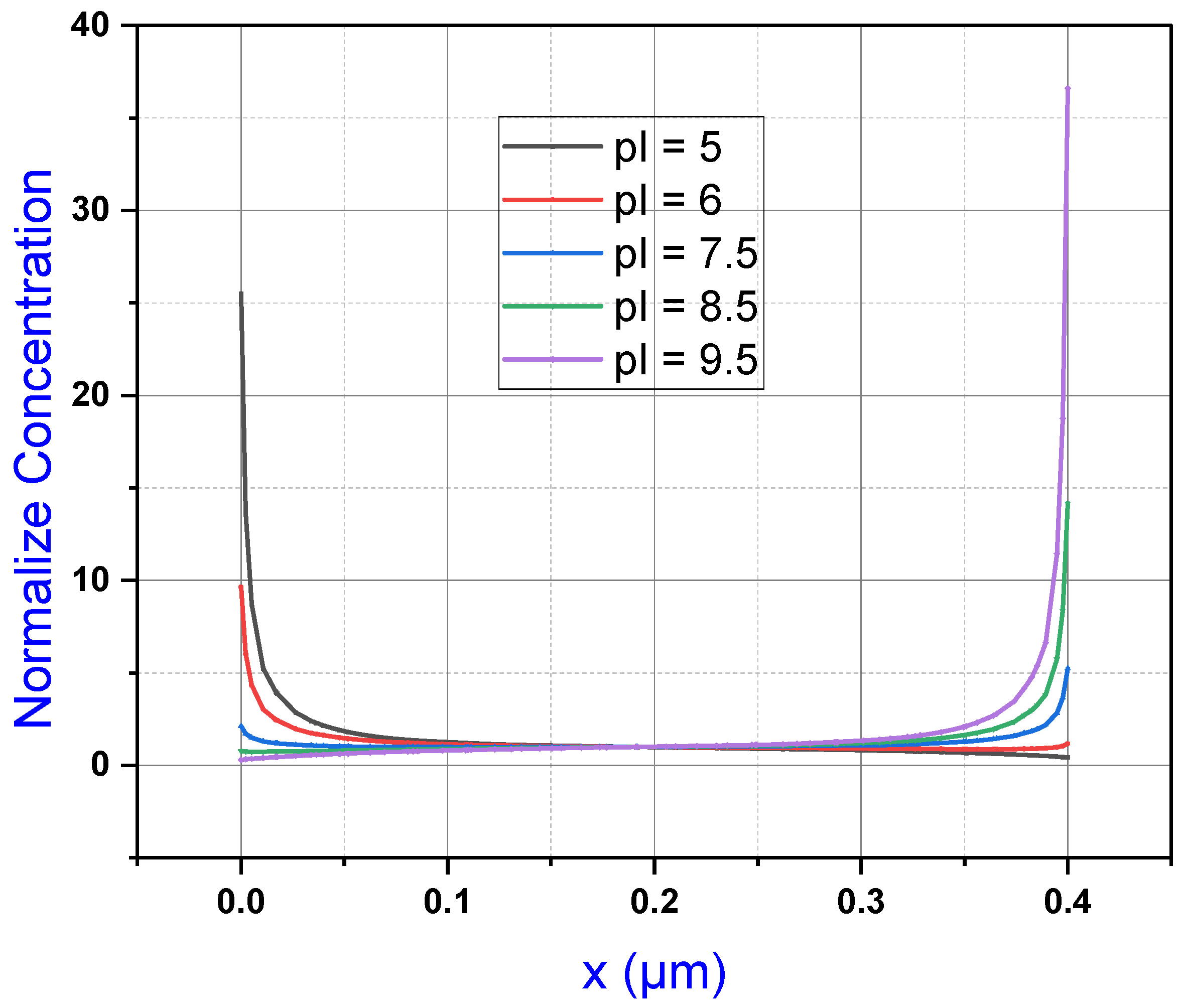
Figure 9, we see that all five protein solution concentrations visualized in
Figure 8 are superimposed on one another. Note that the scope of the graph in
Figure 9 consists of molar concentration (mol/m
3) on the
y-axis and the distance vertically across the microchannel from the bottom of the channel to the top. Locating protein 1 on this graph represented by the dark blue line, we can see the how the concentration of the solution arcs in a negative parabolic curve from 0.209 mol/m
3 toward a concentration of roughly 5 mol/m
3, recorded as 4.99 mol/m
3 at 0.17 μm. From this point, the parabolic curve changes to a positive parabolic curve up to the maximum concentration for protein 1 at 11.72 mol/m
3.
The graphic of the protein 2 (pI = 6.0) solution from
Figure 8 shows an orientation of concentration like that of protein 1 with high concentration at the anode and low concentration at the cathode, but with a much different distribution. Here, we can see that the high concentration of 5.71 mol/m
3 gathers around the anode. This high concentration is located at height of 0.34 from the bottom of the channel with a small region of lower concentration of 3.95 mol/m
3 at 0.4 μm. Concentration remains relatively high moving toward the cathode. Concentration is 4.92 at the 0.17 μm midpoint of the channel as can be seen in
Figure 9. Following the orange line for protein 2, we see how this concentration gradient takes on a more linear relationship down toward the cathode until 0.064 μm where it begins to decline at a parabolic rate down to 0.67 mol/m
3 at 0 μm. The visualization of protein 2 in
Figure 8 expresses this increased rate of change by the tighter regions of decreasing concentrations.
Protein 3 (pI = 7.5) mimics the drops in concentration at the electrodes as seen in protein 2 but does so more abruptly. We can also note that the high concentrations occurred in the middle of the microchannel and were between 0.03 μm and 0.34 μm. The concentration in the protein 3 simulation does not drop below 4 mol/m
3.
Figure 9 gives a clear visualization of this restricted distribution as the gray line for protein 3 is a more horizontal line contained within a more neutral pH region. Protein 3 shares the point of equal concentrations, as do other solutions at 0.17 μm in the microchannel where its concentration is 4.91 mol/m
3. Points of low concentration occur at each electrode with 0.75 mol/m
3 at a height of 0 μm with the cathode and 1.02 mol/m
3 at 0.4 μm with the anode.
Protein 4 (pI = 8.5) demonstrated notable regions of low concentration at the anodes. Here, the low was measured at 0.25 mol/m
3 at 0.4 where the anode is located. The highest pH value occurred at the cathode with a concentration of 9.53 mol/m
3 at a height of 0 μm. The surface graphic in
Figure 8 for protein 4 displays regions of semicircular high and low regions at the respective electrodes. Concentration declines moving toward the center of the microchannel, giving way to a large region of lower concentration where 6.89 mol/m
3 was recorded at a height of 0.17 μm. In
Figure 9, we can see this decline in concentration for protein 4 as indicated by the yellow line mimicking a parabolic rate moving from the cathode at 0 toward the 0.17 midpoint where the line then assumes a negative parabolic-like arc toward the lowest concentration at 0.4 μm.
Protein 5 (pI = 9.5) demonstrated unique behavior at the electrode array where most of the concentration was focused on the cathode at 0 μm with a recorded high of 18.58 mol/m
3. The surface graphic in
Figure 8 indicates this sudden elevation in concentration with very small semicircles of high concentration at cathode sites, which quickly give way to a large region of much lower concentration across the channel to the anode at 0.4 μm. The graph in
Figure 9 gives a clearer indication of this dramatic distribution of concentration, as indicated by the light blue line for protein 5. Here, we can see the dramatic parabolic drop in concentration from 18.58 mol/m
3 at a height of 0 μm to the shared midpoint where concentration was measured at 5.03 mol/m
3 at 0.17 μm. Like other simulations, the concentration readings within the mid-channel region, around 0.17 μm, demonstrate a much gentler gradient. From here, the curve takes on a notably more gradual negative parabolic arc toward a measured concentration of 0.11 mol/m
3 at 0.4 μm.
Here, in the presence of a buffer solution, having a pH slightly less than 7.0, the protein solutions accumulate at cathode or anode depending on their corresponding pI values and how they deviate from the overall pH of the solution. Depending on the predominant net charge on the protein, the plausible half-reactions are very similar to the cases of weak acids and weak bases at the electrodes. A protein with a net negative or positive charge (Pr− or PrH2+) equilibrates with the net neutral protein molecule (HPr) in the buffer solution having electrolytes.
If pH > pI, the protein solution is negatively charged in the presence of a buffer:
If pH < pI, the protein solution is positively charged in the presence of a buffer:
However, the difference between the pI and the overall pH of the solution dictates the dissociation equilibrium of a protein in the solution; hence, it guides the accumulation of protein at corresponding electrodes. As a result, the distribution of the concentration of a given protein solution along the microchannel and at the electrode surfaces, as quantitatively shown in molar concentrations in
Figure 9, can primarily be estimated from simulation results shown in
Figure 8. The higher concentration of protein at cathode indicates more positively charged proteins from the solution are discharged near the electrode (Protein 5 > Protein 4 > Protein 3). At anode, such accumulation indicates more negatively charged protein are discharged from the solution (Protein 1 > Protein 2). However, Protein 2 and Protein 3 rather have a uniform distribution in between the electrodes and minimal accumulation around the anode and cathode surface, respectively, indicating a small quantity of negatively charged and positively charged proteins are generated, respectively, as their pI is closer to the overall pH. For other three cases, the overall pH is either far less or far greater than their pI s; hence, depending on the deviation of pI from the overall pH, given proteins are found to be more concentrated around the corresponding electrode surface. These estimations can be validated from the simulation, by measuring the surface concentration of a given protein in the solution and at the electrodes.
Anode reaction for a protein solution having pH > pI:
Cathode reaction for a protein solution having pH < pI:
Therefore, the fundamental goal of these simulations is whether a given protein solution, based on its corresponding pI, can be identified and separated in a micro-channel by tuning the overall pH using buffer solutions. Dense accumulation of protein around the cathode or anode surface indicates a relatively high concentration of negatively charged or positively charged proteins are generated in the solution, indicating
pIs far from the overall pH of the solution and behave as a relatively stronger base or acid, respectively. For Protein 1 (pI = 5.0), as evident from
Figure 8, the accumulation around the anode in a denser concentration identifies the protein solution was mostly negatively charged in the solution, as shown in Equation (36). Protein 1 generates the conjugate base in the solution, transported to anode and accumulated around the surface of the anode according to Poisson and Nerst–Plank equations. Protein 2, having pI (=6.0) closer to overall pH of the solution, is dissociated in a lower amount of negatively charged protein in the solution and deposited in a lesser amount around the anode surface. A rather uniform spread of protein distribution from the middle of the micro-channel up to around the anode, as seen in
Figure 8, supports the inference. In case of Protein 3–5, as seen in
Figure 8, there is an inverse trend of accumulations around the electrodes, clearly indicating that they were mostly dissociated as a cation in the solution, and a dense accumulation around cathode indicates higher pI than the overall pH of the solution. Protein 3 (pI = 7.5) solution shows a uniform spread of protein concentration along the micro-channel up to the anode, indicating the protein weakly dissociated into its’ positively charged counter ion (see Equation (38)) and rest is mostly undissociated along the microchannel. Protein 4 with pI = 8.5 is dissociated more in cations compared to Protein 3 in the solution, hence comparatively higher accumulation of the protein at the cathode and less concentration of protein in solution is observed within the microchannel. Protein 5, having pI = 9.5, as also evident from
Figure 8, mostly ionizes into cationic counterpart and mostly deposited around the cathode. In this case, the protein leaves almost no trace in the solution in between the microchannel, rather found as a thick deposition around the cathode.
The distribution of the concentration of the corresponding protein solution along the microchannel and at the electrode surfaces is quantitatively shown in molar concentrations in
Figure 9. It supports the simulation results shown in
Figure 8. The concentration of Protein 1 (pI = 5.0) is highest at the anode followed by Protein 2 (pI = 6.5) and almost zero around the cathode, indicating both proteins were predominantly negatively charged in the solution. On the other hand, Protein 5 (pI = 9.5) has the highest concentration around cathode, followed by Protein 4 (pI = 8.5) and Protein 3 (pI = 7.5), and almost no trace is found around the anode, indicating these proteins were predominantly cationic in the solution. As discussed, this is due to the differences between pI of the individual proteins and overall pH of the solution. Protein 3 (pI = 7.5), being very close to the pH of the buffer solution, was found to have minimal accumulation around the cathode. With the change of anode voltage from 0.02 V to 0.14 V, the qualitative features of the accumulation of protein solutions around the electrodes remain same; however, the accumulation becomes significant for each protein solution with the increase in the anode voltage (see surface figure comparison shown in
Figures S2–S4 in the Supplementary Information).
3.8. Isoelectric Point
Despite how individual proteins react to simulated conditions at the electrode array or their maximum recorded molar concentration, the superimposed line graphs of proteins 1 through 5, in
Figure 9, reveals a shared point of concentration mid-channel at roughly 0.17 μm. This convergence of concentrations reveals the isoelectric point in the protein solutions where molecules hold no electrical charge. Isoelectric separation is occurring via electrophoresis at this point in the channel between the anodes and cathodes in the array. Around the middle of the microchannel (0.2 μm), all the ions from the dissociation of electrolyte, weak acid and weak base buffer, and proteins stay in equilibrium and move freely within any direction of the microchannel, helping an efficient ion transport within the microchannel. Hence, all the proteins have the same concentration around the mid region of the microchannel.
We can make an important observation about the relationship between protein pH and concentration behavior at the electrode array here. We can see that proteins 1 and 2 exhibit similar overall behavior with higher concentrations at anode while lower concentrations occur at cathode. The fact that each of these proteins with different pH values share a common point mid-channel, as shown in
Figure 9, indicates as shared neutrality in molecular charge where isoelectric separation will occur for all proteins in the study, regardless of maximum concentration. It can also be understood by looking at the behavior of the lines in the
Figure 9 graph that higher maximum concentrations lead to more dramatic parabolic curves at the electrodes. Solutions with lower maximum pH values display a more linear distribution of concentration across the channel with only slightly parabolic trends at the electrodes.
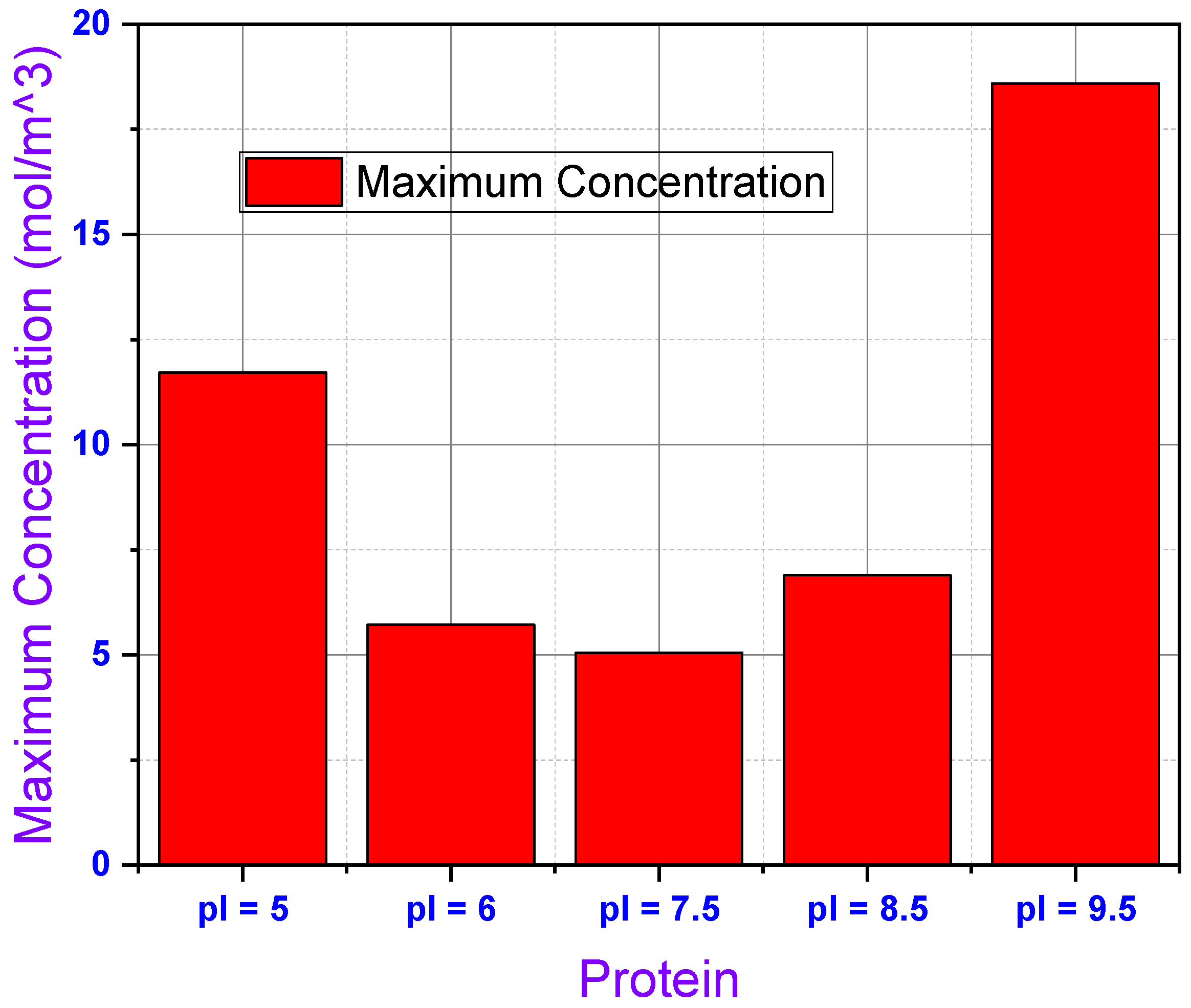
Lastly,
Figure 10 provides an estimate of the surface concentration of proteins at corresponding electrodes and the microchannel in terms of a bar graph of maximum molar concentrations recorded anywhere in the microchannel for the five proteins in this study. This graph highlights the different capacities for surface concentration for the five proteins studied in this simulation. We can see that Protein 1 had the second highest concentration recorded in the microchannel at 11.29 mol/m
3. Protein 2 and 3 had similar maximum concentrations at 5.67 mol/m
3 and 5.04 mol/m
3, respectively, with Protein 3 returning the lowest recorded maximum concentration for the experiment. Protein 4 showed the third highest concentration at 6.75 mol/m
3 and Protein 5 had the highest recorded concentration of the experiment at 17.01 mol/m
3. As the difference between the pI and overall pH of the solution dictates the strength of dissociation of proteins into their anionic/cationic counterparts, Protein 1 and Protein 5 show maximum accumulation due to a large disparity between their corresponding pI and pH of the solution. On the other hand, Protein 2, Protein 3, and Protein 4 show accumulation at almost similar concentration due to small differences between their pI and the overall pH of the solution. Protein 3 has the closest pI with respect to the pH of the solution, hence, the accumulation of this protein at the surface is the minimum.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}