“Click” Chemistry on Gold Electrodes Modified with Reduced Graphene Oxide by Electrophoretic Deposition

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
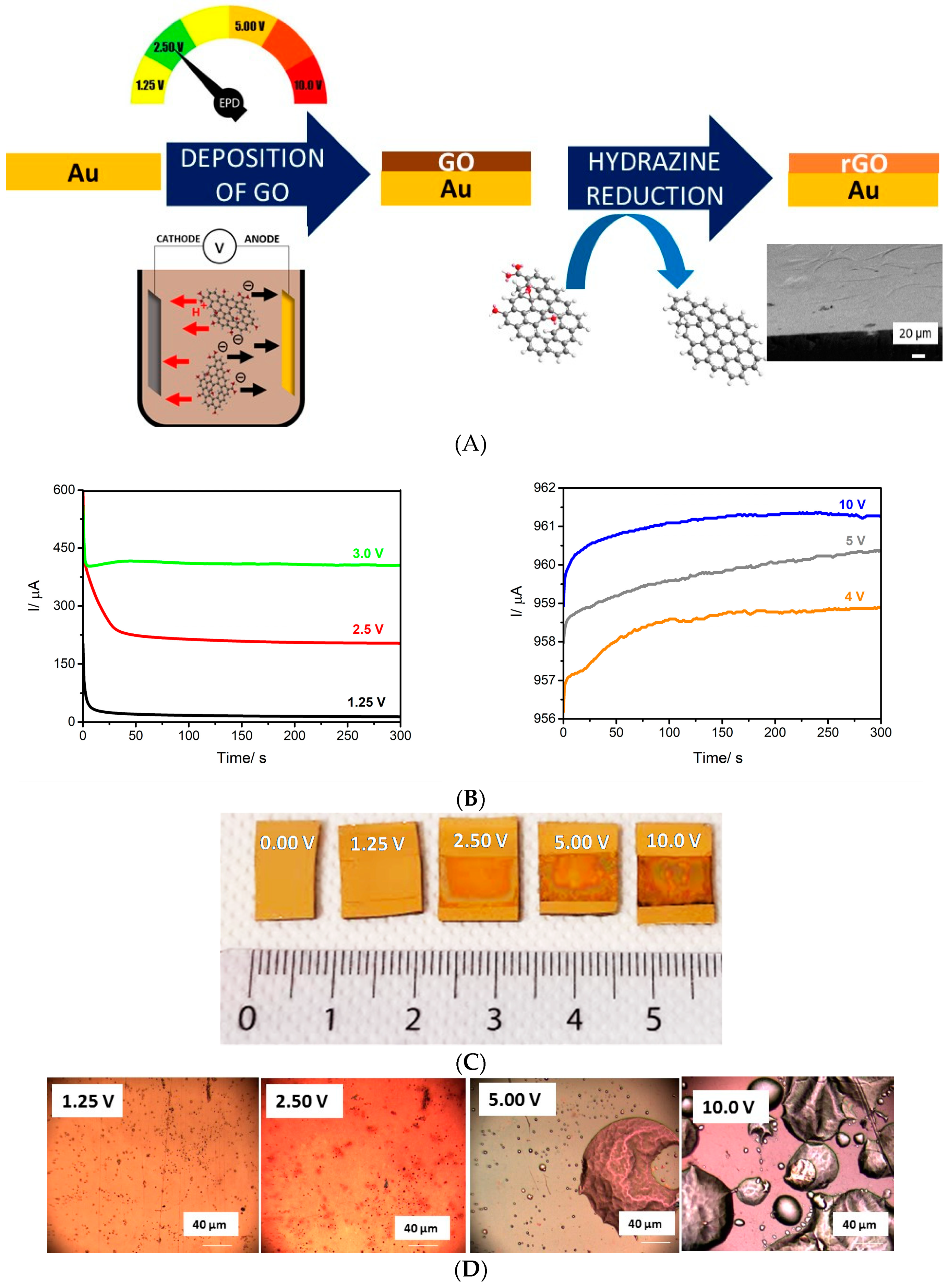
2.2. Electrophoretic Deposition
2.3. Surface Modification
2.3.1. Diazonium Chemistry
2.3.2. “Click” Chemistry
2.4. Surface Characterization Techniques
2.4.1. Scanning Electron Microscopy (SEM)
2.4.2. X-Ray Photoelectron Spectroscopy (XPS)
2.4.3. Electrochemical Measurements
2.4.4. Micro-Raman Analysis
2.4.5. Atomic Force Microcopy (AFM)
2.4.6. Profilometry
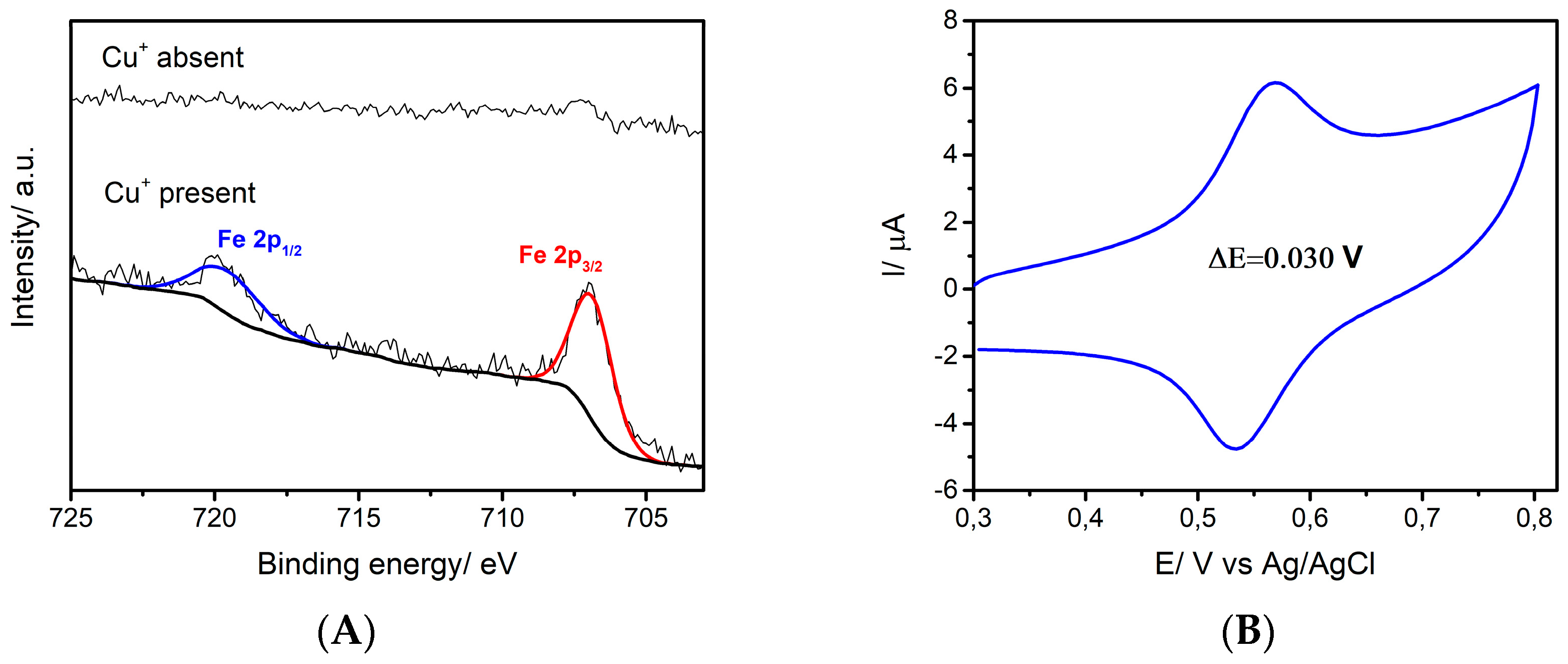
3. Results
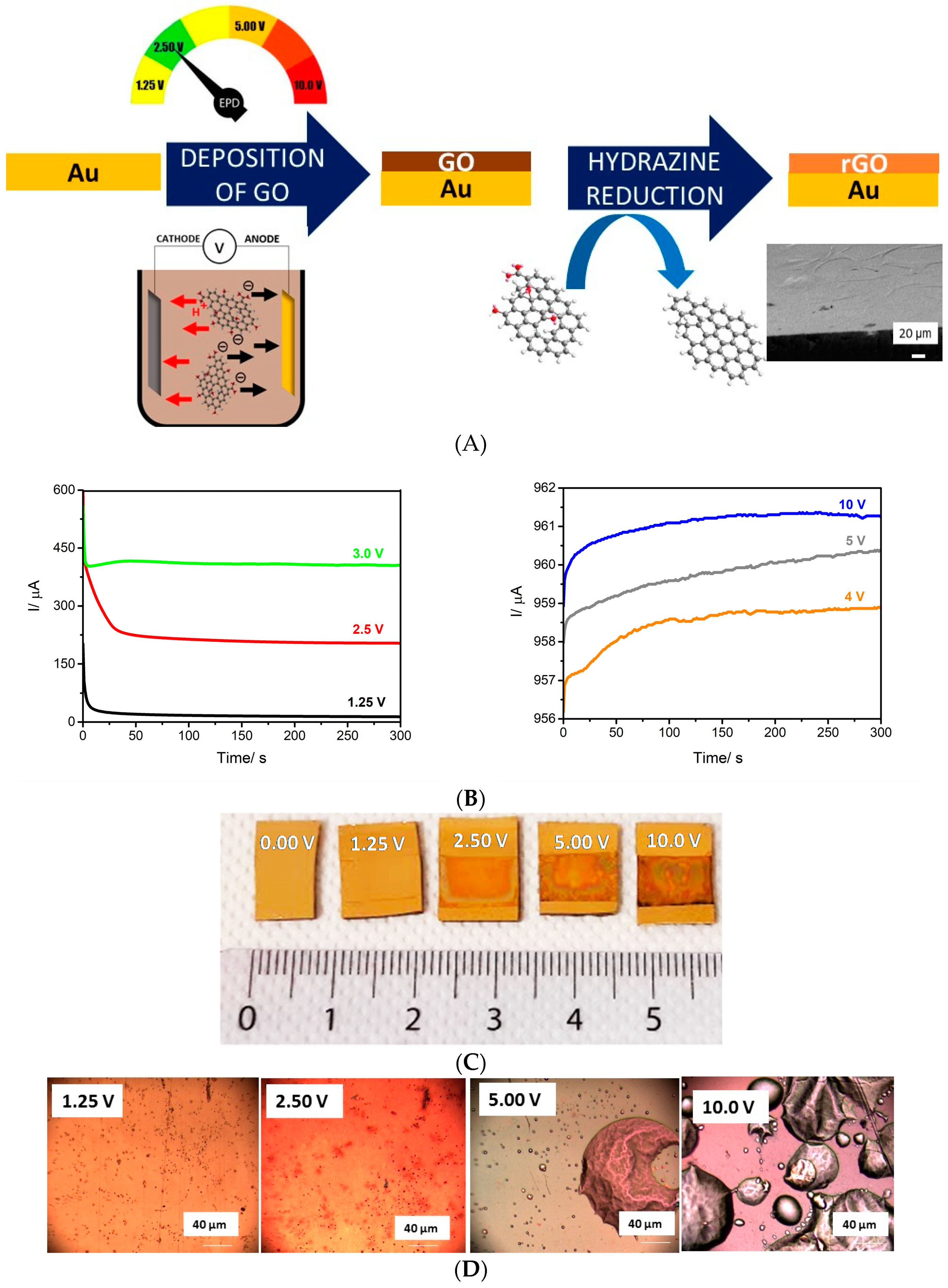
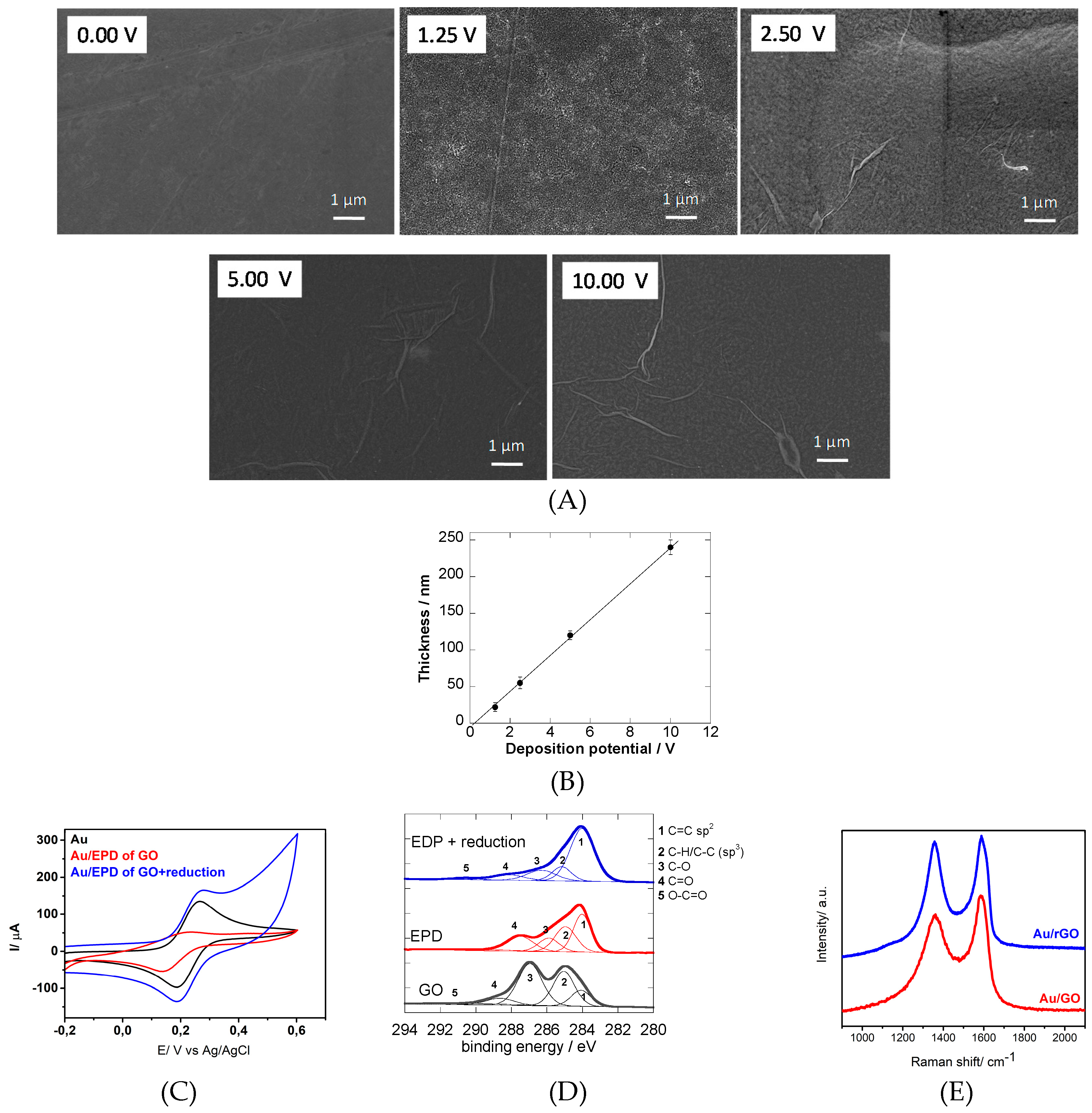
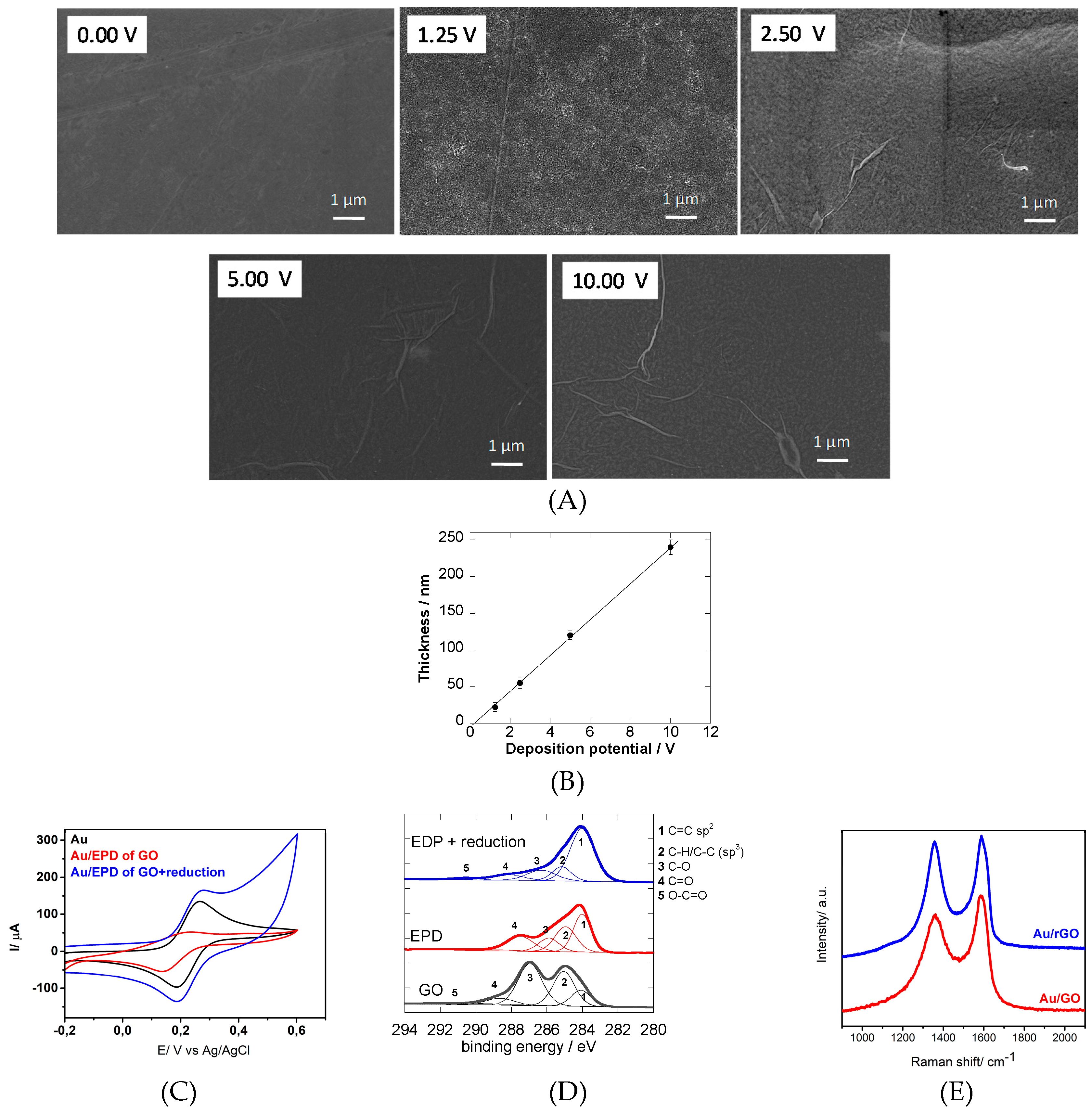
3.1. Electrophoretic Deposition
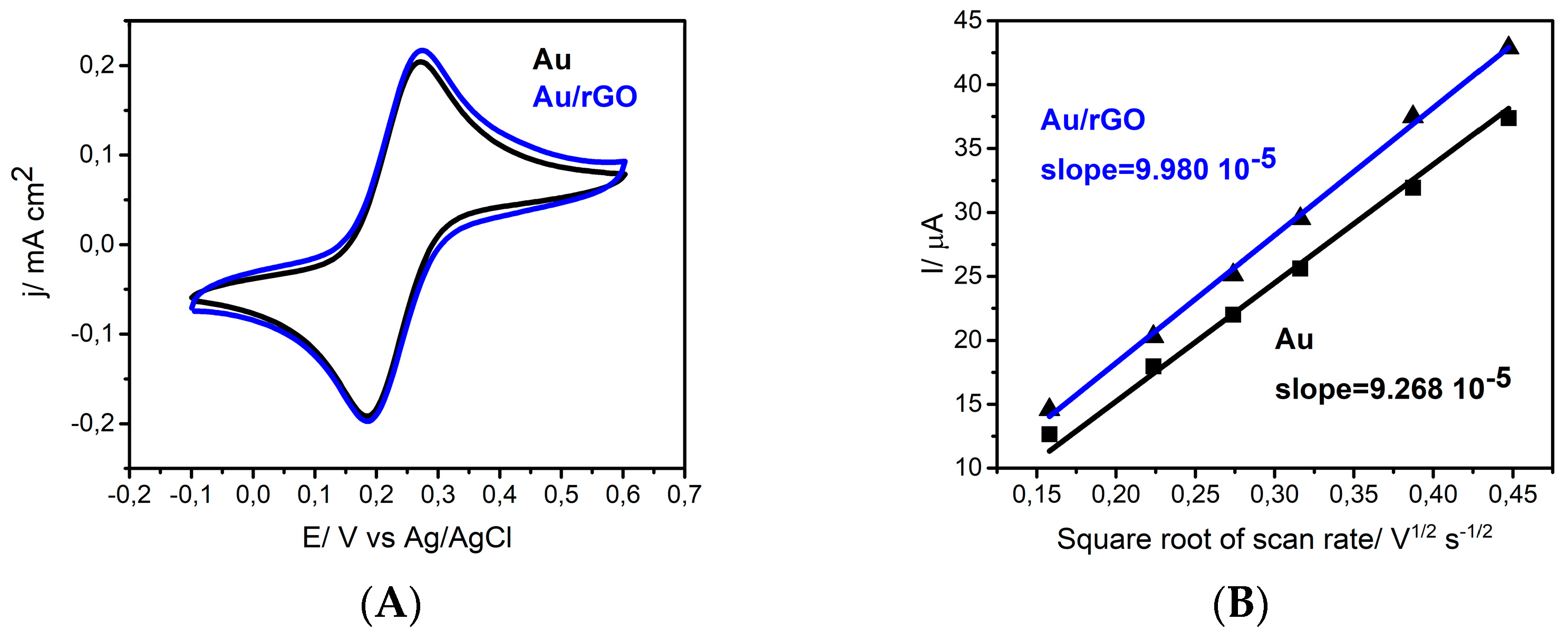
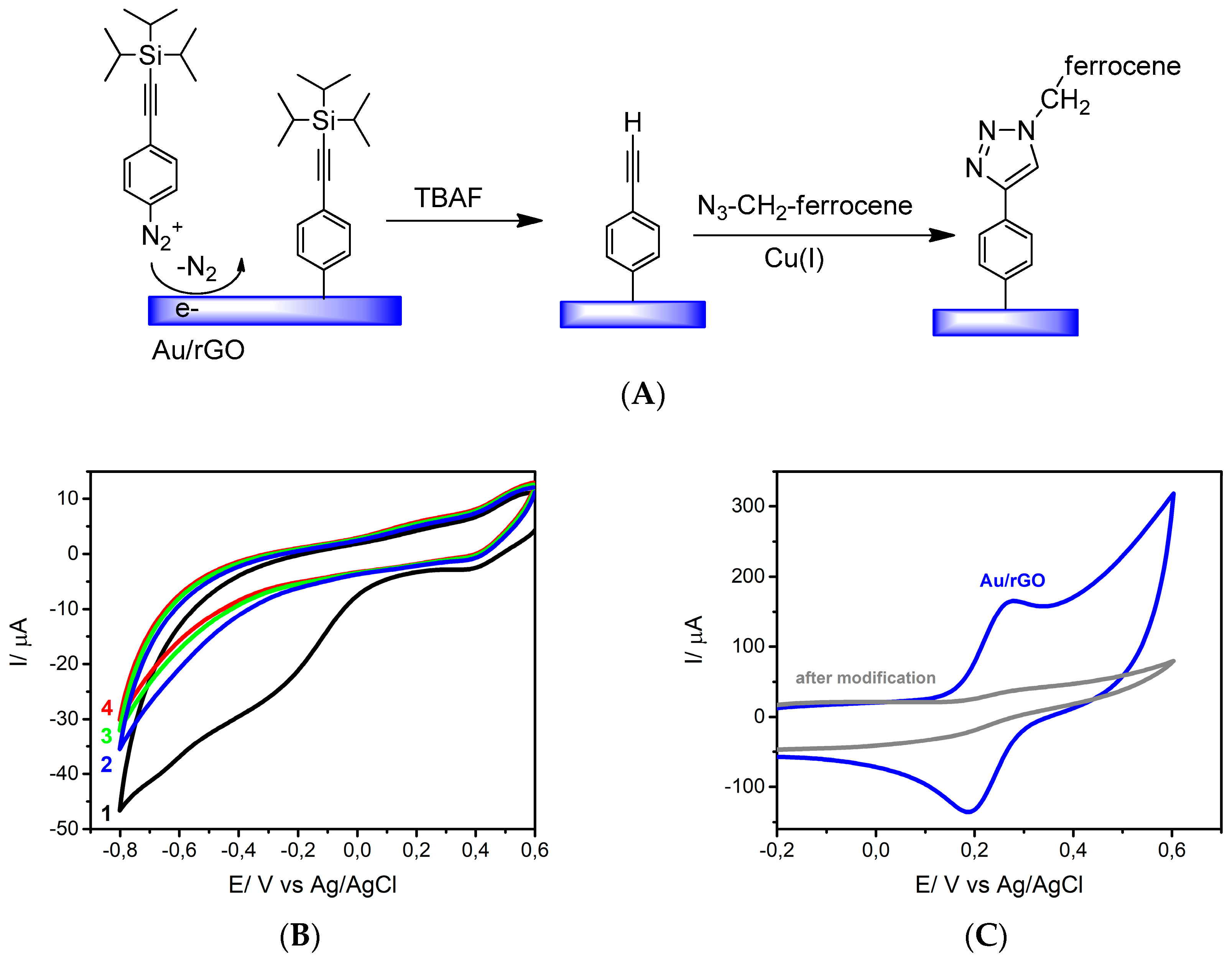
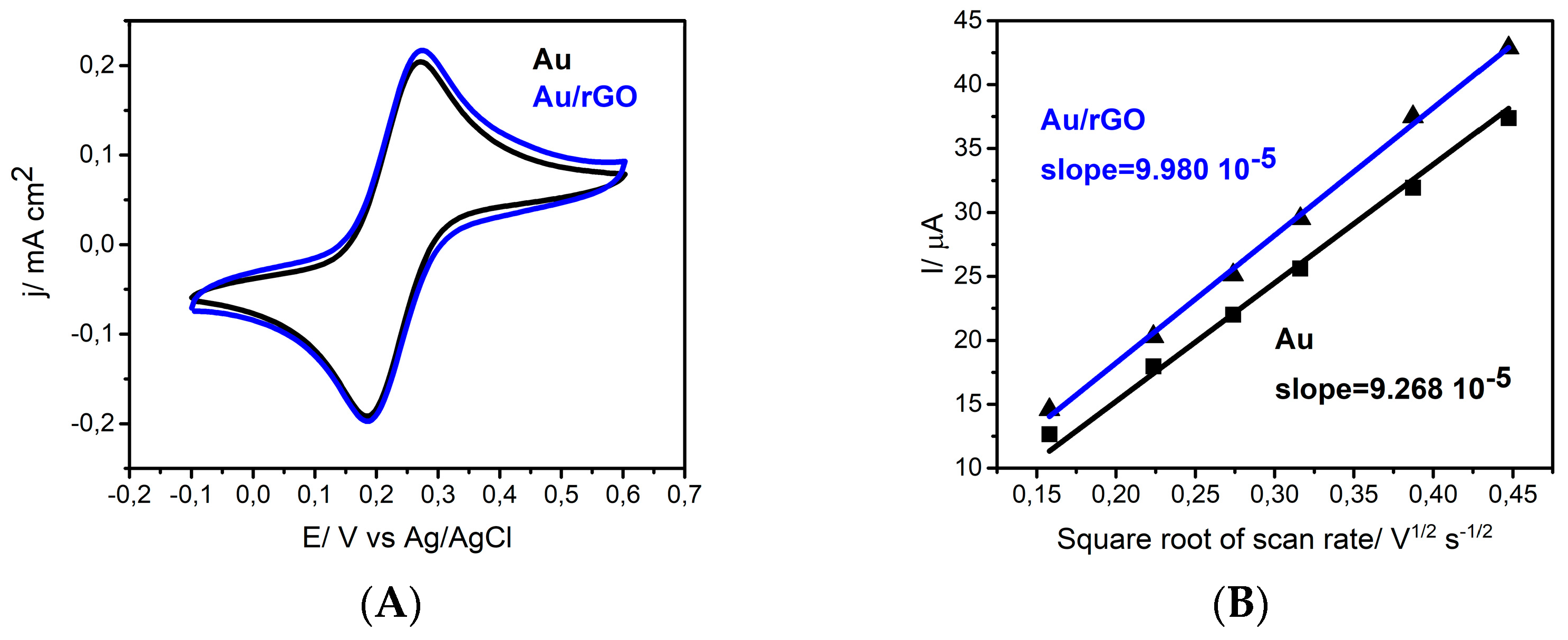
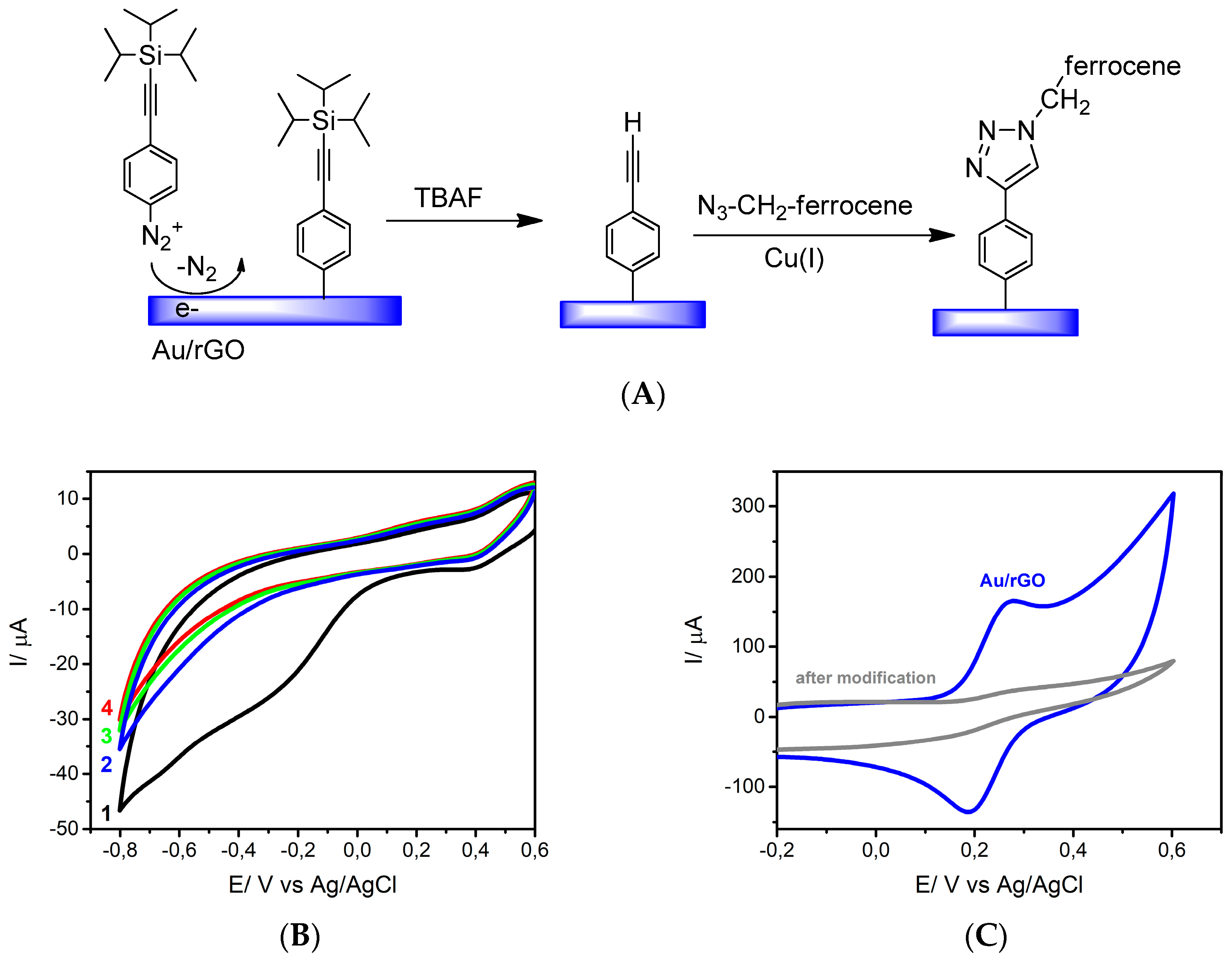
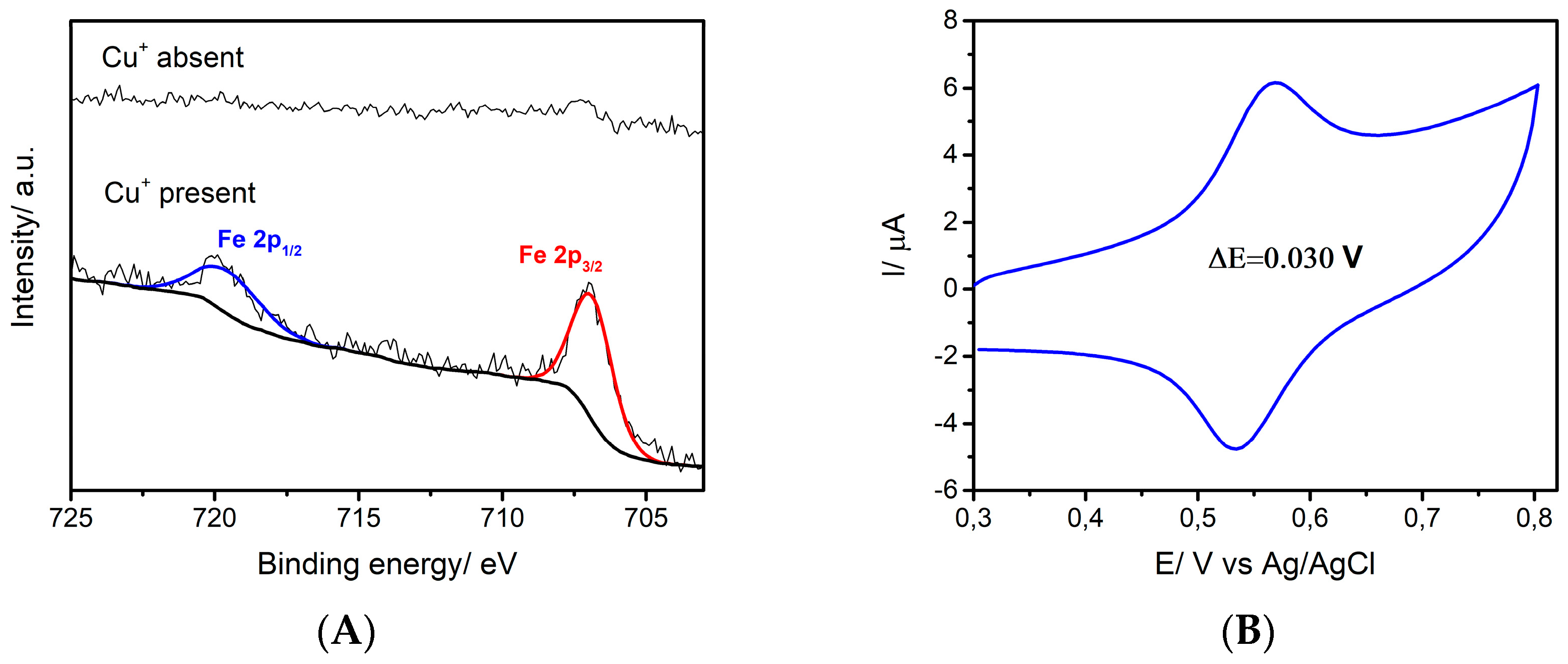
3.2. Surface Modification Using Diazonium Electrografting
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Reina, G.; González-Domínguez, J.M.; Criado, A.; Vázquez, E.; Bianco, A.; Prato, M. Promises, facts and challenges for graphene in biomedical applications. Chem. Soc. Rev. 2017, 46, 4400–4416. [Google Scholar] [CrossRef]
- Szunerits, S.; Boukherroub, R. Graphene-based bioelectrochemistry and bioelectronics: A concept for the future? Curr. Opin. Electrochem. 2018, 12, 141–147. [Google Scholar] [CrossRef]
- Ambrosi, A.; Chua, C.K.; Latiff, N.M.; Loo, A.H.; Wong, C.H.; Eng, A.Y.; Bonanni, A.; Pumera, M. Graphene and its electrochemistry—An update. Chem. Soc. Rev. 2016, 45, 2458–2493. [Google Scholar] [CrossRef] [PubMed]
- Peña-Bahamonde, J.; Nguyen, H.N.; Sofia, K.; Rodrigues, D.F. Recent advances in graphene-based biosensor technology with applications in life sciences. J. Nanobiotechnol. 2018, 16, 75. [Google Scholar] [CrossRef] [PubMed]
- Szunerits, S.; Boukherroub, R. Electrochemistry of graphene: The current state of the art. Electrochemistry 2013, 12, 211–242. [Google Scholar]
- Szunerits, S.; Boukherroub, R. Graphene-based nanomaterials in innovative electrochemistry. Curr. Opin. Electrochem. 2018, 10, 24–30. [Google Scholar] [CrossRef]
- Chekin, F.; Vasilescu, A.; Jijie, R.; Singh, S.K.; Kurungot, S.; Iancu, M.; Badea, G.; Boukherroub, R.; Szunerits, S. Sensitive electrochemical detection of cardiac troponin I in serum and saliva by nitrogen-doped porous reduced graphene oxide electrode. Sens. Actuators B Chem. 2018, 262, 180–187. [Google Scholar] [CrossRef]
- Jijie, R.; Kahlouche, K.; Barras, A.; Yamakawa, N.; Bouckaert, J.; Gharbi, T.; Szunerits, S.; Boukherroub, R. Reduced graphene oxide/polyethylenimine based immunosensor for the selective and sensitive electrochemical detection of uropathogenic Escherichia coli. Sens. Actuators B. 2018, 260, 255–263. [Google Scholar] [CrossRef]
- Wang, Q.; Vasilescu, A.; Wang, Q.; Coffinier, Y.; Li, M.; Boukherroub, R.; Szunerits, S. Electrophoretic Approach for the Simultaneous Deposition and Functionalization of Reduced Graphene Oxide Nanosheets with Diazonium Compounds: Application for Lysozyme Sensing in Serum. ACS Appl. Mater. Interfaces 2017, 9, 12823–12831. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, P.; Lesniewski, A.; Kaminska, I.; Vlandas, A.; Vasilescu, A.; Niedziolka-Jonsson, J.; Pichonat, E.; Happy, H.; Boukherroub, R.; Szunerits, S. Lysozyme detection on aptamers functionalized graphene-coated SPR interfaces. Biosens. Bioelectron. 2013, 50, 239–243. [Google Scholar] [CrossRef]
- Subramanian, P.; Barka-Bouaifel, F.; Bouckaert, J.; Yamakawa, N.; Boukherroub, R.; Szunerits, S. Graphene-coated surface plasmon resonance interfaces for studing the interactions between cells and surfaces. ACS Appl. Mater. Interfaces 2014, 6, 5422–5431. [Google Scholar] [CrossRef] [PubMed]
- Diba, M.; Garcia-Callastegui, A.; Klupp Taylor, R.N.; Pishbin, F.; Ryan, M.P.; Shaffer, M.S.P.; Boccaccini, A.R. Quantitative evaluation of electrophoretic deposition kinetics of graphene oxide. Carbon 2014, 67, 656–661. [Google Scholar] [CrossRef]
- An, S.J.; Zhu, Y.; Lee, S.H.; Stoller, M.D.; Emilsson, T.; Park, S.; Velamakanni, A.; An, J.; Ruoff, R.S. Thin Film Fabrication and Simultaneous Anodic Reduction of Deposited Graphene Oxide Platelets by Electrophoretic Deposition. J. Phys. Chem. Lett. 2010, 1, 1259–1263. [Google Scholar] [CrossRef]
- Besra, L.; Liu, M. A review on fundamentals and applications of electophoretic depositon (EDP). Prog. Mater. Sci. 2007, 52, 1–61. [Google Scholar] [CrossRef]
- Ammam, M. Electrophoretic deposition under modulated electric fields: A review. RSC Adv. 2012, 2, 7633–7646. [Google Scholar] [CrossRef]
- Ata, M.S.; Liu, Y.; Zhitomirsky, I. A review of new methods of surface chemical modification, dispersion and electrophoretic deposition of metal oxide particles. RSC Adv. 2014, 4, 22716–22732. [Google Scholar] [CrossRef]
- Boccaccini, A.R.; Cho, H.; Roether, J.A.; Thoimasz, B.J.C.; Minay, E.J.; Shaffer, M.S.P. Electrophoretic deposition of carbon nanotubes. Carbon 2006, 44, 3149–3160. [Google Scholar] [CrossRef]
- Chavez-Valdez, A.; Shaffer, M.S.P.; Boccaccini, A.R. Applications of Graphene Electrophoretic Deposition. A Review. J. Phys. Chem. B 2013, 117, 1502–1515. [Google Scholar] [CrossRef]
- He, L.; Sarkar, S.; Barras, A.; Boukherroub, R.; Szunerits, S.; Mandler, D. Electrochemically stimulated drug release from flexible electrodes coated electrophoretically with doxorubicin loaded reduced graphene oxide. Chem. Commun. 2017, 53, 4022–4025. [Google Scholar] [CrossRef]
- Kahlouche, K.; Jijie, R.; Hosu, I.; Barras, A.; Gharbi, T.; Yahiaoui, R.; Herlem, G.; Ferhat, M.; Szunerits, S.; Boukherroub, R. Controlled modification of electrochemical microsystems with polyethylenimine/reduced graphene oxide using electrophoretic deposition: Sensing of dopamine levels in meat samples. Talanta 2018, 178, 32–440. [Google Scholar] [CrossRef]
- Maaoui, H.; Singh, S.K.; Teodorescu, F.; Coffinier, Y.; Barras, A.; Chtourou, R.; Kurungot, S.; Szunerits, S.; Boukherroub, R. Copper oxide supported on three-dimensional ammonia-doped porous reduced graphene oxide prepared through electrophoretic deposition for non-enzymatic glucose sensing. Electrochim. Acta 2017, 224, 346–354. [Google Scholar] [CrossRef]
- Wang, Q.; Li, M.; Szunerits, S.; Boukherroub, R. Preparation of reduced graphene oxide-Cu composites through electrophoretic deposition: Application for nonenzyamtic glucose sensing. RSC Adv. 2015, 5, 15861–15869. [Google Scholar] [CrossRef]
- Subramanian, P.; Niedziolka-Jonsson, J.; Lesniewski, A.; Wang, Q.; Li, M.; Boukherroub, R.; Szunerits, S. Preparation of reduced graphene oxide-Ni(OH)2 composites by electrophoretic deposition: Application for non-enzymatic glucose sensing. J. Mater. Chem. A 2014, 2, 5525–5533. [Google Scholar] [CrossRef]
- Wang, Q.; Vasilescu, A.; Wang, Q.; Li, M.; Boukherroub, R.; Szunerits, S. Electrophoretic approach for the modification of reduced graphene oxide nanosheets with diazonium compounds: Application for electrochemical lysozyme sensing. ACS Appl. Mater. Interfaces 2017, 9, 12823–12831. [Google Scholar] [CrossRef]
- Pei, S.; Cheng, H.-M. The reduction of graphene oxide. Carbon 2012, 50, 3210–3228. [Google Scholar] [CrossRef]
- Leroux, Y.R.; Fei, H.; Noel, J.-M.; Roux, C.; Hapiot, P. Efficient Covalent Modification of a carbon surface: Use of a silyl protecting group to form an active monolayer. J. Am. Chem. Soc. 2010, 132, 14039–14041. [Google Scholar] [CrossRef] [PubMed]
- Casas-Solva, J.M.; Vargas-Berenguel, A.; Captian-Vallvey, L.F.; Santoyo-Gonzalez, F. Convenient Methods for the Synthesis of Ferrocene−Carbohydrate Conjugates. Org. Lett. 2004, 6, 3687–3690. [Google Scholar] [CrossRef]
- Park, S.; An, J.; Jung, I.; Piner, R.D.; An, S.J.; Li, X.; Velamakanni, A.; Ruoff, R.S. Colloidal suspensions of highly reduced graphene oxide in a wide variety of organic solvents. Nano Lett. 2009, 9, 1593–1597. [Google Scholar] [CrossRef]
- Hamaker, H.C. Formation of a deposite by electrophoresis. Trans. Farady Soc. 1940, 35, 279–287. [Google Scholar] [CrossRef]
- Ngamchuea, K.; Eloul, S.; Tschulik, K.; Compton, R.G. Planar diffusion to macro disc electrodes—What electrode size is required for the Cottrell and Randles-Sevcik equations to apply quantitatively? J. Sol. State Electrochem. 2014, 18, 3251–3257. [Google Scholar] [CrossRef]
- Nielsen, L.T.; Vase, K.H.; Dong, M.; Besenbacher, F.; Pedersen, S.U.; Daasbjerg, K. Electrochemical Approach for Constructing a Monolayer of Thiophenolates from Grafted Multilayers of Diaryl Disulfides. J. Am. Chem. Soc. 2007, 129, 1888–1889. [Google Scholar] [CrossRef]
- Combellas, C.; Jiang, D.-E.; Kanoufi, F.; Pinson, J.; Podvorica, F.I. Steric Effects in the Reaction of Aryl Radicals on Surfaces. Langmuir 2009, 25, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Ciampi, S.; Le Saux, G.; Harper, J.B.; Gooding, J.J. Optimization of Click Chemistry of Ferrocene Derivatives on Acetylene-Functionalized Silicon(100) Surfaces. Elecroanalysis 2008. [Google Scholar] [CrossRef]
- Chidsey, C.E.D.; Bertozzi, C.R.; Putvinski, T.M.; Mujsce, A.M. Coadsorption of ferrocene-terminated and unsubstituted alkanethiols on gold: Electroactive self-assembled monolayers. J. Am. Chem. Soc. 1990, 112, 4301–4306. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mishyn, V.; Aspermair, P.; Leroux, Y.; Happy, H.; Knoll, W.; Boukherroub, R.; Szunerits, S. “Click” Chemistry on Gold Electrodes Modified with Reduced Graphene Oxide by Electrophoretic Deposition. Surfaces 2019, 2, 193-204. https://doi.org/10.3390/surfaces2010015
Mishyn V, Aspermair P, Leroux Y, Happy H, Knoll W, Boukherroub R, Szunerits S. “Click” Chemistry on Gold Electrodes Modified with Reduced Graphene Oxide by Electrophoretic Deposition. Surfaces. 2019; 2(1):193-204. https://doi.org/10.3390/surfaces2010015
Chicago/Turabian StyleMishyn, Vladyslav, Patrik Aspermair, Yann Leroux, Henri Happy, Wolfgang Knoll, Rabah Boukherroub, and Sabine Szunerits. 2019. "“Click” Chemistry on Gold Electrodes Modified with Reduced Graphene Oxide by Electrophoretic Deposition" Surfaces 2, no. 1: 193-204. https://doi.org/10.3390/surfaces2010015
APA StyleMishyn, V., Aspermair, P., Leroux, Y., Happy, H., Knoll, W., Boukherroub, R., & Szunerits, S. (2019). “Click” Chemistry on Gold Electrodes Modified with Reduced Graphene Oxide by Electrophoretic Deposition. Surfaces, 2(1), 193-204. https://doi.org/10.3390/surfaces2010015