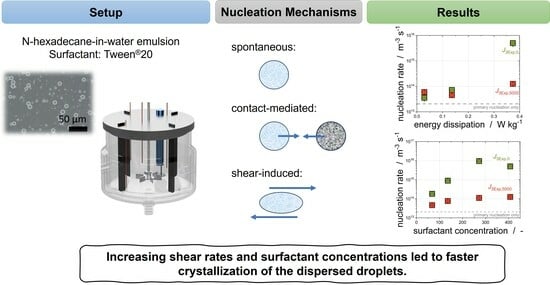
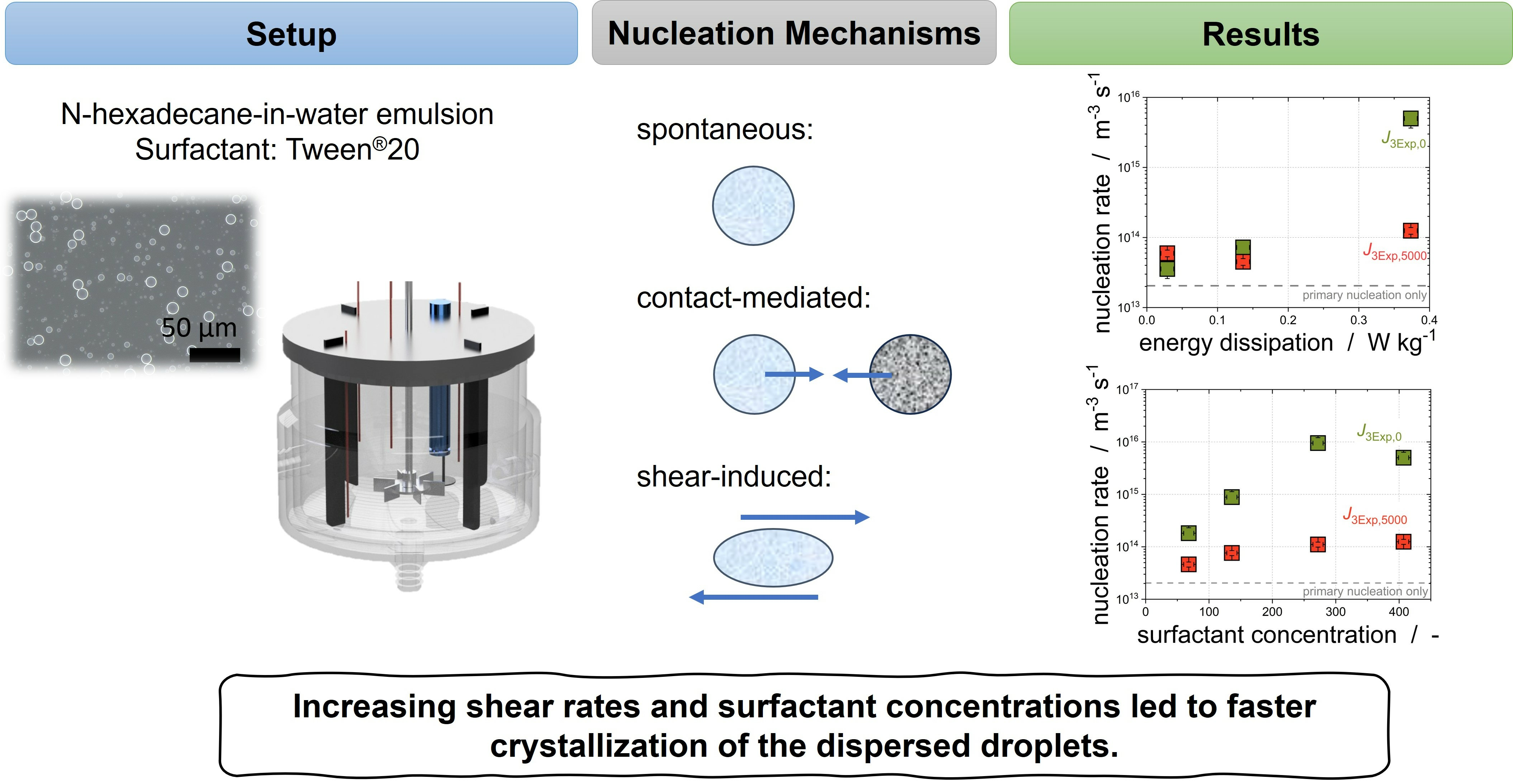
3.2. Models for Describing Droplet Crystallization in Stirred Emulsions
To the best of our knowledge, no models have yet been developed to describe the crystallization of emulsions in a stirred vessel. Such a model is needed to describe the overlapping effects of primary nucleation, shear-induced nucleation, and CMN.
A first model of the time-dependent solid fraction
of subcooled liquids was established by Turnbull [
27,
28,
29]. Dickinson along with McClements and coworkers proved the general application of this model for describing
for oil-in-water emulsions [
10,
30,
31,
32,
33,
34]. The nucleation rate
in emulsions for primary nucleation at a constant subcooling can be estimated by modelling
. New nuclei are formed exclusively in liquid droplets; therefore,
is proportional to the fraction of remaining liquid droplets
and decreases as the crystallization time progresses:
The kinetic constant for primary nucleation
can be expressed as a function of
[
11]. If homogeneous nucleation takes place inside the volume of the droplet,
is proportional to the droplet volume
and is provided by
where
x hereby represents the droplet diameter.
Solving the differential Equation (
4) with the assumption that at time
there are no solid droplets present
leads to
This model is referred to as the 1Exp-model. It does not consider the DSD.
The nucleation volume is not constant, and each droplet size class exhibits a changing nucleation rate due to the different induction times; therefore, the DSD must be considered as well [
35]:
where
is the volumetric density distribution of the droplet diameter at the beginning of crystallization. Large droplets crystallize at lower subcooling than small droplets; therefore, the density distribution of the liquid droplets changes as crystallization progresses. This temporal change of the density distribution is described by
The crystallization of emulsions due to acceleration by means of the number of catalytic impurities
can be described by [
11]:
with
Here, refers to the initial nucleation rate.
Contrary to the primary nucleation kinetics, CMN can be described by [
32]
Here, represents the kinetic constant for CMN, in analogy to .
None of the presented models have been able to describe for stirred vessel crystallization with satisfactory accuracy ( > 0.85) for all subcoolings tested.
Assuming a superposition of primary and secondary nucleation mechanisms, a model with a time-dependent nucleation rate
would be required for the description of
. Therefore, we chose a statistical approach and modified Equation (
6) (1Exp model) for this purpose as follows, based on the Weibull probability density function:
In the following, this approach is referred to as the 3Exp-model. The first fitting parameter
(sometimes called the shape parameter) describes the temporal change of the nucleation rate. A nucleation rate decreasing over time is expressed by
and a nucleation rate increasing over time by
. For
, the 3Exp-model equals the 1Exp-model with a constant nucleation rate [
36]. The second fitting parameter,
, is the kinetic constant for the emulsion crystallization in the stirred vessel at time
, and is referred to as the scale parameter of the Weibull distribution. The third parameter,
, denotes the solid fraction of the dispersed phase at
. The time-dependent nucleation rate is calculated using
Equation (
12) is valid under the condition that the emulsion droplets have the same droplet size. If the droplet size is distributed, this is calculated analogously to Equation (
8) via
The 3Exp model is applied to characterize the progressive increase in solid fraction within the emulsion droplets during their crystallization within the stirred vessel. Specifically, we emphasize two distinct nucleation rates. First, represents the initial nucleation rate. This rate assumes the absence of CMN due to the unavailability of solid particles that could induce nucleation. Thus, is solely influenced by either primary or shear-induced nucleation. Second, denotes the nucleation rate at 5000 s after reaching a constant subcooling state. Here, we anticipate that CMN will predominantly influence the nucleation process. A comprehensive discussion elaborating on this aspect is provided in the subsequent data evaluation.
For the evaluation of the CMN, the following needs to be taken into account. A collision between a particle and a liquid droplet must happen for the the CMN to occur; however, a successful inoculation process must occur as well [
11]. Because the CMN does not take place every time a particle collides with a liquid droplet, the inoculation efficiency is defined, which is derived from coalescence theory according to [
37]. The inoculation efficiency is influenced by three factors: the collision rate
, the contact force
, and the contact time
.
depends on the volume-based number of liquid and solid droplets
and
:
The volume-based number of solid droplets
is calculated with
and the number of liquid droplets with
The collision kernel is calculated regarding the flow regime (laminar, turbulent, or transient). The equations used to calculate the collision kernel include the respective droplet sizes of the particles and droplets ; therefore, the collision rate depends on the droplet size.
The collision kernel of a laminar flow is defined as
and the collision kernel of a turbulent flow as
We suggested earlier that the fraction of collisions that trigger secondary nucleation can be described by the nucleation efficiency
and the kinetic constant
[
38]:
According to the coalescence theory [
37], the rate of secondary nucleation
can be determined by multiplying the nucleation efficiency by the collision rate [
39]:
3.3. Impact of Surfactant Concentration
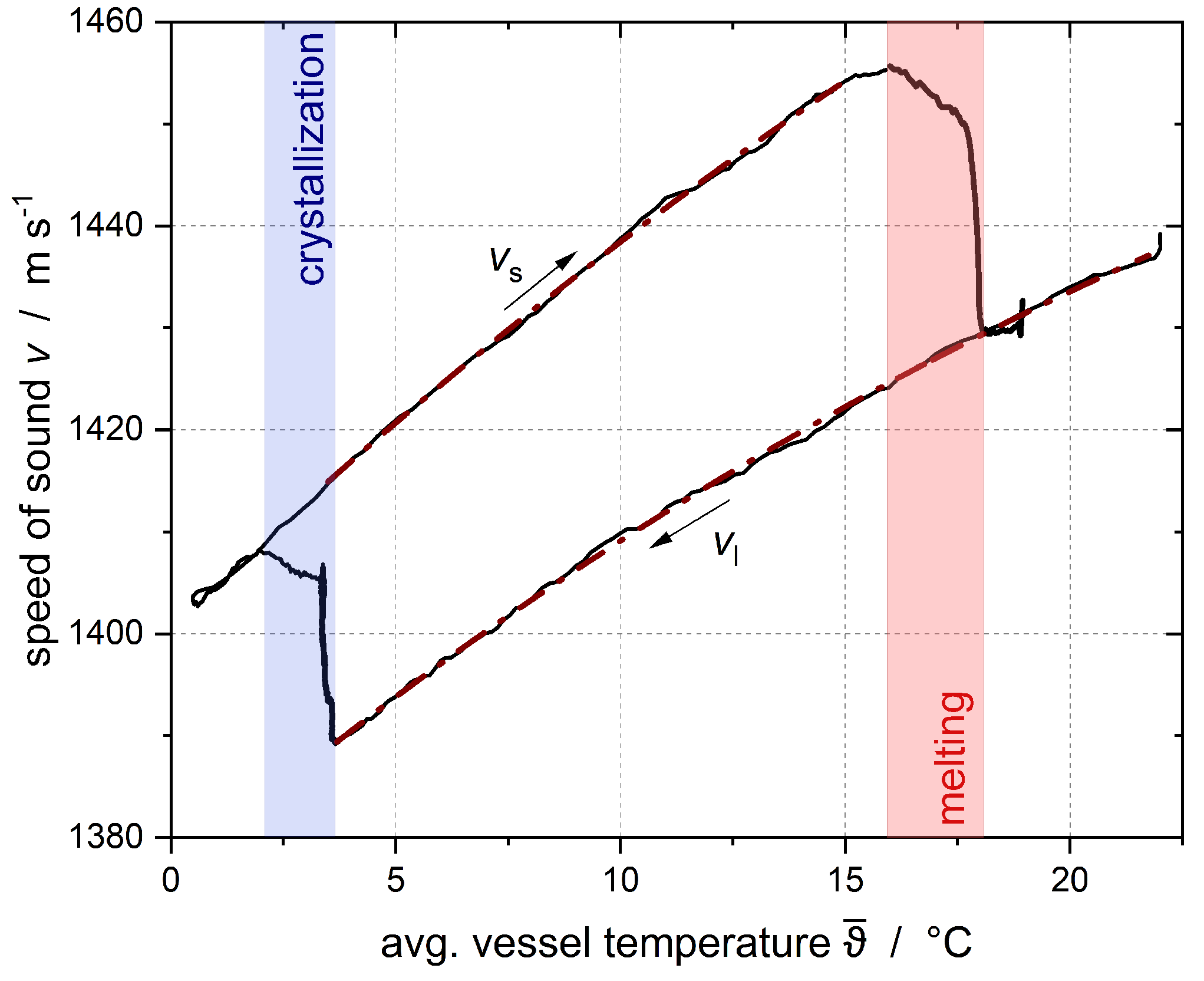
Crystallization tests were carried out for both emulsions with different subcooling at variable emulsifier concentrations.
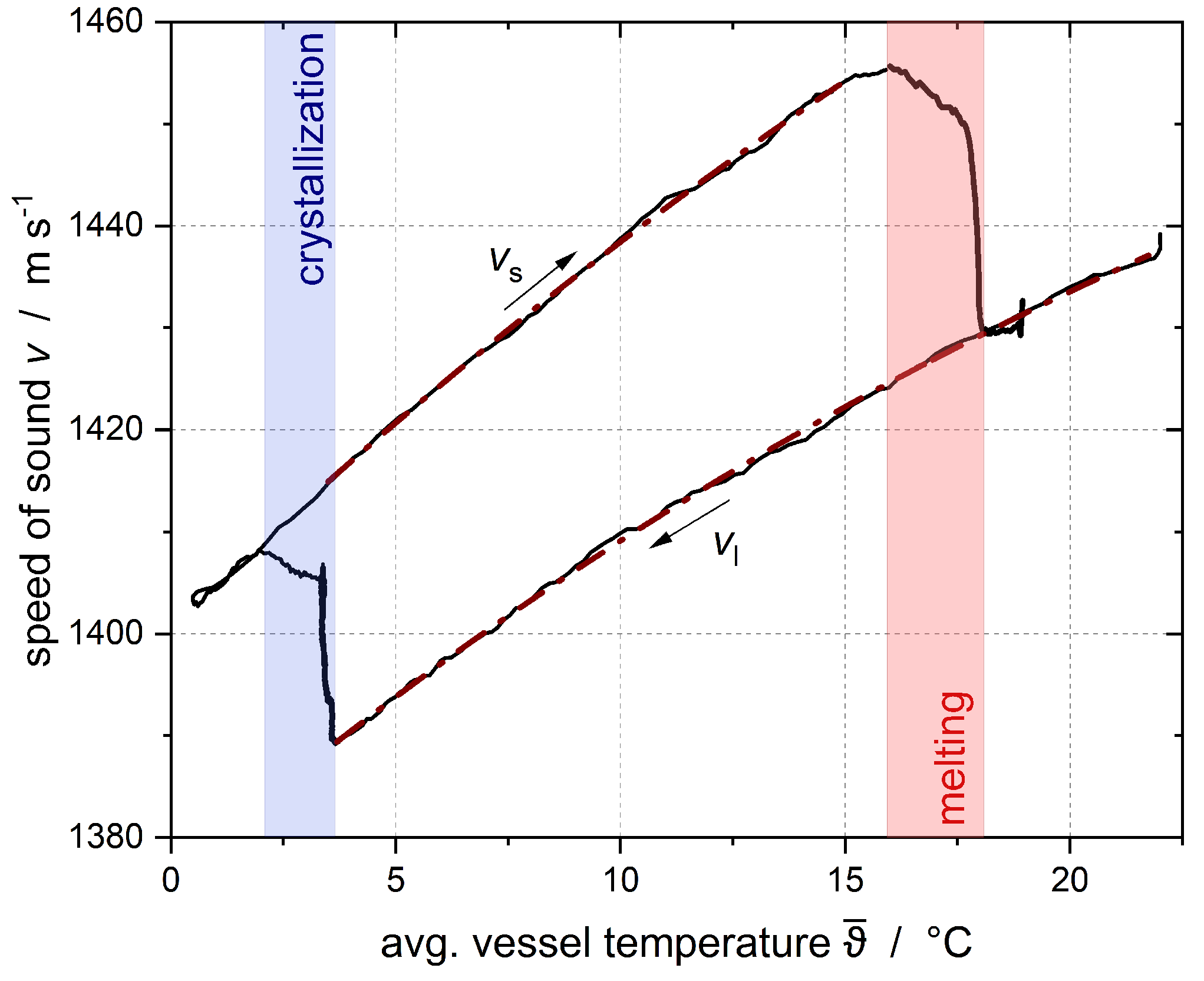
was recorded using an ultrasonic sensor, and the solid fraction was calculated according to Urick [
23] (Equation (
1)).
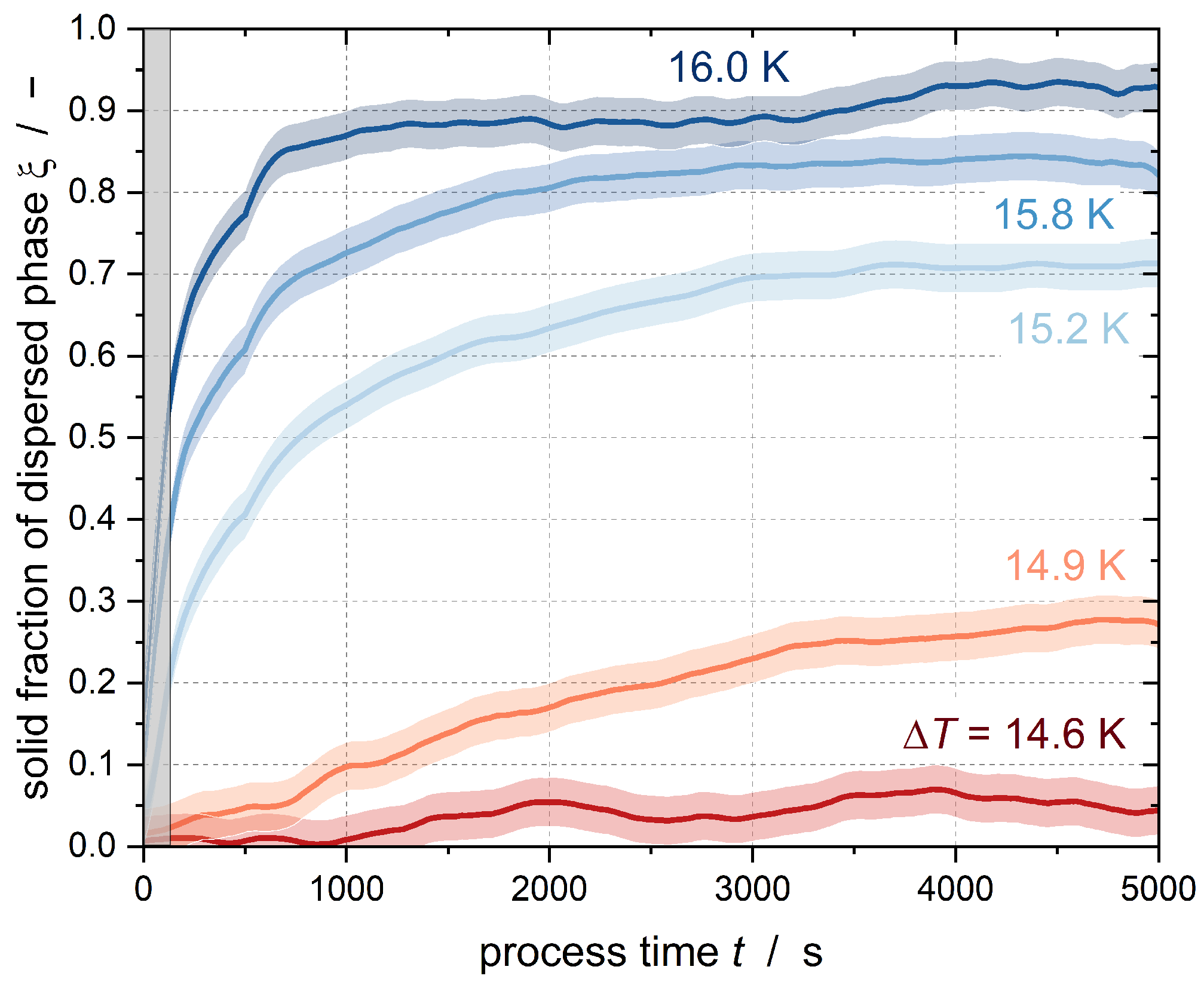
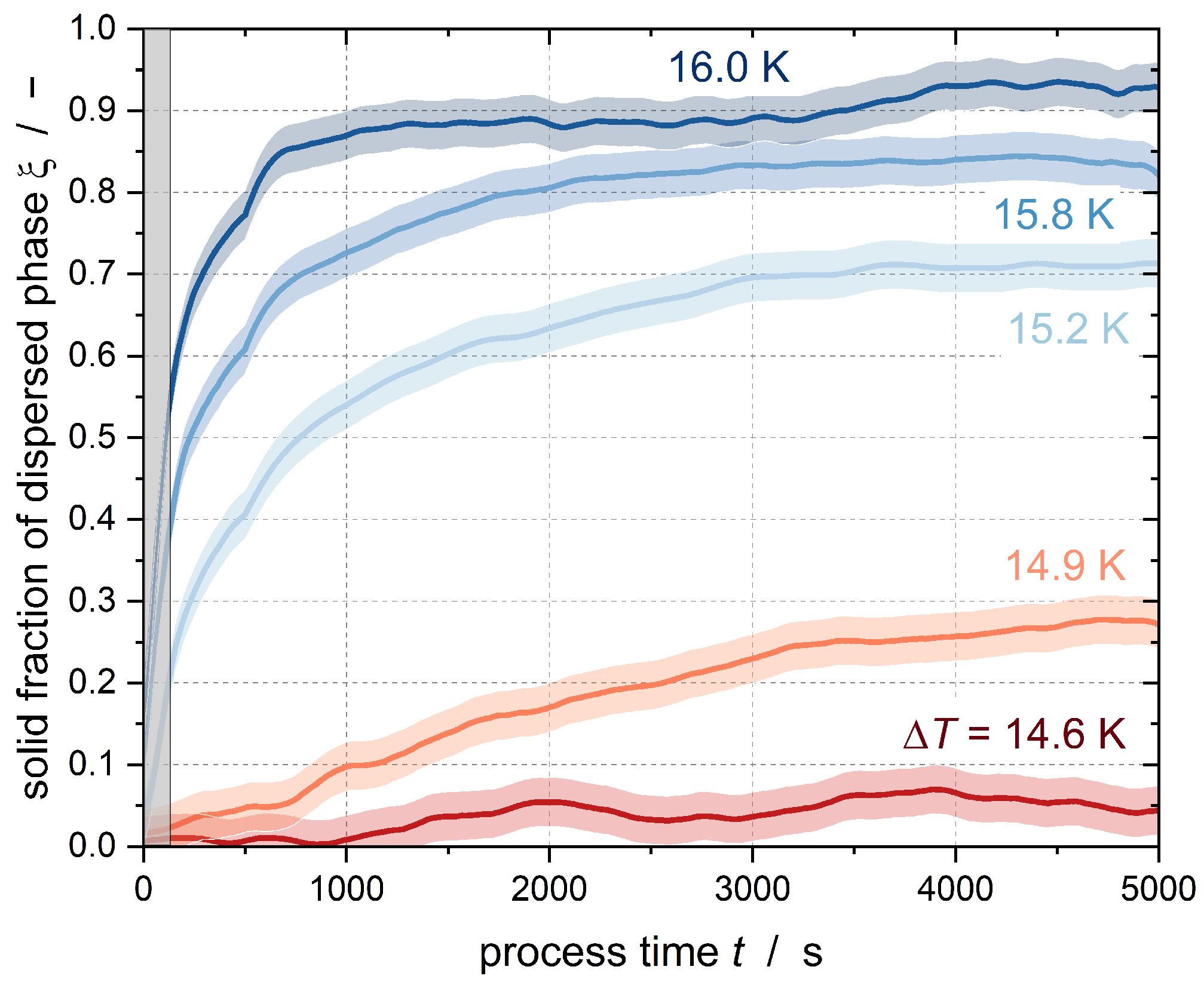
Figure 3 shows the results of the crystallization experiments with Emulsion I at
N = 350 min
−1 (equaling a mean energy dissipation
of 0.37 W kg
−1) for
= 16 mol m
−3. The gray area represents the time at which a constant temperature had not yet been fully established. The error intervals of the results shown in this publication are the result of three repeated tests of two subcoolings per emulsion. The variations were then applied to all other subcoolings. The 95% confidence interval for the solid fraction of the n-hexadecane droplets was calculated as ±0.03 for Emulsion I and ±0.05 for Emulsion II.
Figure 3 shows different curve shapes. In the case of high subcooling, there is a sharp increase in the solid content in the stirred tank immediately after reaching constant subcooling. This abrupt increase is due to heterogeneous or homogeneous nucleation in the large droplets of the emulsion. Primary homogeneous nucleation is typically observed in the initial phase of a crystallization process without seed crystals, and decreases after the formation of sufficient crystals [
9]. Droplets with a larger volume have a higher probability of primary nucleation than small droplets. This is because the induction time for primary nucleation is reduced due to a higher probability of the formation of growable clusters in a larger volume [
29]. Therefore, the large droplets already crystallize due to homogeneous nucleation, whereas no stable clusters can be formed in the small droplets and the subcooled droplets do not change their aggregation state.
The slope of the curve decreases after the fast increase at t → 0, although the fraction of solids continues to increase in the course of further measurement. This slower increase is due to secondary nucleation. The collision of the already solidified droplets with the still-liquid subcooled droplets results in CMN, meaning that some droplets crystallize that may not crystallize by pure homogeneous nucleation at the given subcooling. Moreover, the efficiency of the CMN reaches a maximum when there are equal numbers of liquid and solid droplets, i.e., a solid fraction of = 0.5 is reached. As the solid fraction continues to increase, the efficiency decreases because the probability of a liquid droplet colliding with a particle is reduced. In addition, the nucleation triggered by the shear of the droplets in the flow field is another nucleation-triggering mechanism. A statement about the occurrence of shear-induced nucleation cannot be made based on tests at an identical stirrer speed (and identical shear rate). If homogeneous nucleation does not occur at the beginning of the process time due to insufficient subcooling, there is no discernible jump in the course of the solids content; instead, crystalline nuclei are formed in the emulsion droplets by heterogeneous or shear-induced nucleation, which can be seen from the constant slow increase in the solids content. As soon as particles are present, CMN takes place.
Although a constant subcooling was set for approximately 90 min, none of the curves shown here reached a solid content of
= 1, i.e., not all droplets were crystalline. One possible reason for this is that the experimental period was too short. Hindle et al. [
11] achieved complete crystallization of stirred n-hexadecane emulsions only after an experimental period of 16 days. The slightly positive slope of
after 90 min in
Figure 3 suggests that if the experiments were conducted for a longer period of time, then complete crystallization of the emulsion by secondary nucleation would be possible.
Conclusions can be drawn about the influence of the emulsifier concentration on nucleation by combining the curves at the same stirring speed and subcooling.
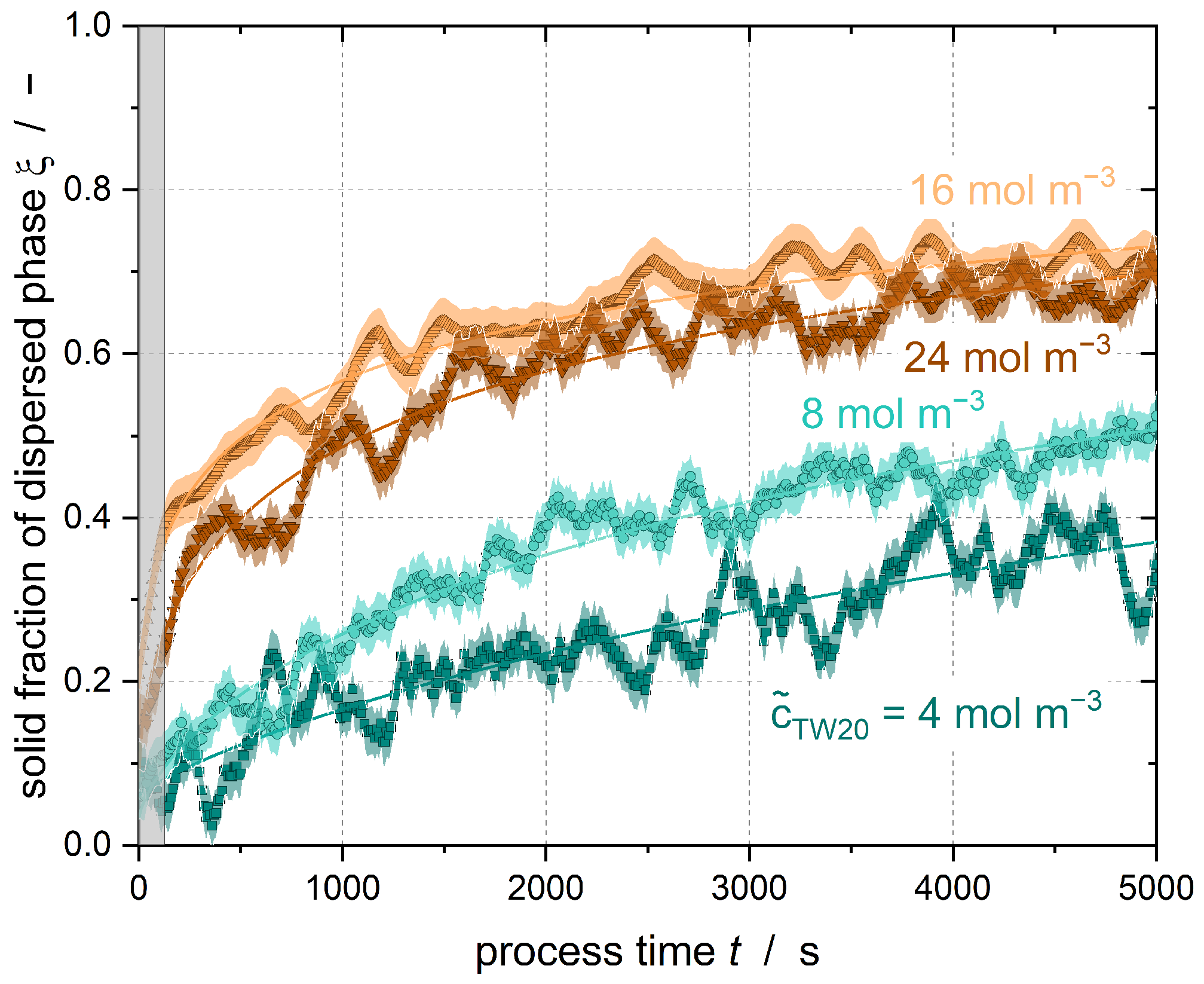
Figure 4 shows the solid content of Emulsion I at varying emulsifier concentrations. The test conditions were identical for all curves shown.
Figure 4 shows that the solid content increases faster with increasing emulsifier concentration. After 5000 s, the solid content of the emulsion with
= 24 mol m
−3 is
= 0.66 and that with
= 16 mol m
−3 is
= 0.71, while the emulsion with
= 8 mol m
−3 has a solid content of
= 0.50 and that with
= 4 mol m
−3 is
= 0.39. Thus, the crystallization of the emulsions is accelerated with increasing emulsifier concentration. Whether further acceleration of crystallization by increasing the emulsifier concentration of
≥ 16 mol m
−3 is possible cannot be determined from the data shown here. It is possible that the underlying mechanism reaches a plateau or maximum state with respect to further increases in the surfactant concentration.
The 3Exp-model (Equation (
14)) was fitted to the experimental data (the line shown in
Figure 4).
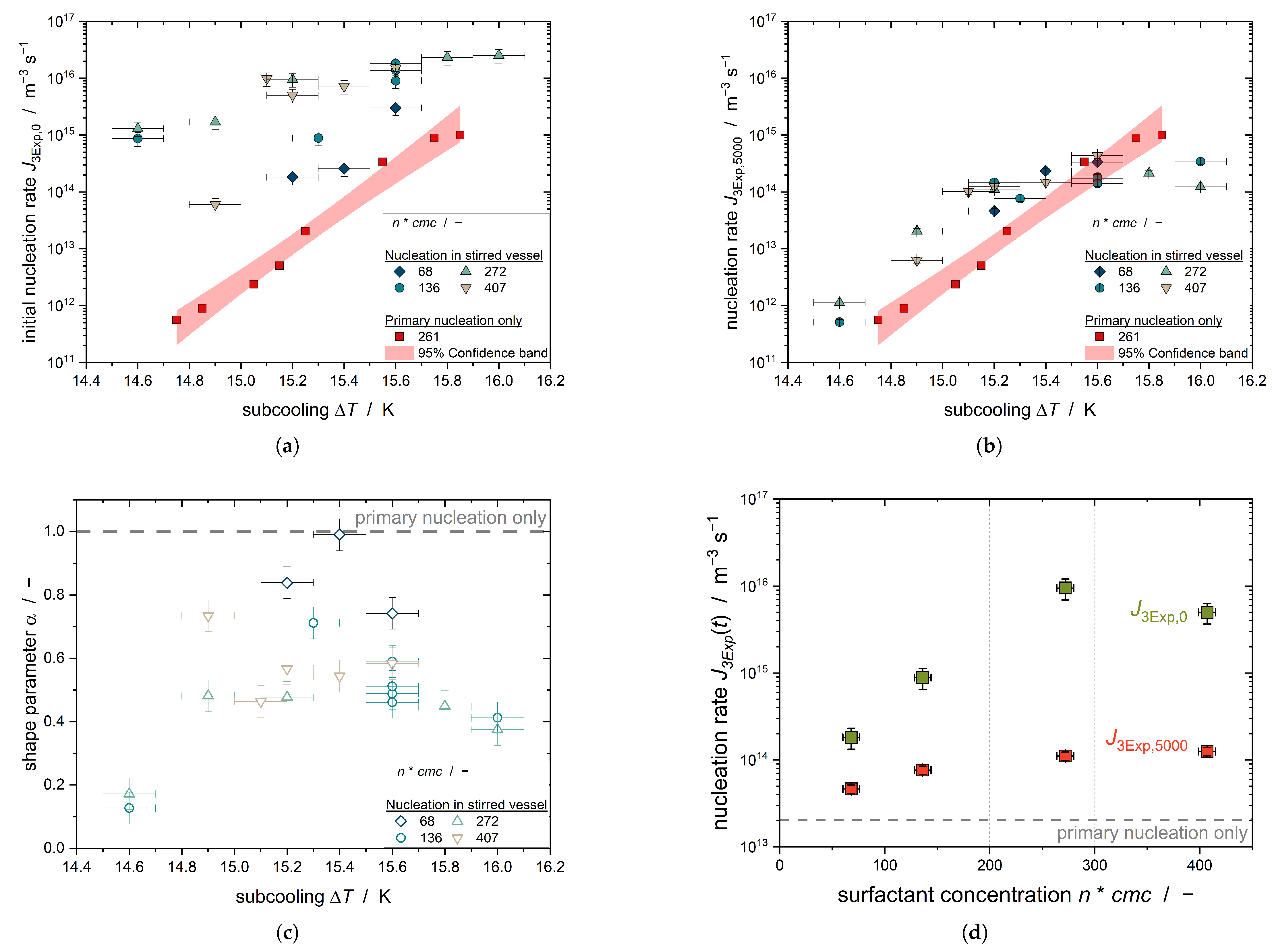
Figure 5 shows the initial nucleation rate
, the shape parameter
of the 3Exp-model, and the nucleation rate after 5000 s
for the subcoolings investigated for all tested surfactant concentrations. Additionally, the primary nucleation rate
is shown.
was determined using spectroscopic NMR measurements according to [
38]. By fitting the time-dependent solid fraction of the emulsion droplets according to Equation (
8),
was determined for Emulsion I without the influence of shear at a given subcooling.
The plot shows that the nucleation rate increases with increasing subcooling, and as such with increase in the driving force of crystallization. This relationship is evident for all emulsifier concentrations. Furthermore,
increases with increasing emulsifier concentration. This confirms the relationship seen in
Figure 4 that the initial steep increase in the solid content of n-hexadecane droplets is dependent on the emulsifier concentration. Comparison with
clearly shows that
is higher than
for all experiments in the stirred tank due to secondary nucleation. The difference between the purely primary nucleation and the nucleation in the stirred tank decreases with increasing subcooling, as the primary nucleation at high subcooling is the dominant nucleation mechanism in the emulsion droplets.
The time variation of the nucleation rate is expressed by the shape parameter
.
Figure 5c shows that
< 1 for all subcooling and TW20 concentrations; thus, the nucleation rate of all settings decreases with time. Regarding
= 4 mol m
−3,
takes the highest value, i.e., for the lowest emulsifier concentration, the time decrease in the nucleation rate is the smallest. The two values for
= 14.6 K are very small compared to the rest of the values, with
< 0.2, which is due to the fact that the two curves are poorly fitted by the 3Exp-model (with a goodness of fit of
< 0.1). The application of second-order kinetics would be more appropriate for these experiments, as presumably primary nucleation does not occur with the low subcooling and secondary nucleation is present.
The nucleation rate after 5000 s, calculated from
and
using Equation (
13), shows that the nucleation rate in the stirred tank approaches the purely primary nucleation rate. Observation of the fits at different emulsifier concentrations shows that the nucleation rate increases with the free emulsifier concentration in the aqueous phase; thus, the solid fraction of the n-hexadecane droplets increases more strongly. Consequently, a nucleation-promoting mechanism of the micelles must exist.
Dickinson et al. [
10] found through a variation of emulsifier concentrations and types that there was an increase in nucleation frequency with increasing emulsifier concentration independent of the chemical nature of the emulsifier. Only CMN was observed, primary nucleation was excluded, and no shear was applied. The authors determined the kinetics constant
at
= 12.6 K using the second-order kinetics. The corresponding nucleation rate
was determined taking the droplet volume into account. When the TW20 concentration was increased from
= 6 mol m
−3 to
= 18 mol m
−3, the nucleation rate increased from
= 6.41
m
−3s
−1 to
= 1.96
m
−3s
−1, which is slightly lower than the nucleation rate determined in this work without inoculation crystals. In addition, the emulsions were not stirred and the mass fraction of the hexadecane was 12.5% above the mass fraction of this work, which is 5%.
McClements and Dungan [
26] observed an increase in the nucleation frequency with increasing emulsifier concentration in an n-hexadecane-in-water emulsion with TW20 as the surfactant. They used n-hexadecane-in-water emulsions with an n-hexadecane mass fraction of 30%, which they studied at
= 12.6 K. When increasing
= 2 mol m
−3 to
= 14 mol m
−3, the emulsions showed an increase in the nucleation rate from
= 9.72
m
−3s
−1 to
= 2.82
m
−3s
−1. The authors mentioned a lowering of the interfacial tension with increasing emulsifier concentration, the template effect, and depletion flocculation as possible reasons for this increase [
26]. The possibility of accelerated nucleation due to lower interfacial tension can be excluded for the emulsions used in this work, as the emulsifier concentration of all emulsions was clearly above the
; thus, the increase of interfacial surfactant molecules should be negligible [
40]. The template effect is the acceleration of nucleation due to the similarity of the hydrophobic surfactant tail and the dispersed phase. When the similarity is increased, the likeliness of the dispersed phase crystallizing is increased as well, because the tails increase the structure of the dispersed phase and consequently lower the energy barrier needed to overcome nucleation [
41]. Because n-hexadecane molecules and the hydrophobic part of TW20 molecules have low structural similarity, the promotion of crystallization due to the template effect can be excluded as the reason for the acceleration of nucleation [
42].
McClements and Dungan [
26] put forward their own explanatory approach involving depletion flocculation. They excluded the influence of the emulsifier concentration on nucleation in the individual droplets, as their DSC measurements showed no dependence of the crystallization and melting behavior on the emulsifier concentration. Instead, the authors suspected changed approach behavior on the part of the solid and liquid droplets resulting from the micelle-free volumes around the droplets. If two emulsion droplets approach each other and the micelle-free volumes overlap, this state is energetically more favorable due to the reduced common volume without micelles. This state is mentioned as being energetically more favorable. A biopolymer must be present in the continuous phase that forms a network for this effect to occur in TW20-stabilized emulsions. Thus, depletion flocculation can be ruled out for the emulsions studied in this work, which lacked a a biopolymer.
We showed in our previous study of the interfacial occupancy of liquid and solid droplets that there are fewer TW20 molecules on the surface of a crystalline n-hexadecane droplet than on a liquid droplet [
43]. During crystallization, TW20 molecules desorb from the interface. The results of our small-angle X-ray scattering (SAXS) measurements indicated that n-hexadecane molecules were entrapped in the center of TW20 micelles [
43]. While these nanodroplets occupy a small volume, they have a very large surface area and are present in great numbers. When these small droplets crystallize, they contribute strongly to the CMN, as the collision frequency is increased. The subcooling of
13 K required for homogeneous nucleation in nanodroplets with a diameter of
x = 10 nm was achieved in all experiments conducted in this work [
44]. The nanodroplets had such a small volume that the increase in the solid fraction during the crystallization of the nanodroplets due to the change in the sound velocity was probably not measurable. Because no further acceleration of nucleation is evident when increasing the emulsifier concentration from
= 16 mol m
−3 to
= 24 mol m
−3, it is reasonable to assume that the number of nanodroplets has reached its maximum and that the equilibrium distribution has been reached.
3.4. Impact of Shear on Crystallization
We have shown previously that laminar shear can be used to trigger crystallization up to a specific shear rate [
14]. At this shear rate, the crystallization efficiency reaches its maximum depending on, inter alia, the fraction of the dispersed phase and the shear flow (laminar, turbulent). However, not only the CMN is impacted by shear; as has already been described for polymer solutions, shear can trigger crystallization by increasing the structure of the molecules and consequently decreasing the energy barrier. Whereas the impact of shear on the droplet increases with increasing stirring rate, and should fasten the droplet crystallization without any limitations, the CMN might reach a maximum at a given shear rate and lose its ability to promote the crystallization progress of the dispersed phase. We assume that the impact of shear on the droplets, referred to in the following as shear-induced nucleation, should be especially visible at the beginning of the crystallization of the bigger droplets. Therefore,
is used to evaluate the impact of shear-induced nucleation. The CMN needs solid particles which have already been formed in order to occur; thus,
is used to determine the possibility of fastening the crystallization of the dispersed phase via CMN.
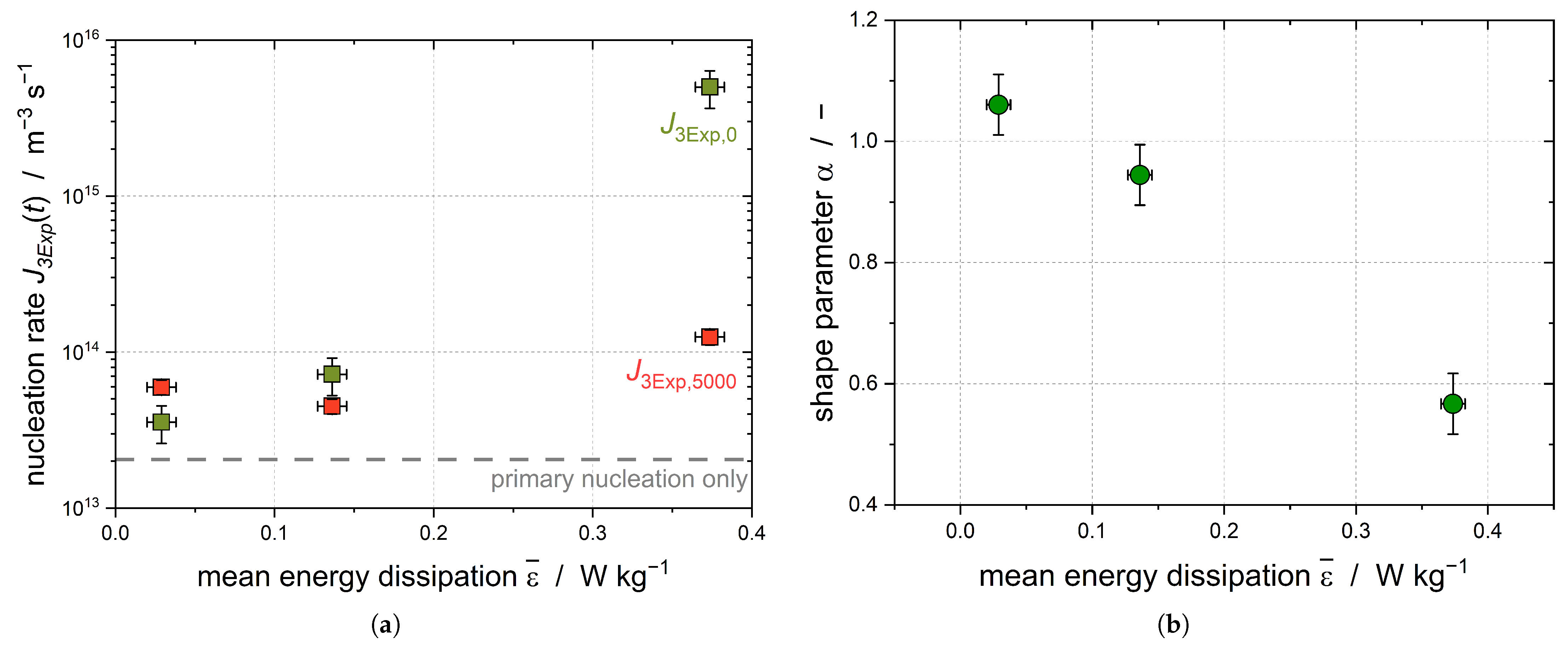
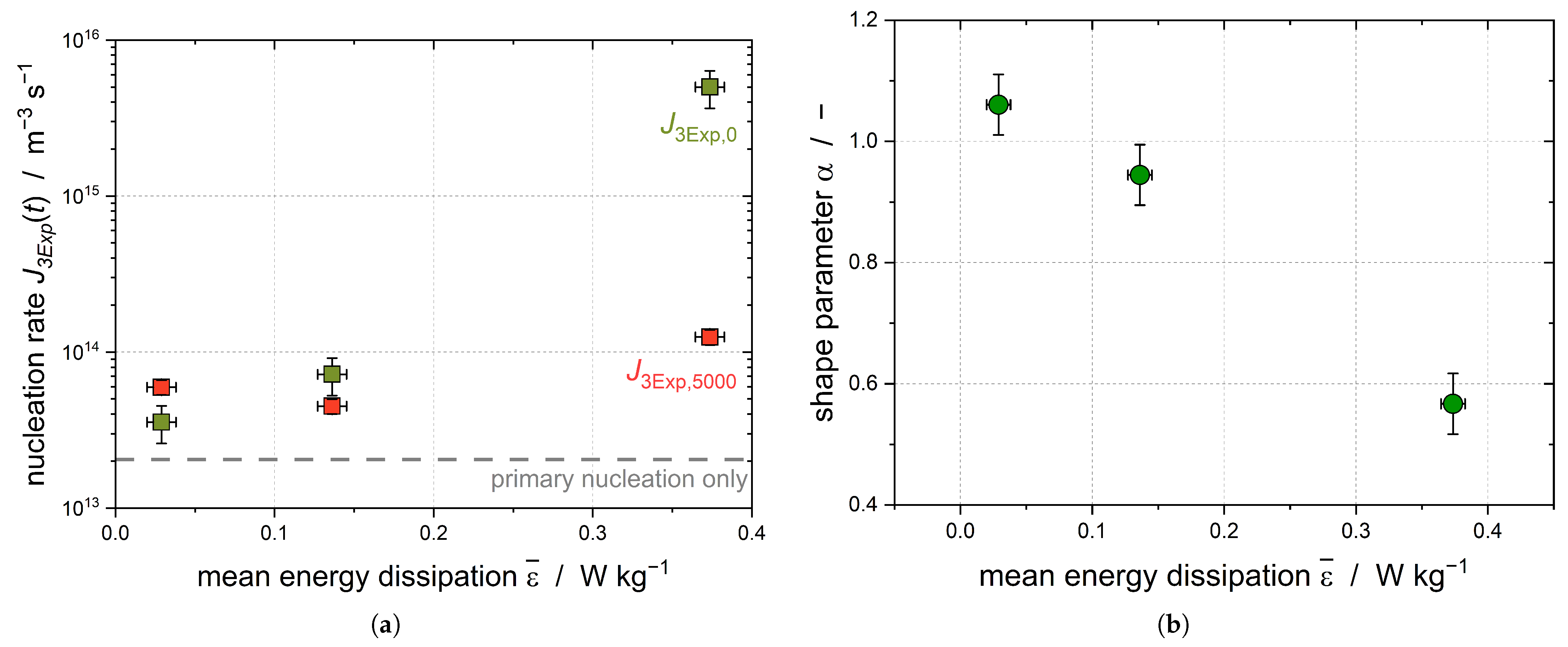
increases with increasing stirring rate, and as such with increasing mean energy dissipation (
Figure 6a). Because the subcooling was kept constant for all experiments, this increase can show the impact of the shear-induced nucleation, as primary nucleation should be independent of the applied shear.
As the stirring rates increase, the shear parameter
diminishes, leading to a growing disparity in the nucleation rates over the course of the experiment (
Figure 6b). In order to guarantee that the higher energy input did not lead to a better temperature homogeneity, the temperature difference of the four integrated temperature sensors was compared and found to be independent of the stirring rate. It can be assumed that CMN is primarily responsible for the increase in the solid droplet fraction at later process times (
t > 2000 s), leading to
=
.
The CMN is mainly influenced by three factors: the collision frequency
, contact time, and contact force. Whereas
and the contact force increase with increasing
, the contact time decreases simultaneously. This is why an increasing application of shear forces does not necessarily result in a faster crystallization progress of the dispersed phase. We were able the show in previous works that the crystallization kinetic factor for the laminar Couette flow increases with increasing mean shear rate up to a maximum, after which the contact time becomes limiting and the complete crystallization of the dispersed phase requires longer process times [
14,
38]. The shear field in the stirred vessel is widely distributed; therefore, droplets and particles move to different relative velocities independent of their location in the vessel itself. This provides an explanation of why there was no limitation of the contact time detectable in these experiments.
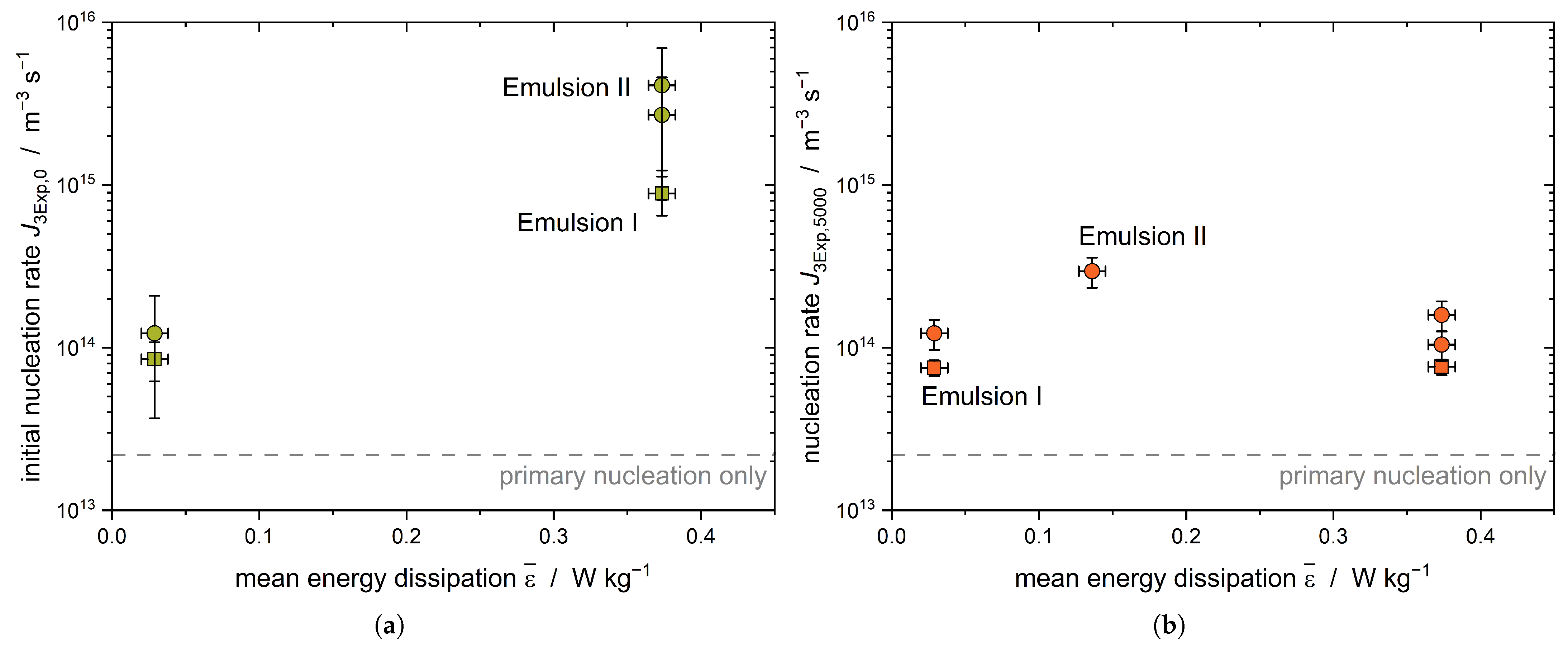
A second emulsion with a bimodal DSD was used to further investigate the effect of shear-induced nucleation. As larger droplets are exposed to higher deforming forces, an increase in
must be visible (
Figure 7a).
A clear increase in the initial nucleation rate
with increasing energy dissipation rate is evident for Emulsion II (
= 14.8
m). The crystallization of the latter is triggered by the shearing of the large droplets; therefore, an increase in the nucleation rate is evident. When increasing the energy dissipation rate from
= 0.03 W kg
−1 to
= 0.38 W kg
−1, this effect is less pronounced for Emulsion I, as only smaller droplets are present compared to the droplet sizes in Emulsion II. The nucleation rate of Emulsion II after 5000 s decreases to about the same value as the nucleation rate of Emulsion I (
Figure 7b). Assuming that the largest droplets of Emulsion II crystallized due to shear during the first part (up to 2000 s) of the crystallization process, shear-induced nucleation no longer plays a major role at later process times (
t > 2000 s) and CMN dominates the secondary fraction of the crystallization kinetics.
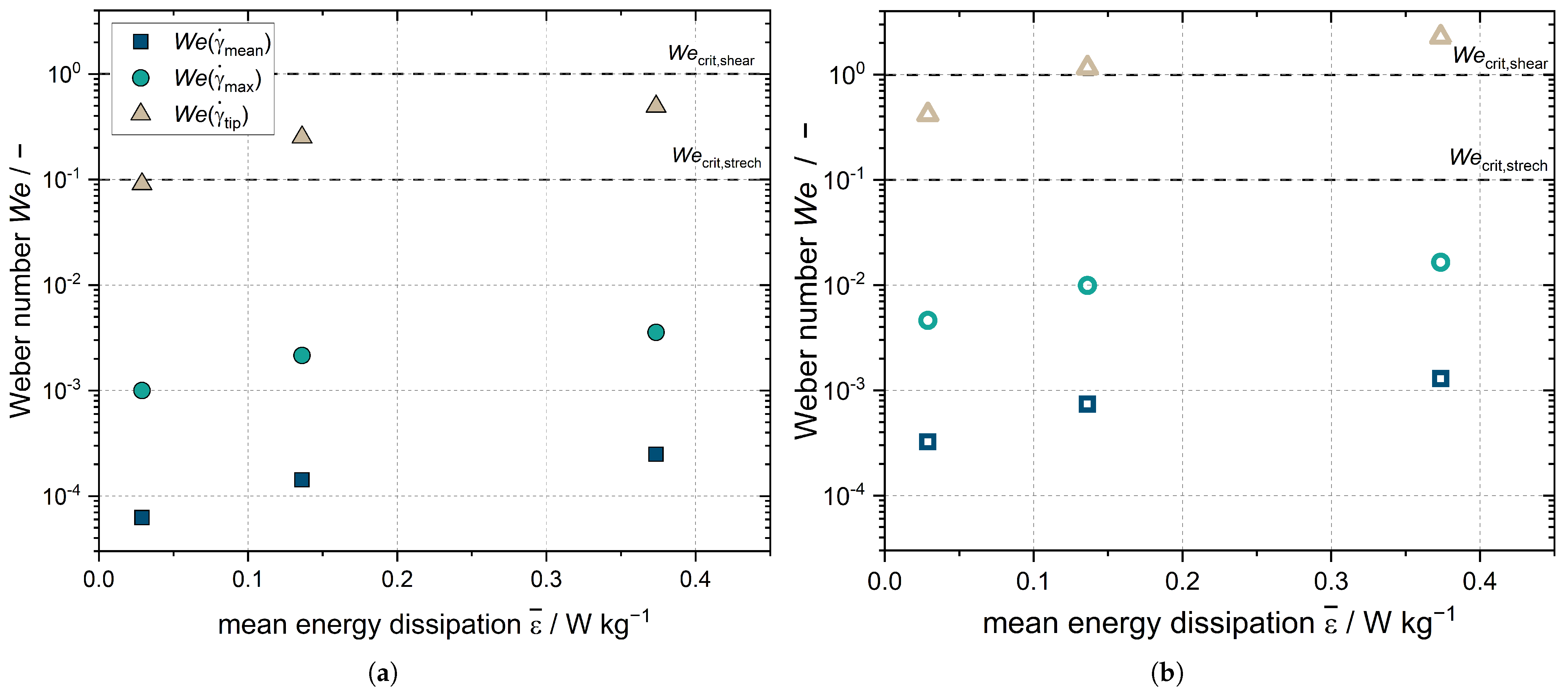
To assess the impact of shear on the droplets, the analysis involves defining the deformation of the droplets through a dimensionless parameter known as the Weber number (
). This numerical value represents the ratio between the disruptive hydrodynamic forces and the stabilizing force of surface tension. Thus, the
number serves as an indicator to determine whether kinetic or surface tension energy plays a dominant role. In the context of a spherical droplet, the
number can be calculated by comparing the droplet’s kinetic energy to its surface energy. The
number is calculated according to Equation (
22):
where
refers to the viscosity of the continuous phase and
to the interfacial tension of the liquid–liquid interface of the droplet. If
>
, droplet breakup occurs. Droplet breakup for n-hexadecane-in-water systems with a viscosity ratio between the dispersed phase (
) and the continuous phase of
3.8 occurs in a laminar stretching flow for
numbers above the critical stretching
number
0.1 and in a laminar shear flow above the critical shear
number
1 [
45]. However, the
number is only applicable to the laminar flow case [
46], which does not exist at any of the three stirrer speeds studied here. Laminar flow regions may exist locally in the stirred tank; thus, the
number is used for a lower estimate of the droplet deformation in the stirred tank. Due to the local velocity and shear peaks in a turbulent flow, it is possible that the real shear velocities are above the ones calculated and that droplets may be locally deformed more than the
number predicts. The maximum droplet deformation is present for the largest droplet fraction, which is
x = 9.86
m for Emulsion I regardless of the emulsifier concentration. The largest droplet diameter for Emulsion II is
x = 45.60
m for all emulsifier concentrations studied. The
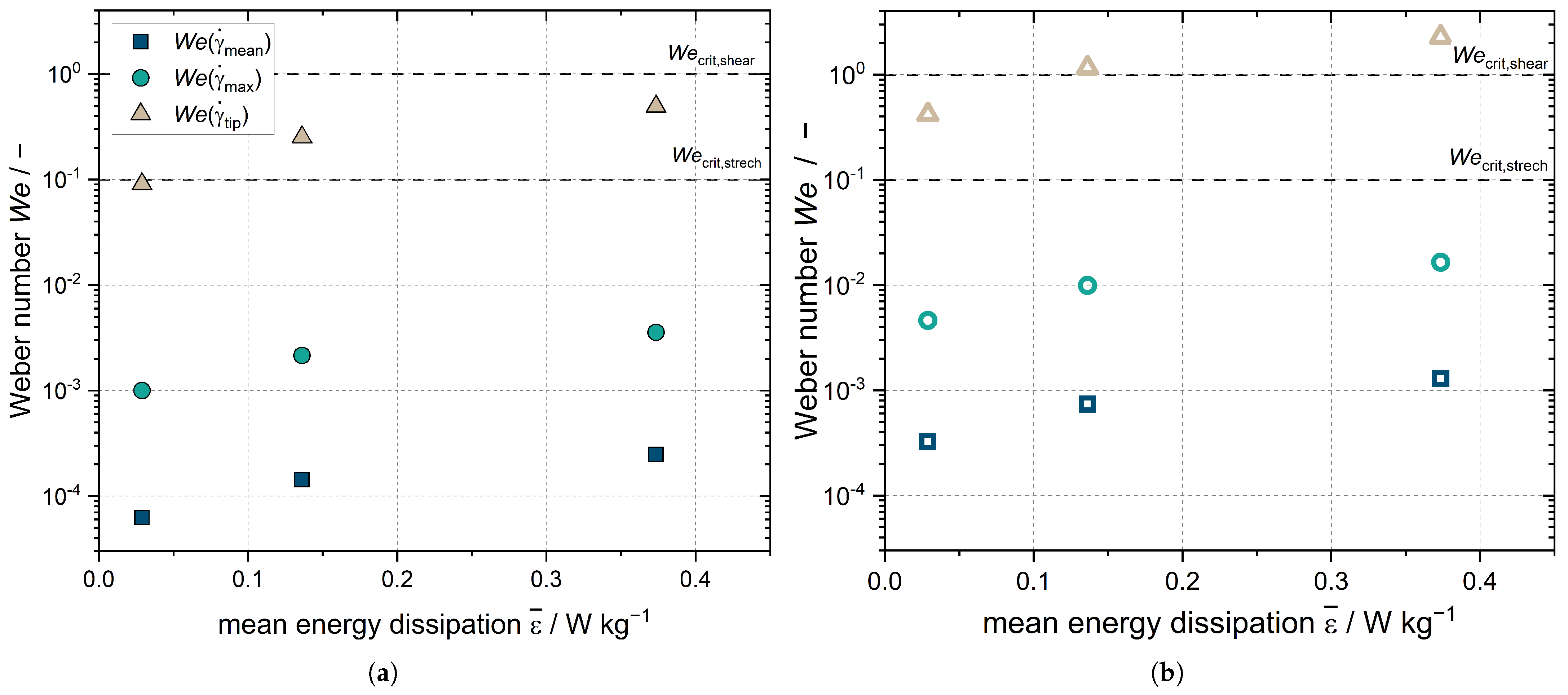
number, and consequently the deformation of the droplets, increases with the increasing energy dissipation rate resulting from increasing stirrer speed (
Figure 8).
is the
number for droplets that are in close proximity to the rotating stirrer blade. This is higher for Emulsion I (
Figure 8a) for the highest stirrer speed set and is higher for Emulsion II (
Figure 8b) than the critical
number
0.1 for all settings. However, this coefficient is only applicable for laminar flow conditions, which do not exist in the vicinity of the stirrer. Reliable statements about the size reduction of the droplets in the stirred tank cannot be made based on the
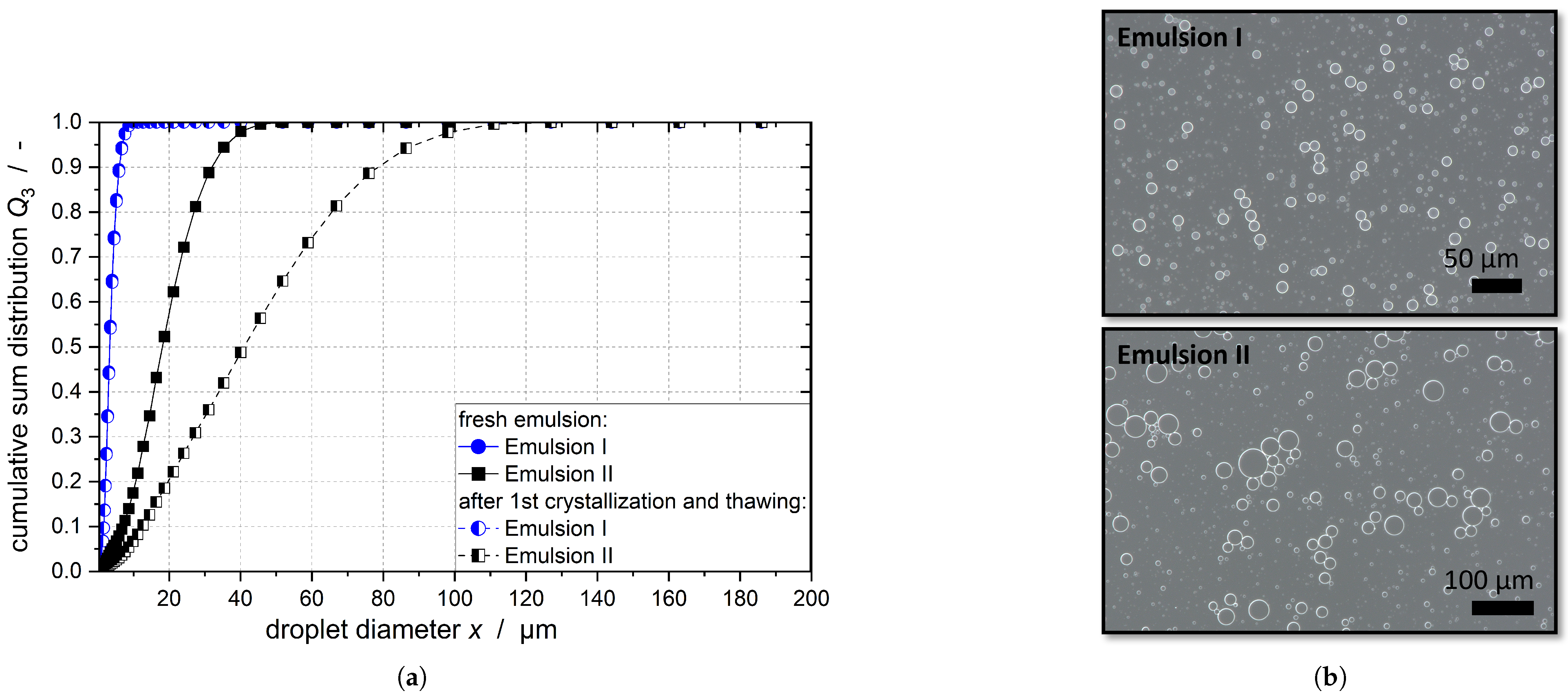
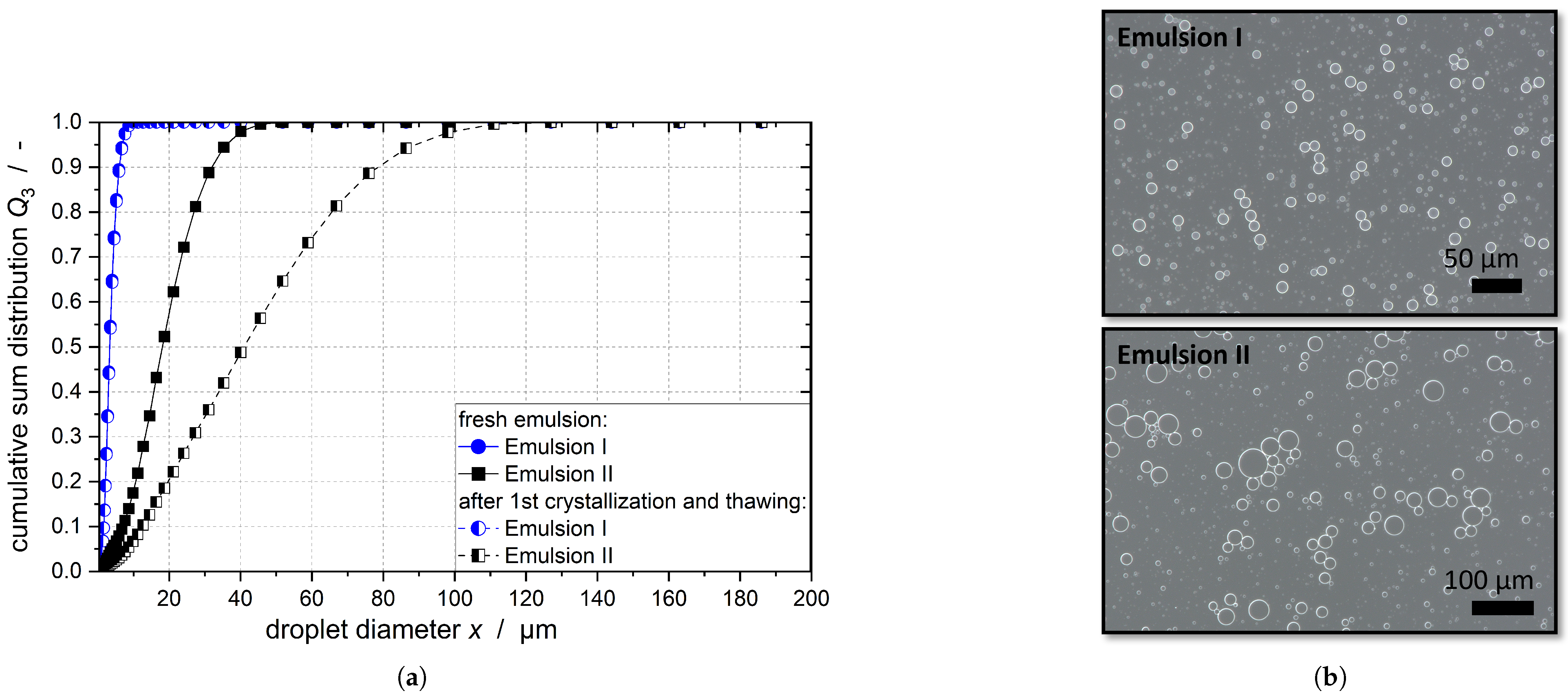
number, and numerical simulations are needed; however, the measurements of the DSD of Emulsion I after crystallization showed no change in the droplets sizes (compare
Figure 1). In the case of Emulsion II, the DSD even increased after crystallization as coalescence of the melted droplets took place (compare
Figure 1). The
numbers for the mean and maximum shear rates in the stirred volume
and
, respectively, were significantly below the critical
numbers and the
number at the stirrer tip. Thus, while the flow in the stirred vessel deforms the droplets, it does not crush them in the process. The acceleration of crystallization is due to shear and CMN, and does not result from the change in DSD.
In addition, the size of the micro-eddies for the turbulent flow must be considered. The average size of the micro-eddies
for
= 0.37 W kg
−1 is ≈95
m, which is double the size of the largest droplets in the two emulsions. Only droplets which are larger or the same size as
are exposed to deformation forces; therefore, the DSD should not be changed by means of
[
47]. However, fluctuations of the velocity may impact the DSD. The droplets can start to oscillate and reach the droplet resonant frequency through fast changes of the local velocity, which can be given in stirred tanks, resulting in droplet breakage [
48].
3.5. Collision Kernel, Collision Rate, and Collision Efficiency
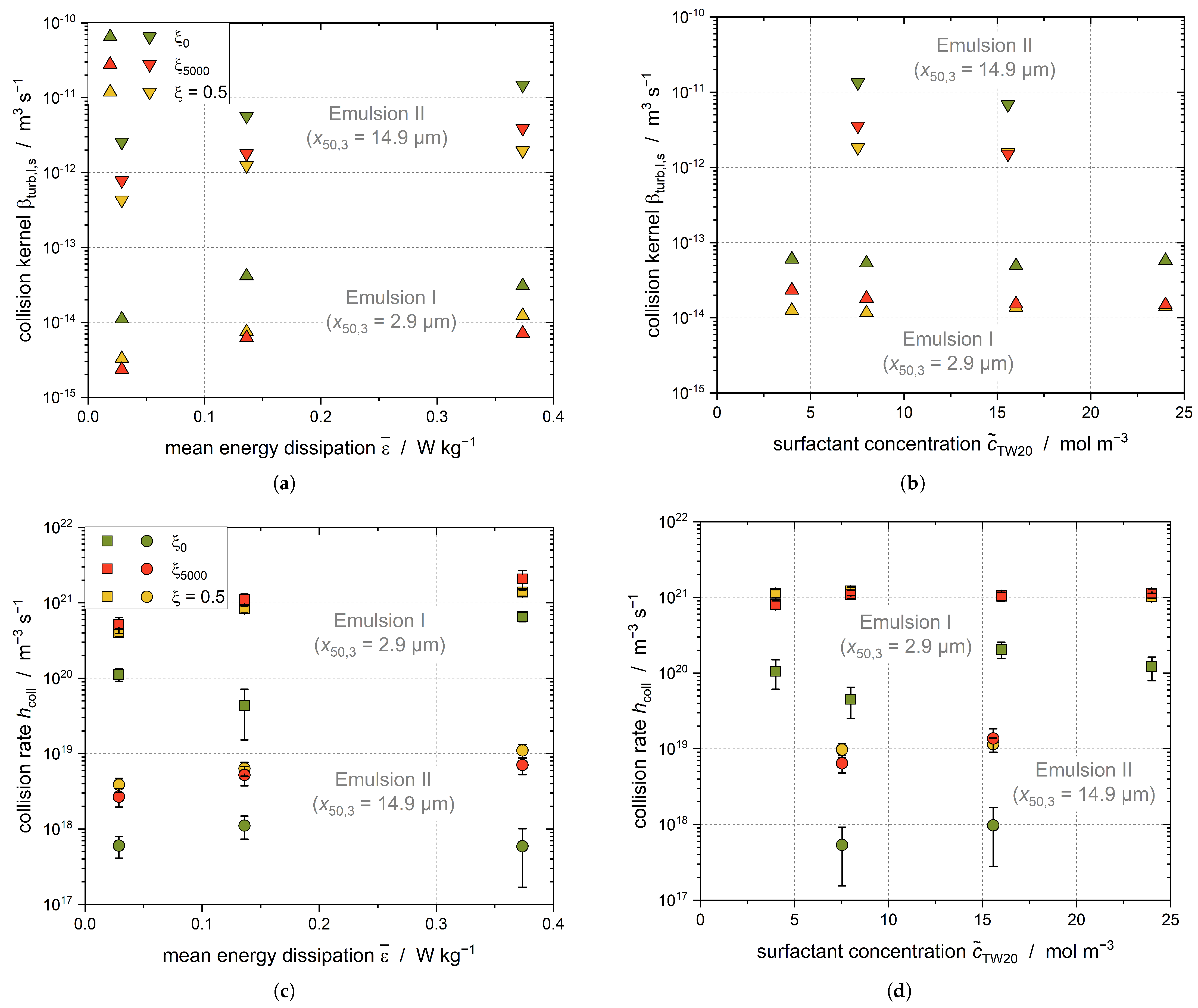
The flow regime in the stirred tank is transient or turbulent for the studied settings; thus, Equation (
19) is used to calculate the collision kernel. The required droplet sizes of the liquid and solid droplets, respectively
and
, were estimated using the measured DSD and the solid content of the dispersed phase determined at three different process times: (1)
t = 0 s (
); (2)
t = 5000 s (
); and (3)
t(
= 0.5) (
Figure 9).
The collision kernel in the stirred tank increases with increasing energy dissipation rate and stirrer speed. The solid fraction increases and the size of the remaining liquid and the solid droplets decreases during the crystallization process. Therefore, the collision kernel of the droplets decreases as the crystallization time progresses, as shown by comparison of the collision kernels at and . The initial droplet sizes of the emulsions did not change significantly with the increasing TW20 concentration; therefore, the collision kernel does not depend on the emulsifier concentration. Due to the larger droplets, the collision kernels for Emulsion II are correspondingly higher for all stirrer speeds and TW20 concentrations.
The collision rate
increases with the increasing energy dissipation rate and stirrer speed because the collision kernel increases. According to Equation (
15), the collision rate depends on the number of liquid and solid droplets, which in turn is determined by the droplet and particle size. Both emulsions used in this work had the same disperse phase fraction while differing in their droplet size, and consequently in their number of droplets. Accordingly, the collision rate is higher in Emulsion I than Emulsion II at all settings due to the higher number density of liquid and solid droplets.
The collision rate after t = 5000 s is higher than at the beginning of the crystallization process, as the volume-based number density of particles increases with the increasing solid fraction. The result when multiplying both number densities reaches a maximum at = 0.5. The magnitude of the collision kernel is much smaller than the magnitude of the number densities; therefore, the temporal decrease of the collision kernel does not affect the collision rate.
There is no dependency of the collision rate from the TW20 concentration visible for Emulsion I and II, which is in accordance with expectations; the initial droplet size does not differ for any TW20 concentrations tested for these emulsions, meaning that there should not be any impact of the DSD on at any time. Nonetheless, only the data from Emulsion I are interpreted in the following, as Emulsion II always had to be prepared from scratch for each test and breakage of the droplets could not be ruled out during the tests. This also explains the larger uncertainties for Emulsion II.
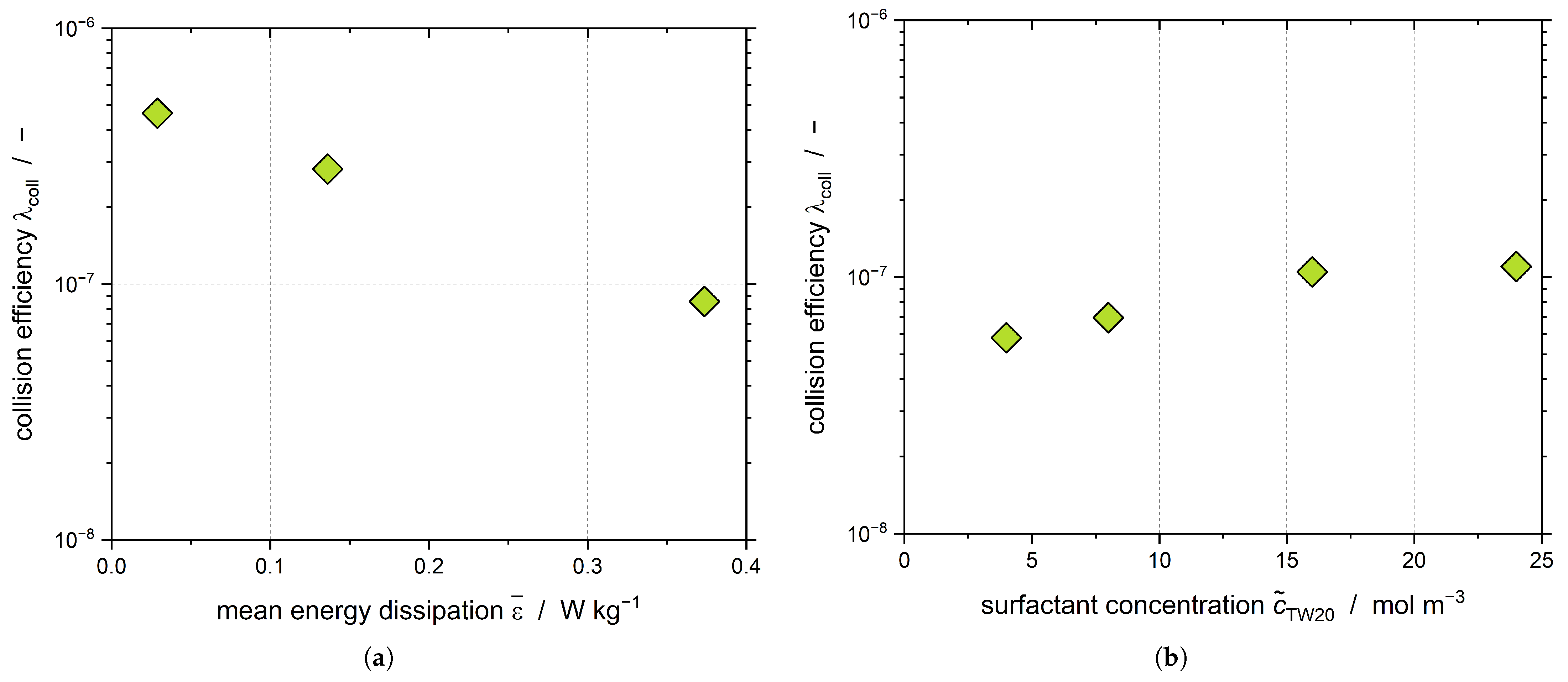
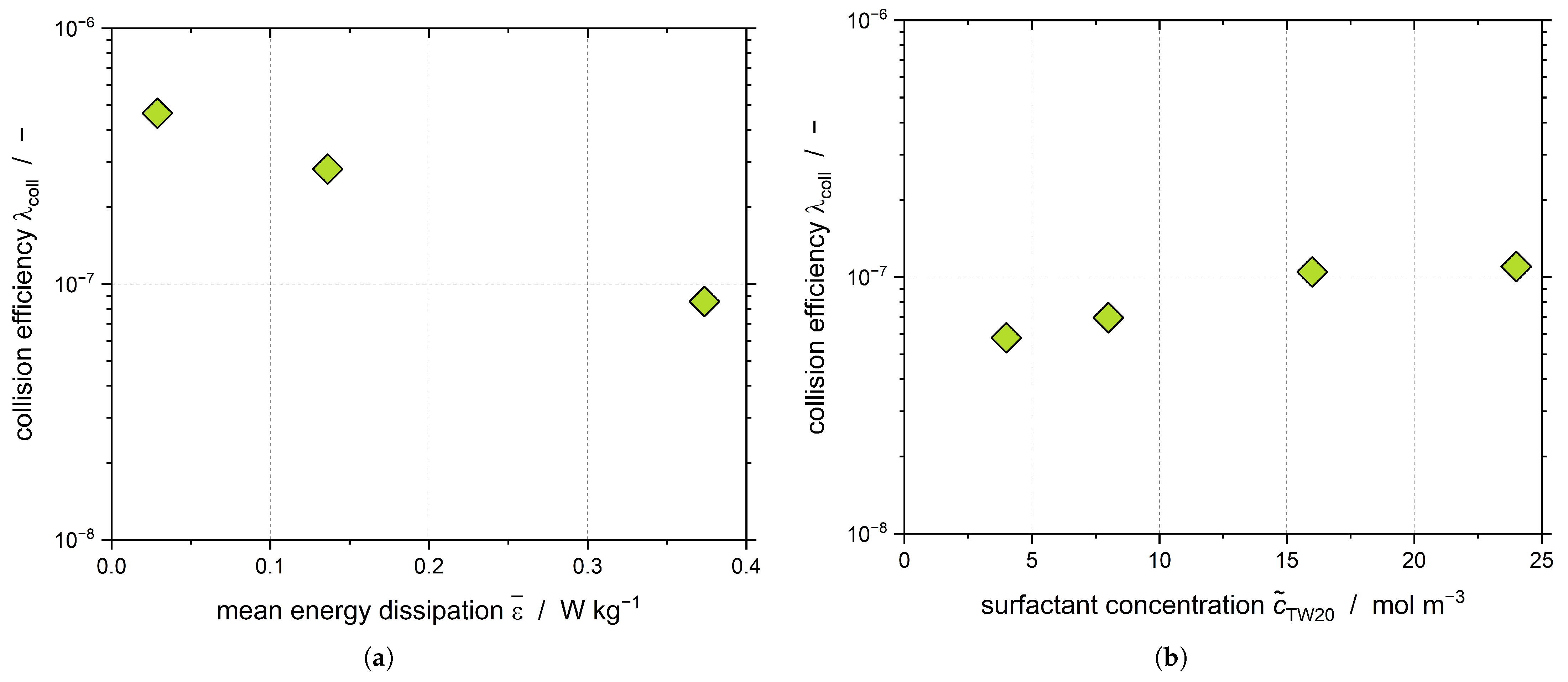
Assuming that CMN only takes place for
t > 5000 s and that both primary and shear-mediated nucleation can be neglected, the collision efficiency of the CMN
can be evaluated (
Figure 10).
We proved in our previous work that
decreases when increasing
(
Figure 10a), which is in good agreement with the data presented here [
38]. As has been mentioned previously, increasing
led to an increase of
(
Figure 4), whereas the evaluation of
did not show a dependency of
from
(
Figure 9c), as the DSD stayed the same. Therefore, the positive influence of the nanoparticles formed during the crystallization process must be visible in the collision efficiency
(
Figure 10b). However, it should be noted that the collisions occurring between nanoparticles and droplets were not considered in the calculation of
. Thus, the visible increase supports the hypothesis that the CMN not only takes place between droplets and particles but between nanoparticles and droplets.
Overall, it can be concluded that a higher energy input (increasing ) leads to higher nucleation rates; however, considering the increasing collision rate, the general efficiency of the collision decreases. If the obtained results are transferred to the industrial production of emulsions, the new findings can now be used to assess whether an increase in the power input with the given faster crystallization of the dispersed phase can compensate for the otherwise longer cooling phase. Moreover, the increase in is one option to guarantee a complete crystallization of the dispersed phase in case further subcooling is either too expensive or simply not feasible. This can be the case when tap water is used for cooling, as the maximum subcooling is limited by the temperature of tap water.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}