
Thermoresponsive Starch Nanoparticles for the Extraction of Bitumen from Oil Sands


Abstract

1. Introduction
2. Materials and Methods
2.1. Chemicals and Materials
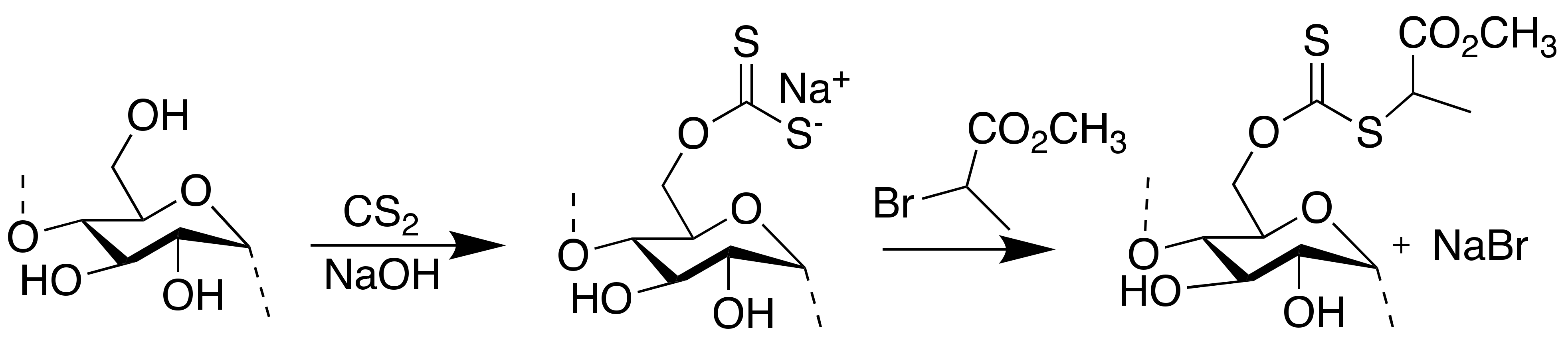
2.2. Synthesis of Xanthanted SNPs
2.3. Synthesis of Starch-Based RAFT Agent
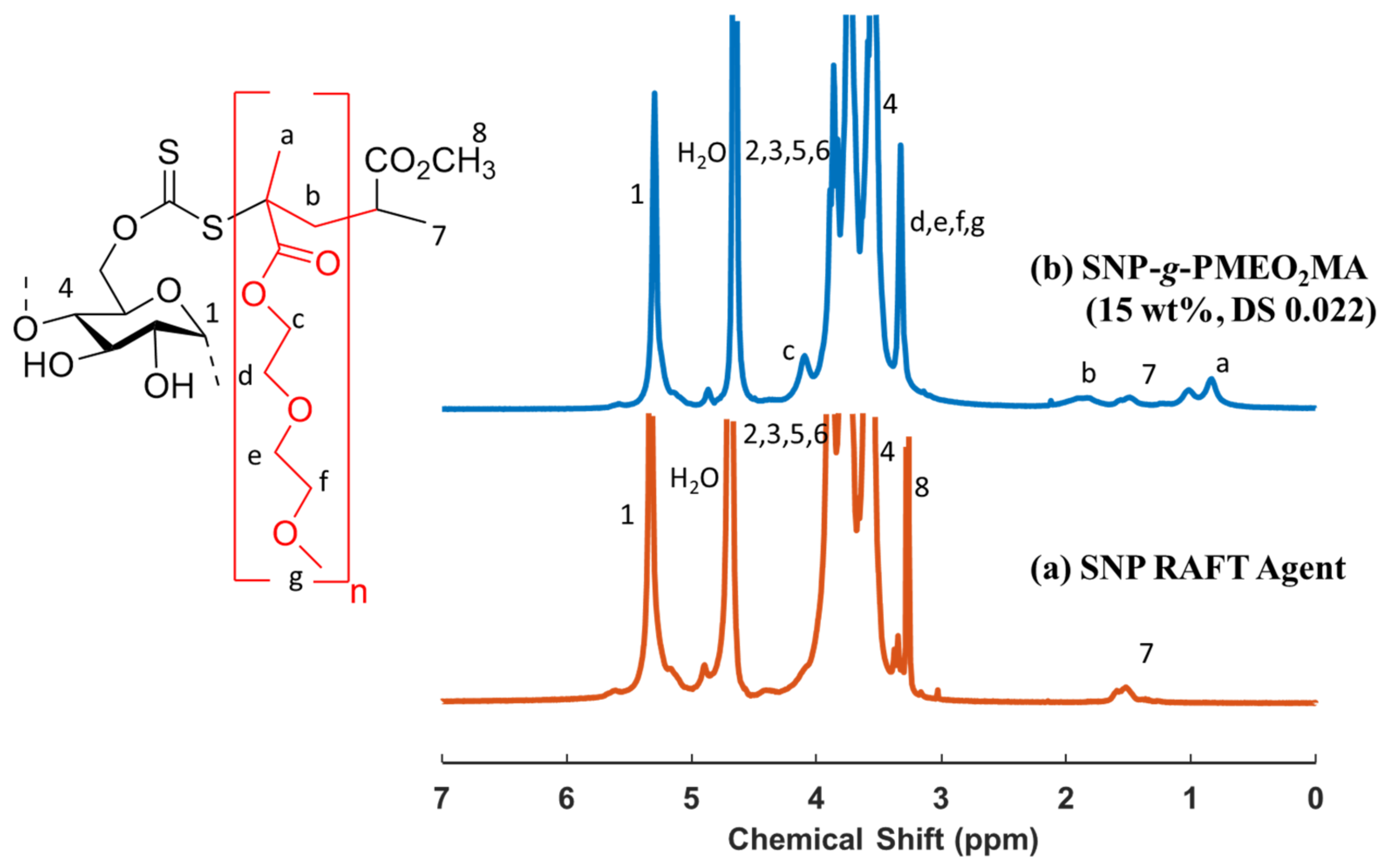
2.4. Synthesis of SNP-g-PMEO2MA
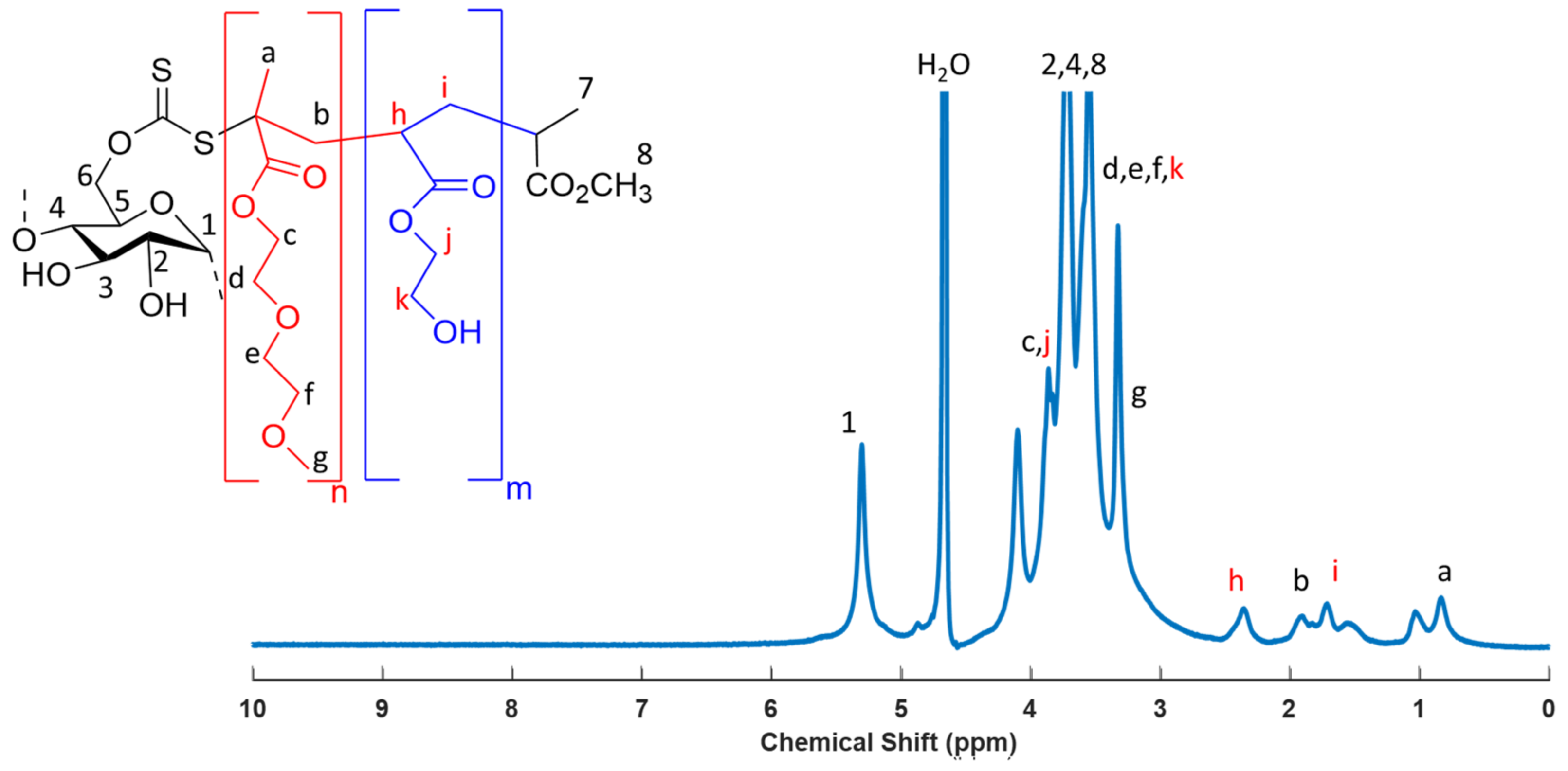
2.5. Synthesis of SNP-g-PMEO2MA-b-PHEA
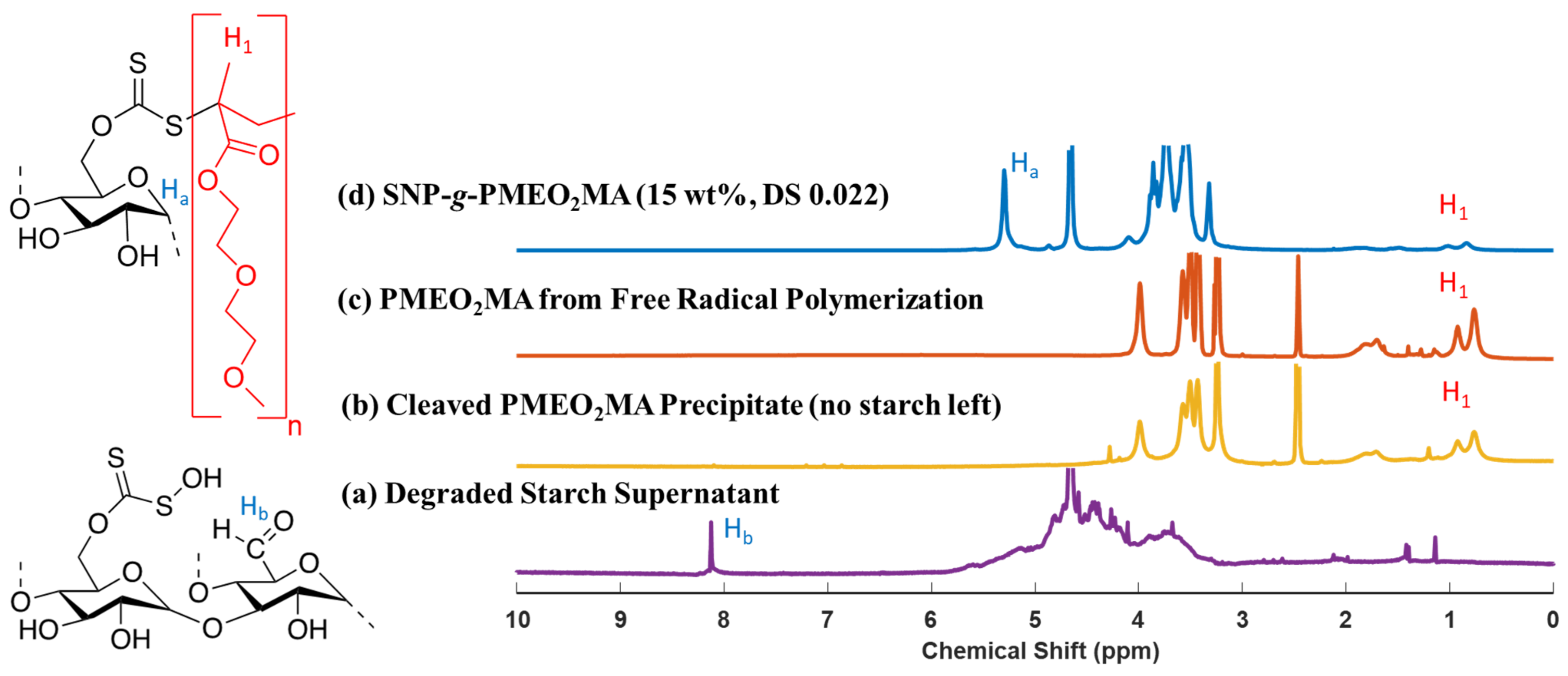
2.6. Cleavage of PMEO2MA from SNP-g-PMEO2MA
2.7. Characterization Techniques
2.7.1. 1H NMR
2.7.2. UV-Visible Spectroscopy
2.7.3. DLS Analysis
2.7.4. Transmission Electron Microscopy (TEM)
2.7.5. Gel Permeation Chromatography
2.7.6. Measurement of Fines and Bitumen Contents in IOos and SBos
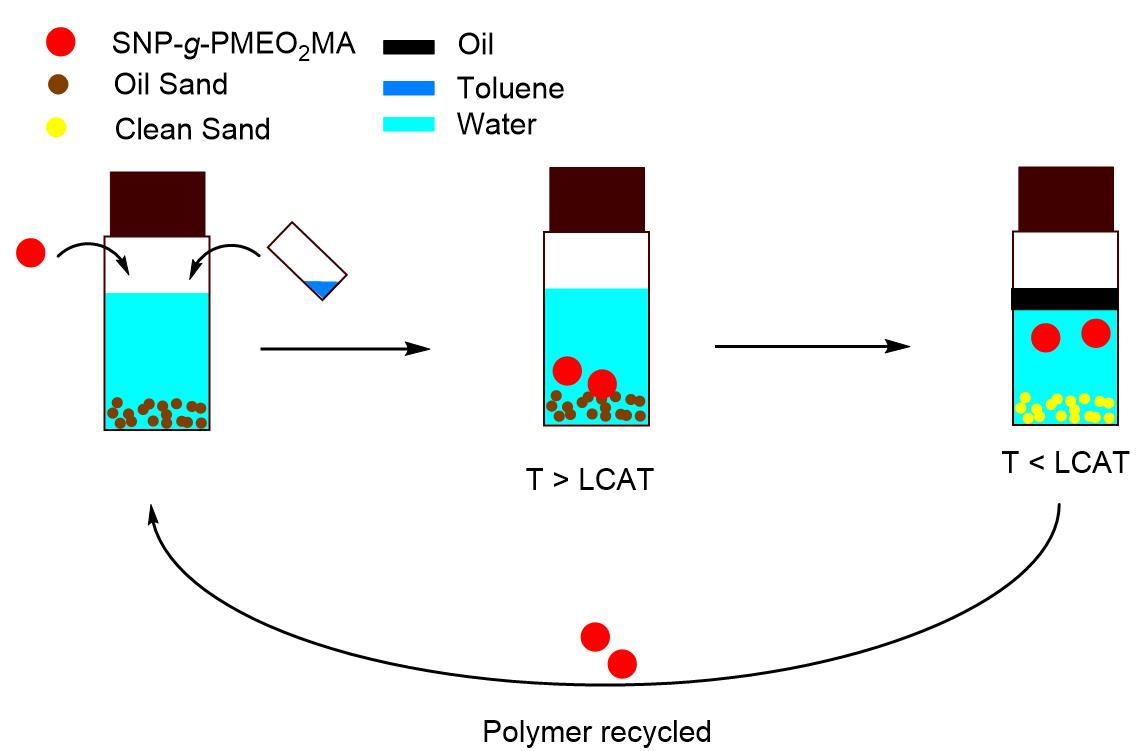
2.8. Bitumen Extraction Procedure
3. Results
3.1. Synthesis of the RAFT Agent and SNP-g-PMEO2MA
3.2. Cleavage of PMEO2MA from SNP-g-PMEO2MA
3.3. Synthesis of SNP-g-PMEO2MA-b-PHEA
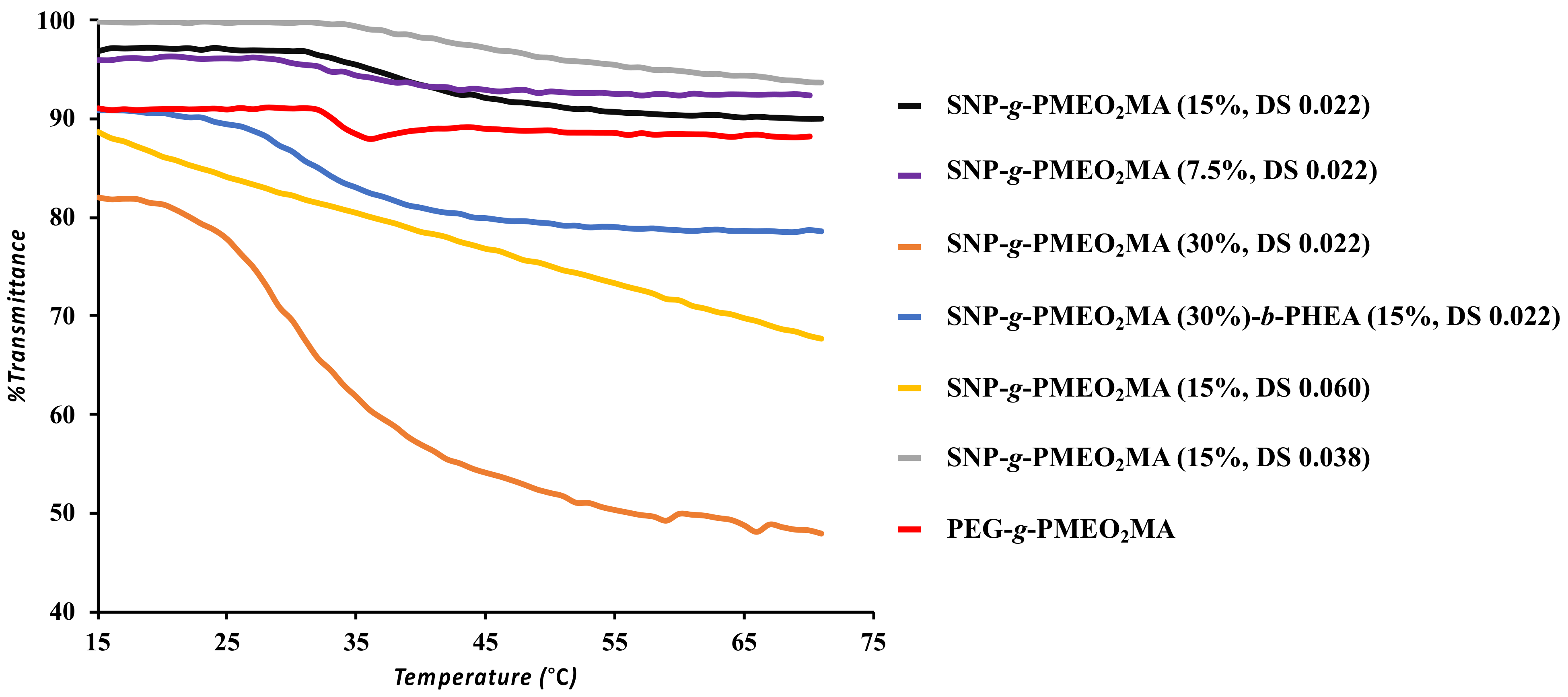
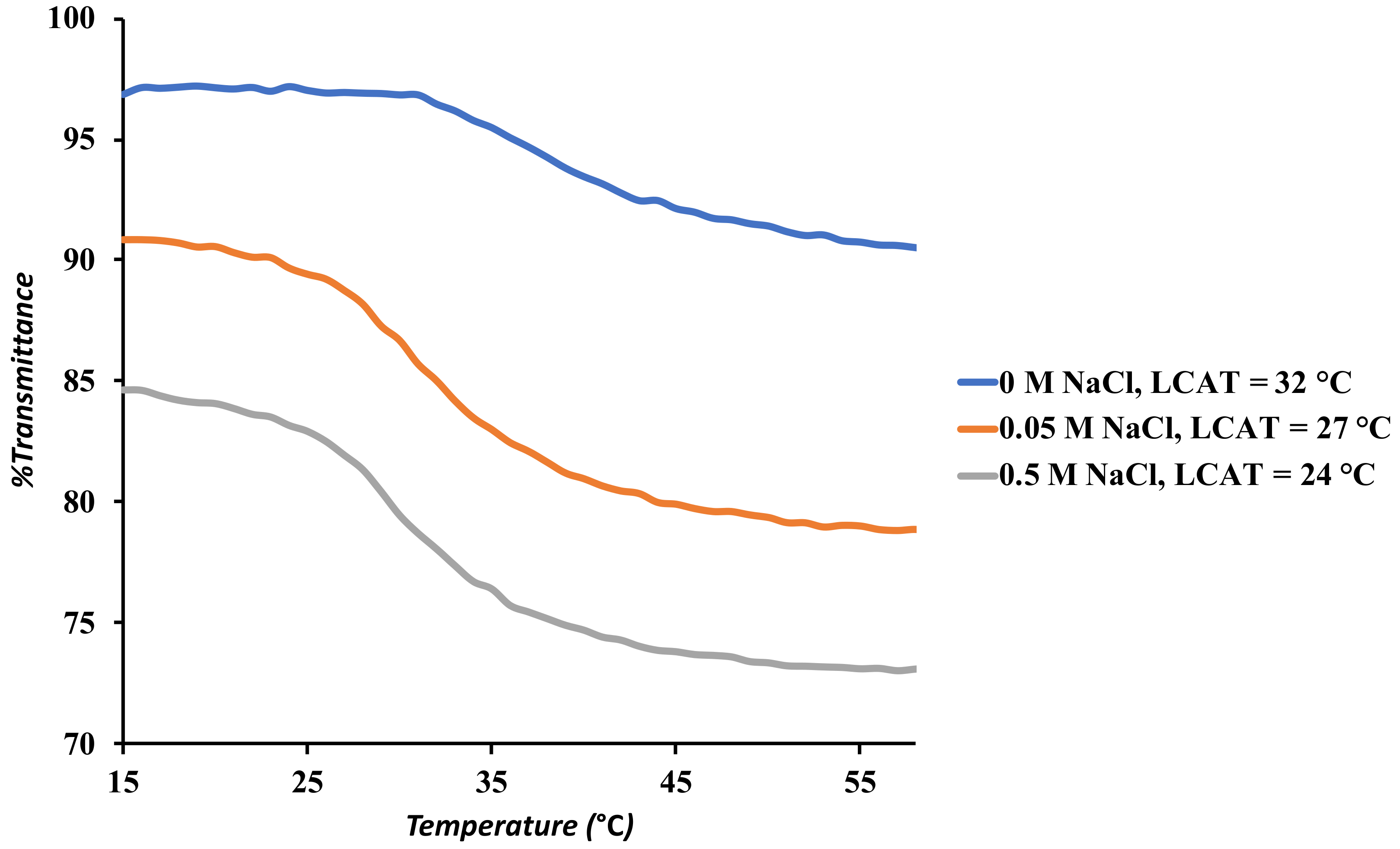
3.4. UV-Visible Spectroscopy
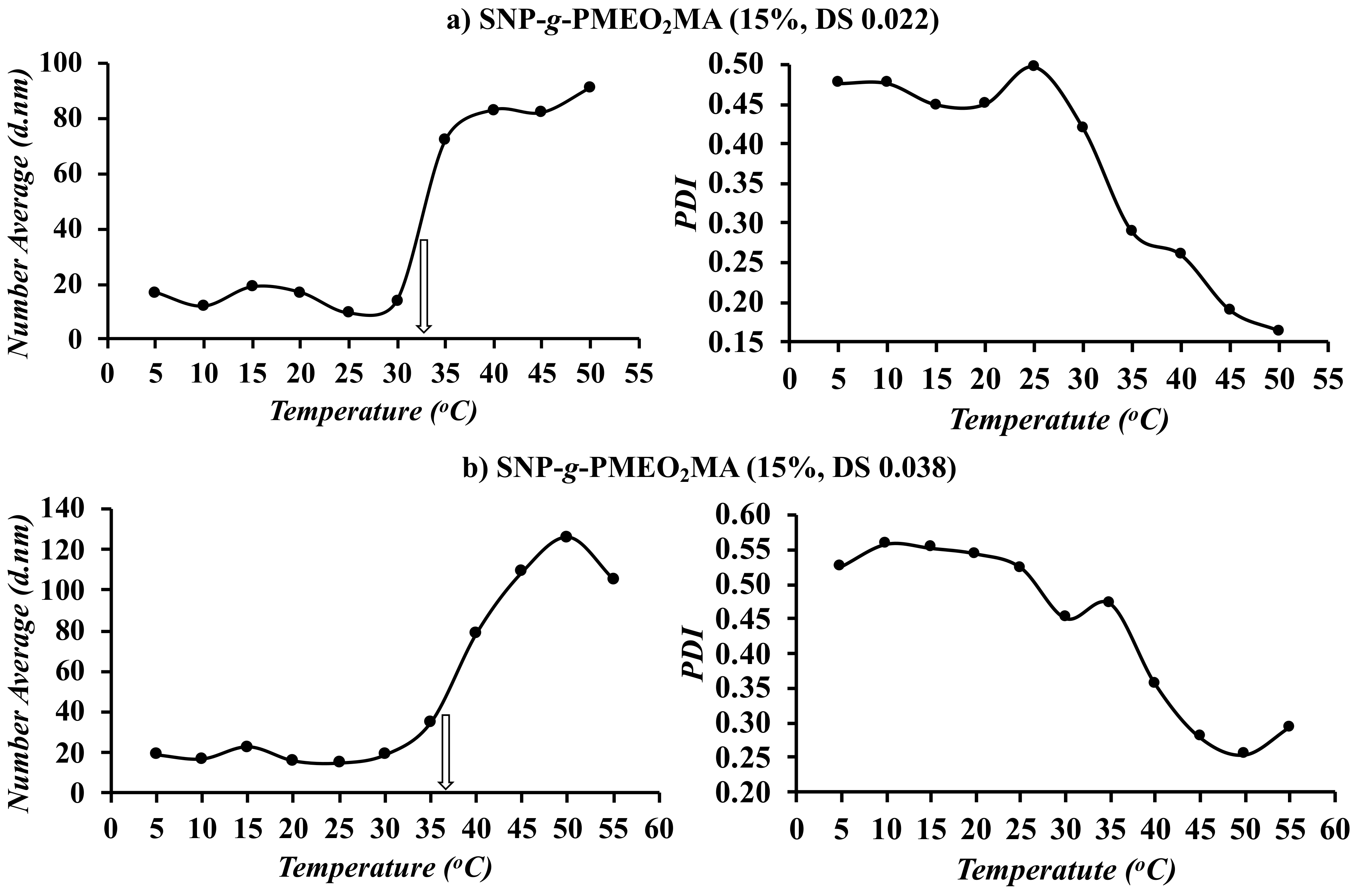
3.5. DLS Analysis
3.6. Transmission Electron Microscopy
3.7. Determination of Bitumen and Fines Contents of IOos and SBos
3.8. Bitumen Extraction
3.8.1. Controls for Efficient Bitumen Extraction
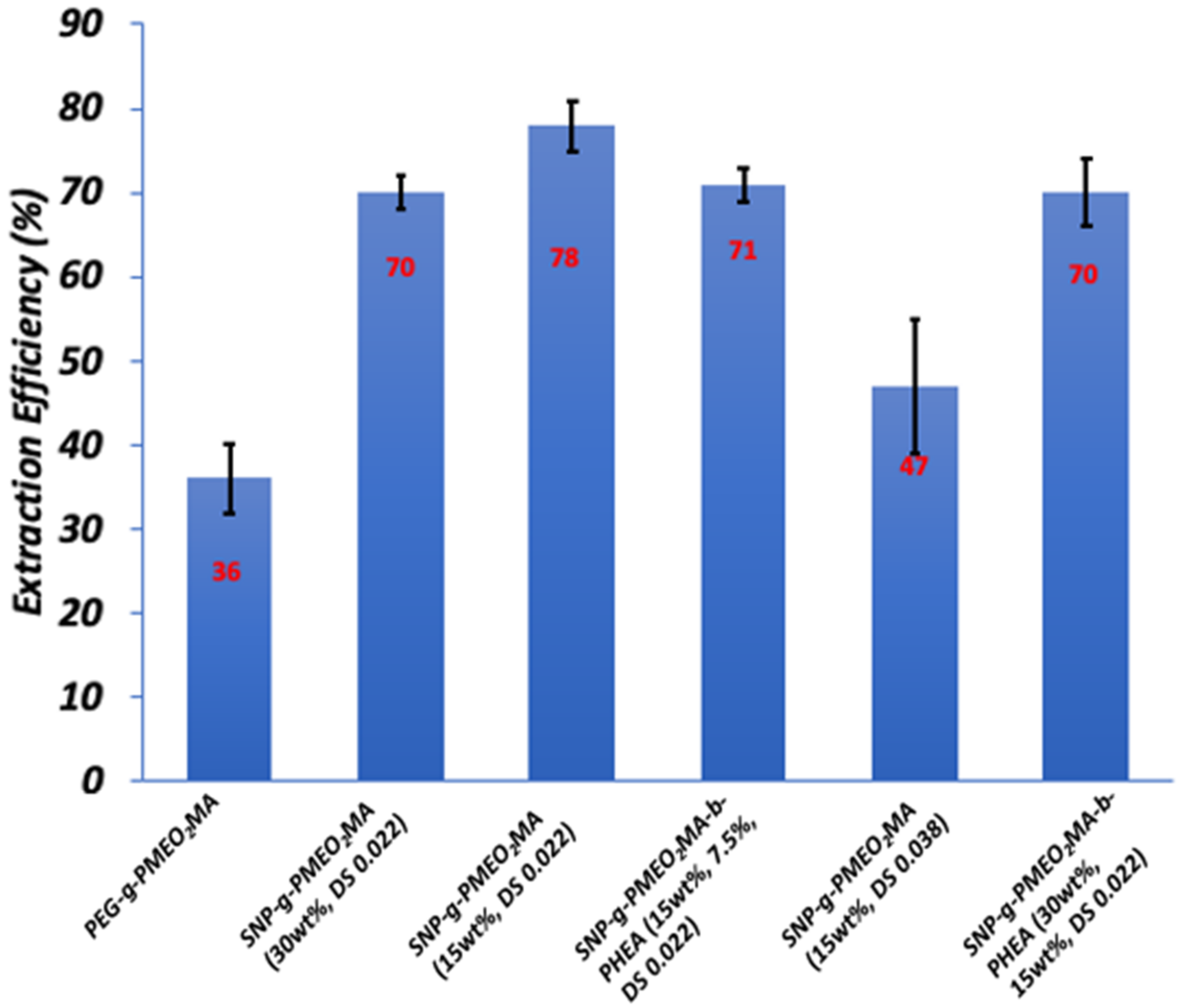
3.8.2. Optimal SNP-g-PMEO2MA Composition and Structure
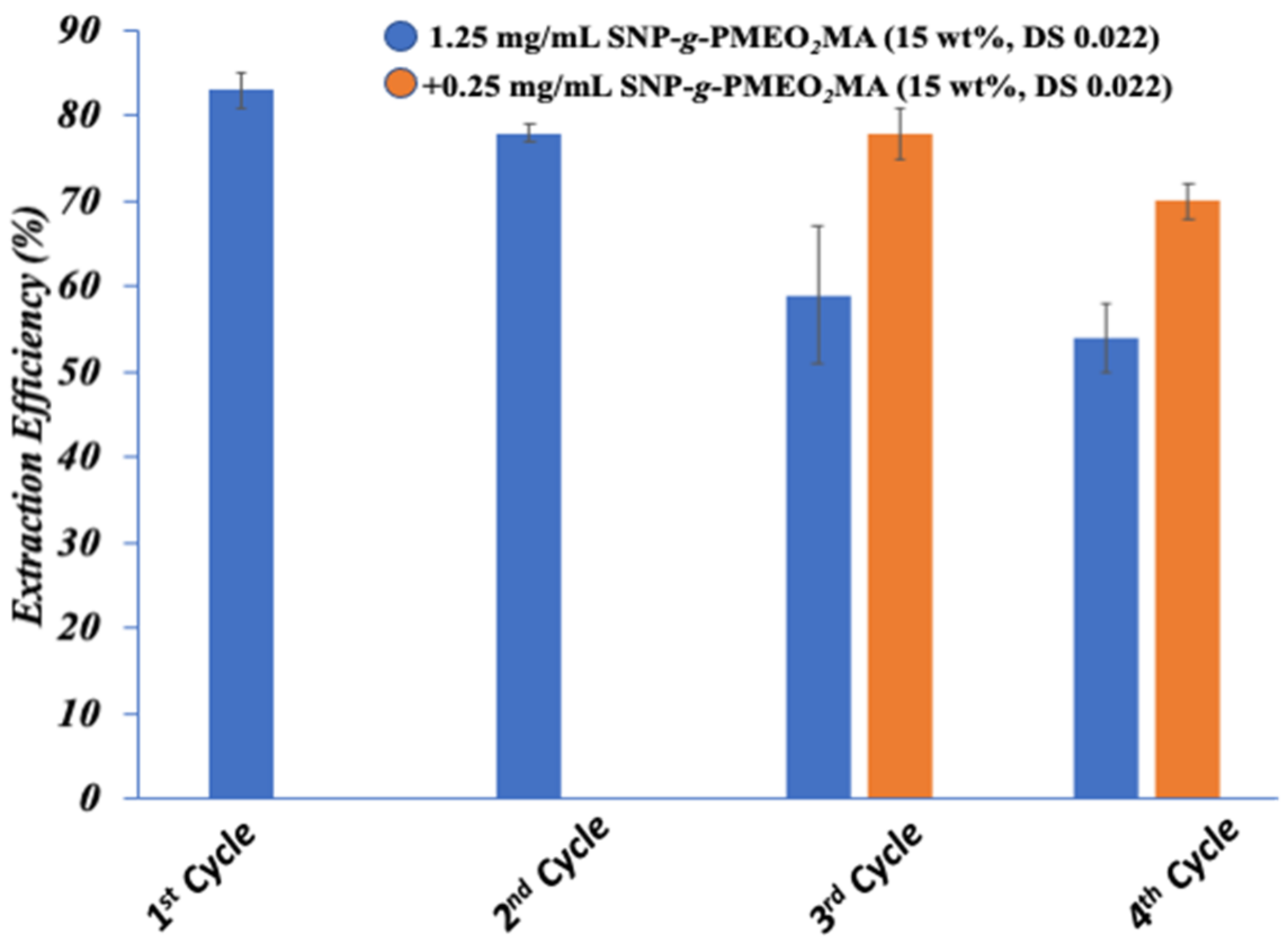
3.8.3. Recycling of Polymer-Grafted SNPs for Bitumen Extraction
3.8.4. Oil Extraction from IOos
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tenenbaum, D.J. Oils Sands Development: A Health Risk Worth Taking? Environ. Health Perspect. 2009, 117, A150–A156. [Google Scholar] [CrossRef] [PubMed]
- Masliyah, J.; Zhou, Z.J.; Xu, Z.; Czarnecki, J.; Hamza, H. Understanding Water-Based Bitumen Extraction from Athabasca Oil Sands. Can. J. Chem. Eng. 2008, 82, 628–654. [Google Scholar] [CrossRef]
- Takamura, K. Microscopic Structure of Athabasca Oil Sand. Can. J. Chem. Eng. 1982, 60, 538–545. [Google Scholar] [CrossRef]
- Czarnecki, J.; Radoev, B.; Schramm, L.L.; Slavchev, R. On the Nature of Athabasca Oil Sands. Adv. Colloid Interface Sci. 2005, 114, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Rao, F.; Liu, Q. Froth Treatment in Athabasca Oil Sands Bitumen Recovery Process: A Review. Energy Fuels 2013, 27, 7199–7207. [Google Scholar] [CrossRef]
- Miller, J.D.; Misra, M. Hot Water Process Development for Utah Tar Sands. Fuel Process. Technol. 1982, 6, 27–59. [Google Scholar] [CrossRef]
- Choung, J.; Walker, J.; Xu, Z.; Masliyah, J. Effect of Temperature on the Stability of Froth Formed in the Recycle Process Water of Oil Sands Extraction. Can. J. Chem. Eng. 2008, 82, 801–806. [Google Scholar] [CrossRef]
- Flury, C.; Afacan, A.; Tamiz Bakhtiari, M.; Sjoblom, J.; Xu, Z. Effect of Caustic Type on Bitumen Extraction from Canadian Oil Sands. Energy Fuels 2014, 28, 431–438. [Google Scholar] [CrossRef]
- Kotlyar, L.S.; Deslandes, Y.; Sparks, B.D.; Kodama, H.; Schutte, R. Characterization of Colloidal Solids from Athabasca Fine Tails. Clays Clay Miner. 1993, 41, 341–345. [Google Scholar] [CrossRef]
- Lo, C.C.; Brownlee, B.G.; Bunce, N.J. Mass Spectrometric and Toxicological Assays of Athabasca Oil Sands Naphthenic Acids. Water Res. 2006, 40, 655–664. [Google Scholar] [CrossRef]
- Long, Y.; Dabros, T.; Hamza, H. Stability and Settling Characteristics of Solvent-Diluted Bitumen Emulsions. Fuel 2002, 81, 1945–1952. [Google Scholar] [CrossRef]
- Joshi, V.; Kundu, D. Ionic liquid promoted extraction of bitumen from oil sand: A review. J. Pet. Sci. Eng. 2021, 199, 108232. [Google Scholar] [CrossRef]
- Lin, F.; Stoyanov, S.R.; Xu, Y. Recent advances in nonaqueous extraction of bitumen from mineable oil sands: A review. Org. Process. Res. Dev. 2017, 21, 492–510. [Google Scholar] [CrossRef]
- Zhou, J.Z. Role of mineral flotation technology in improving bitumen extraction from mined Athabasca oil sands III. Next generation of water-based oil sands extraction. Can. J. Chem. Eng. 2021, 99, 755–777. [Google Scholar] [CrossRef]
- Lu, Y.; Sun, D.; Ralston, J.; Liu, Q.; Xu, Z. CO2-responsive surfactants with tunable switching pH. J. Colloid Interface Sci. 2019, 557, 185–195. [Google Scholar] [CrossRef]
- Han, C.; Li, R.; Lu, Y. Study on synthesized thermoresponsive block copolymer for water-based oil sands extraction. Energy Fuels 2020, 34, 9473–9482. [Google Scholar] [CrossRef]
- Yang, B.; Duhamel, J. Extraction of Oil from Oil Sands Using Thermoresponsive Polymeric Surfactants. ACS Appl. Mater. Interfaces 2015, 7, 5879–5889. [Google Scholar] [CrossRef]
- Góis, J.R.; Popov, A.V.; Guliashvili, T.; Serra, A.C.; Coelho, J.F.J. Synthesis of Functionalized Poly(Vinyl Acetate) Mediated by Alkyne-Terminated RAFT Agents. RSC Adv. 2015, 5, 91225–91234. [Google Scholar] [CrossRef]
- Keddie, D.J.; Moad, G.; Rizzardo, E.; Thang, S.H. RAFT Agent Design and Synthesis. Macromolecules 2012, 45, 5321–5342. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E.; Thang, S.H. A RAFT Tutorial. Strem Chem. 2011, 25, 2–10. [Google Scholar]
- Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef] [PubMed]
- Moad, G.; Rizzardo, E.; Thang, S.H. End-Functional Polymers, Thiocarbonylthio Group Removal/Transformation and Reversible Addition-Fragmentation-Chain Transfer (RAFT) Polymerization. Polym. Int. 2011, 60, 9–25. [Google Scholar] [CrossRef]
- Jesson, C.P.; Pearce, C.M.; Simon, H.; Werner, A.; Cunningham, V.J.; Lovett, J.R.; Smallridge, M.J.; Warren, N.J.; Armes, S.P. H2O2 Enables Convenient Removal of RAFT End-Groups from Block Copolymer Nano-Objects Prepared via Polymerization-Induced Self- Assembly in Water. Macromolecules 2017, 50, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Pfukwa, R.; Pound, G.; Klumperman, B. Facile End Group Modification of Raft Made Polymers, by Radical Exchange with Hydrogen Peroxide. Polym. Prepr. 2008, 49, 117–118. [Google Scholar]
- Masina, N.; Choonara, Y.E.; Kumar, P.; du Toit, L.C.; Govender, M.; Indermun, S.; Pillay, V. A Review of the Chemical Modification Techniques of Starch. Carbohydr. Polym. 2017, 157, 1226–1236. [Google Scholar] [CrossRef]
- Tomasik, P.; Schilling, C.H. Chemical Modification of Starch. Adv. Carbohydr. Chem. Biochem. 2004, 59, 175–403. [Google Scholar]
- Somcynsky, T. The Lower Critical Solution Temperature (LCST) of Non-polar Polymer Solutions: An Introduction. Polym. Eng. Sci. 1982, 22, 58–63. [Google Scholar] [CrossRef]
- Du, H.; Wickramasinghe, R.; Qian, X. Effects of Salt on the Lower Critical Solution Temperature of Poly (N-Isopropylacrylamide). J. Phys. Chem. B 2010, 114, 16594–16604. [Google Scholar] [CrossRef]
- Lutz, J.F.; Akdemir, Ö.; Hoth, A. Point by Point Comparison of Two Thermosensitive Polymers Exhibiting a Similar LCST: Is the Age of Poly(NIPAM) Over? J. Am. Chem. Soc. 2006, 128, 13046–13047. [Google Scholar] [CrossRef]
- Lessard, D.G.; Ousalem, M.; Zhu, X.X. Effect of the Molecular Weight on the Lower Critical Solution Temperature of Poly(N,N-Diethylacrylamide) in Aqueous Solutions. Can. J. Chem. 2001, 79, 1870–1874. [Google Scholar] [CrossRef]
- Zhou, Z.A.; Li, H.; Chow, R.; Adeyinka, O.B.; Xu, Z.; Masliyah, J. Impact of Fine Solids on Mined Athabasca Oil Sands Extraction II. Effect of Fine Solids with Different Surface Wettability on Bitumen Recovery. Can. J. Chem. Eng. 2017, 95, 120–126. [Google Scholar] [CrossRef]
- Luque de Castro, M.D.; Priego-Capote, F. Soxhlet Extraction: Past and Present Panacea. J. Chromatogr. A 2010, 1217, 2383–2389. [Google Scholar] [CrossRef]
- Chen, Q.; Liu, Q. Bitumen coating on oil sands clay minerals: A review. Energy Fuels 2019, 33, 5933–5943. [Google Scholar] [CrossRef]
- Kunz, W.; Henle, J.; Ninham, B.W. About the Science of the Effect of Salts. Curr. Opin. Colloid Interface Sci. 2004, 9, 19–37. [Google Scholar] [CrossRef]
- Eeckman, F.; Amighi, K.; Moës, A.J. Effect of some physiological and non-physiological compounds on the phase transition temperature of thermoresponsive polymers intended for oral controlled-drug delivery. Int. J. Pharm. 2001, 222, 259–270. [Google Scholar] [CrossRef]
- Rocha, J.A.; Baydak, E.N.; Yarranton, H.W.; Sztukowski, D.M.; Ali-Marcano, V.; Gong, L.; Shi, C.; Zeng, H. Role of Aqueous Phase Chemistry, Interfacial Film Properties, and Surface Coverage in Stabilizing Water-in-Bitumen Emulsions. Energy Fuels 2016, 30, 5240–5252. [Google Scholar] [CrossRef]
- Leung, S.S.C.; MacKinnon, M.D.; Smith, R.E.H. Aquatic Reclamation in the Athabasca, Canada, Oil Sands: Naphthenate and Salt Effects on Phytoplankton Communities. Environ. Toxicol. Chem. 2001, 20, 1532–1543. [Google Scholar] [CrossRef]
- Akimoto, J.; Ito, Y. Thermoresponsive Biodegradable Polymeric Materials for Biomedical Application. In Sustainability & Green Polymer Chemistry Volume 1; American Chemical Society: Washington, DC, USA, 2020; Volume 1372, pp. 159–172. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Mn from GPC (kg/mol) | PDI | Theoretical Mn (kg/mol) | Initiator Efficiency |
|---|---|---|---|---|
| SNP-g-PMEO2MA (30%, DS 0.022) | 57.5 | 1.60 | 3.21 | 0.056 |
| SNP-g-PMEO2MA (15%, DS 0.022) | 43.3 | 1.64 | 1.33 | 0.031 |
| SNP-g-PMEO2MA (7.5%, DS 0.022) | 30.8 | 1.59 | 0.61 | 0.020 |
| SNP-g-PMEO2MA (15%, DS 0.012) | 58.6 | 1.86 | 2.41 | 0.041 |
| PMEO2MA-Grafted Samples | LCAT (°C) |
|---|---|
| SNP-g-PMEO2MA (15%, DS 0.022) | 32 |
| SNP-g-PMEO2MA (7.5%, DS 0.022) | 28 |
| SNP-g-PMEO2MA (30%, DS 0.022) | 20 |
| SNP-g-PMEO2MA (30%)-b-PHEA (15%, DS 0.022) | 26 |
| SNP-g-PMEO2MA (15%, DS 0.060) | - |
| SNP-g-PMEO2MA (15%, DS 0.038) | 36 |
| PEG-b-PMEO2MA | 32 |
| # | Trials | Extraction Conditions | Extraction Efficiency (%) |
|---|---|---|---|
| 1 | 3 | No Salt, no polymer, 15 mL water, 60 mg toluene, 45 °C | 20 ± 1 |
| 2 | 3 | No Salt, no polymer, 15 mL water, 70 mg toluene, 45 °C | 34 ± 4 |
| 3 | 3 | 0.5 M NaCl, no polymer, 15 mL water, 60 mg toluene, 45 °C | 31 ± 2 |
| 4 | 3 | 0.5 M NaCl, 15 mg SNP, 15 mL water, 60 mg toluene, 45 °C | 33 ± 4 |
| 5 | 12 | 0.5 M NaCl, SNP-g-PMEO2MA (15%, DS 0.022), 15 mL water, 60 mg toluene, 45 °C | 78 ± 3 |
| 6 | 3 | No Salt, SNP-g-PMEO2MA (15%, DS 0.022), 15 mL water, 60 mg toluene, 45 °C | 48 ± 3 |
| 7 | 3 | 0.5 M NaCl, SNP-g-PMEO2MA (15%, DS 0.022), 15 mL water, 60 mg toluene, 22 °C | 41 ± 5 |
| 8 | 6 | 15 mL recycled SNP-g-PMEO2MA (15%, DS 0.022) solution, 60 mg toluene, 45 °C | 82 ± 3 |
| 9 | 3 | 0.5 M NaCl,15 mL PEG-b-PMEO2MA solution, 60 mg toluene, 45 °C | 36 ± 5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dasgupta, N.; Wang, J.-Z.O.; Nguyen, V.T.A.; Gauthier, M. Thermoresponsive Starch Nanoparticles for the Extraction of Bitumen from Oil Sands. Colloids Interfaces 2022, 6, 67. https://doi.org/10.3390/colloids6040067
Dasgupta N, Wang J-ZO, Nguyen VTA, Gauthier M. Thermoresponsive Starch Nanoparticles for the Extraction of Bitumen from Oil Sands. Colloids and Interfaces. 2022; 6(4):67. https://doi.org/10.3390/colloids6040067
Chicago/Turabian StyleDasgupta, Natun, Jun-Zhi Oliver Wang, Vo Thu An Nguyen, and Mario Gauthier. 2022. "Thermoresponsive Starch Nanoparticles for the Extraction of Bitumen from Oil Sands" Colloids and Interfaces 6, no. 4: 67. https://doi.org/10.3390/colloids6040067
APA StyleDasgupta, N., Wang, J.-Z. O., Nguyen, V. T. A., & Gauthier, M. (2022). Thermoresponsive Starch Nanoparticles for the Extraction of Bitumen from Oil Sands. Colloids and Interfaces, 6(4), 67. https://doi.org/10.3390/colloids6040067