
Relevance of Colloid Inherent Salt Estimated by Surface Complexation Modeling of Surface Charge Densities for Different Silica Colloids



Abstract
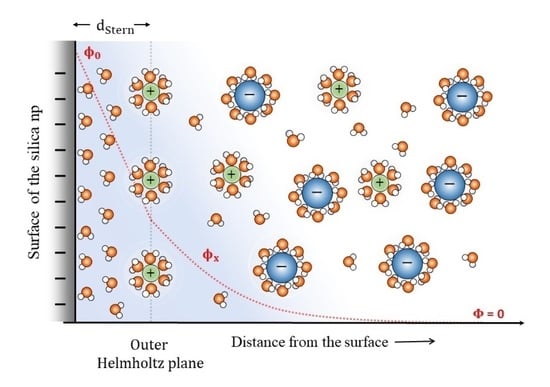
:
{kind=link}
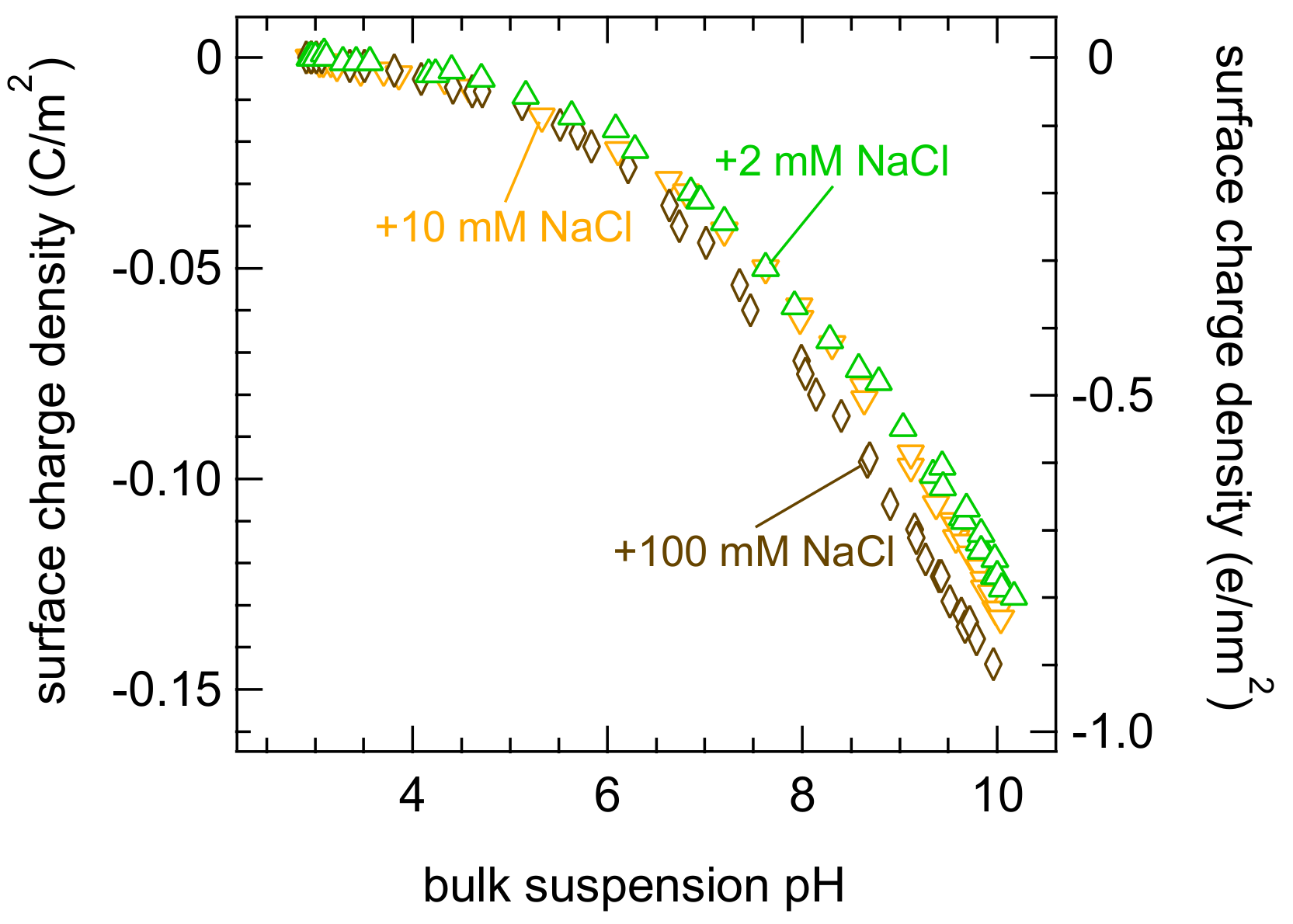{kind=link}
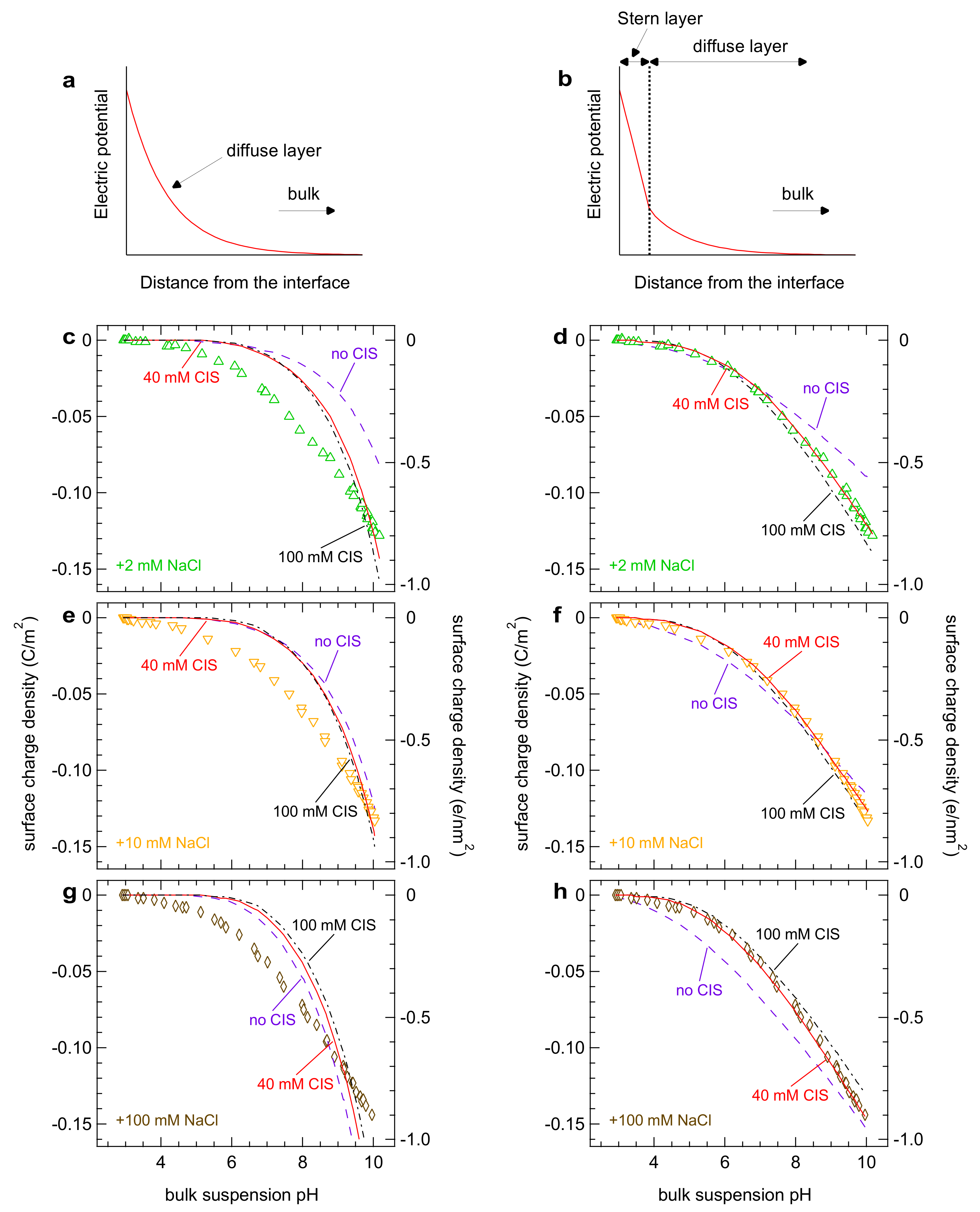
{kind=link}
{kind=link}
{kind=link}
1. Introduction
- Change the location of a given ion by using an electrolyte-specific value for the inner layer capacitance(s);
- Use a unique set of inner layer capacitance(s) involving charge distribution. This places more or less charge of the ion-pair in the inner or outer planes of the simplified interface model.
2. Materials and Methods
2.1. Chemicals
2.2. Potentiometric Titration
2.3. Calculation of Surface Charge Density (SCD)
2.4. Surface Complexation Modeling
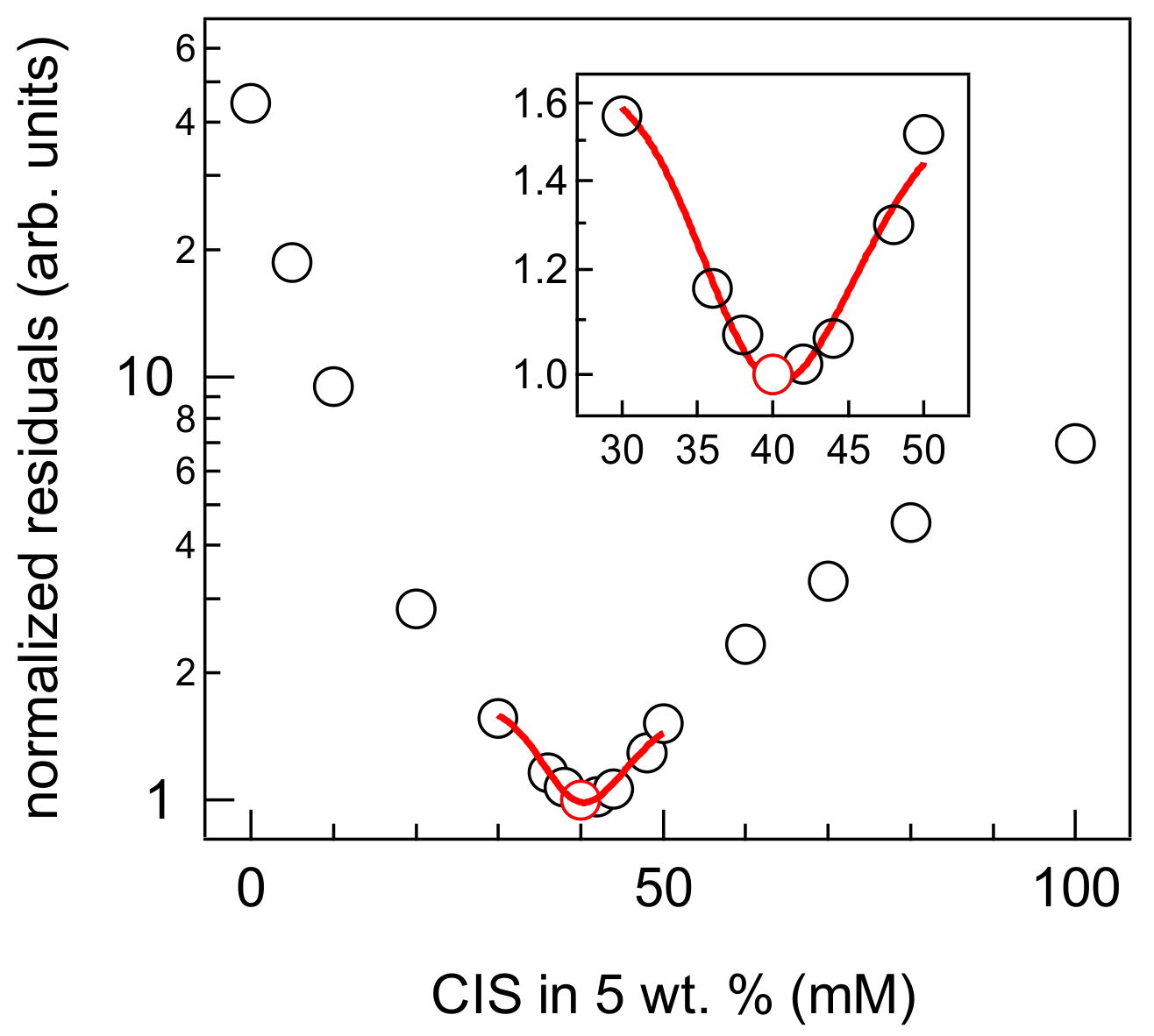
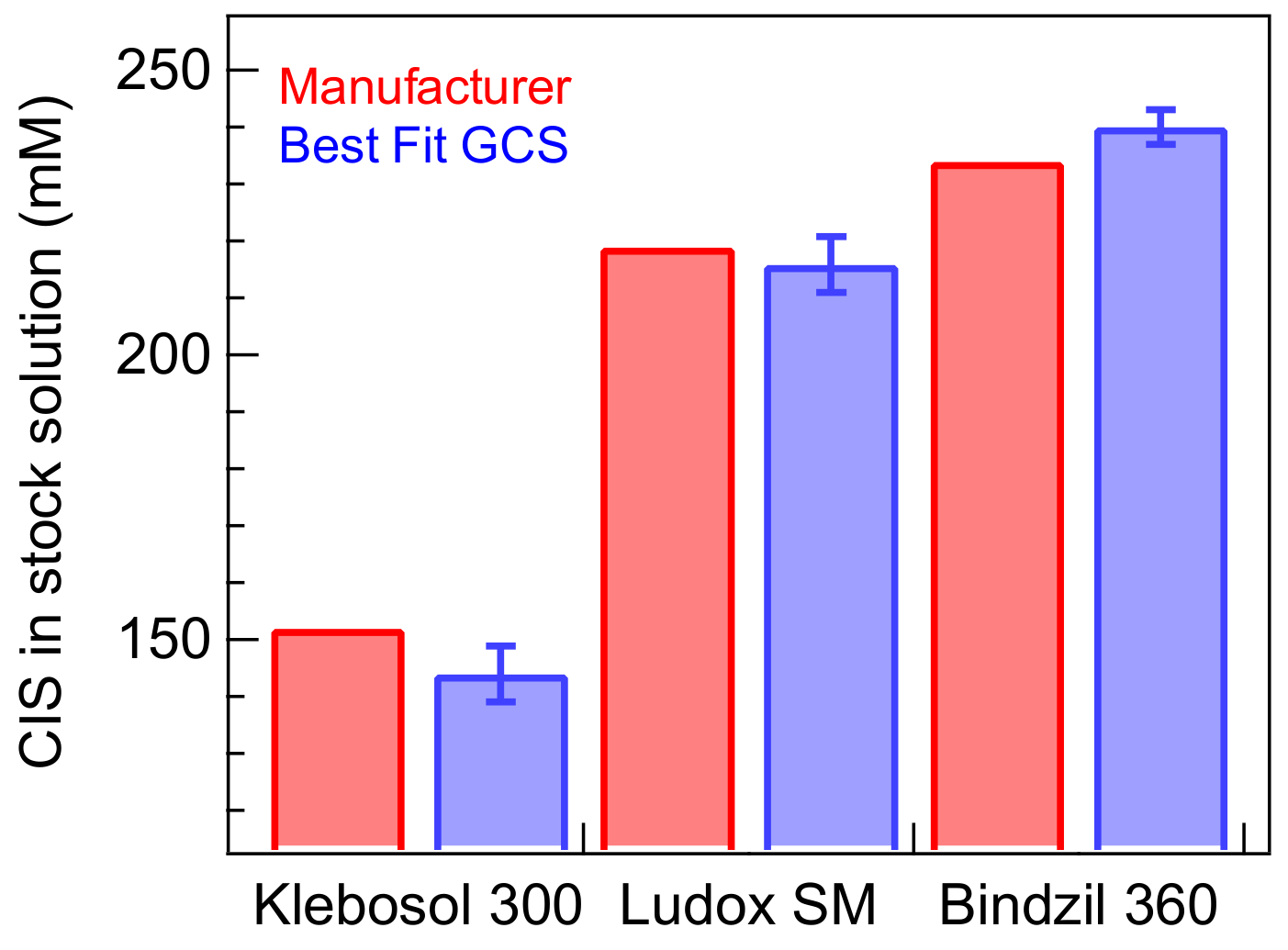
3. Results and Discussions
4. Conclusions
- The models appear to be sufficiently robust so that errors in the total concentration of a relevant chemical component (here Na) can be traced. This would mean that, if such models fail, an error in a mole balance may be the origin (among other possibilities);
- The study of ion-specific effects will be affected by the presence of residual salt (CIS) because there is interaction with ions (here, sodium) that are not necessarily considered;
- The relevance of CIS crucially depends on the conditions (such as the added salt amount and sol concentration);
- For the models, disregarding the residual salt in titrations and adsorption experiments will lead to inaccurate model parameters.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jenne, E.A. Trace Element Sorption by Sediments and Soils: Sites and Processes. In Molybdenum in the Environment; Chappel, W.R., Petersen, K.K., Eds.; Marcel-Dekker: New York, NY, USA, 1977; Volume 2, pp. 425–553. [Google Scholar]
- Stumm, W.; Morgan, J.J. Aquatic Chemistry: An Introduction Emphasizing Chemical Equilibria in Natural Waters; Wiley: New York, NY, USA, 1981. [Google Scholar]
- Schindler, P.W.; Sposito, G. Surface complexation at (Hydr)oxide surfaces. In Interactions at the Soil Colloid—Soil Solution Interface; Bolt, G.H., De Boodt, M.F., Hayes, M.H.B., McBride, M.B., De Strooper, E.B.A., Eds.; Springer: Dordrecht, The Netherlands, 1991; Volume 190, pp. 115–145. [Google Scholar]
- Hiemstra, T.; Van Riemsdijk, W.H. On the relationship between charge distribution, surface hydration, and the structure of the interface of metal hydroxides. J. Colloid Interface Sci. 2006, 301, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Hayes, K.F.; Leckie, J.O. Modeling ionic strength effects on cation adsorption at hydrous oxide/solution interfaces. J. Colloid Interface Sci. 1987, 115, 564–572. [Google Scholar] [CrossRef]
- Garcia, D.; Lützenkirchen, J.; Petrov, V.; Siebentritt, M.; Schild, D.; Lefèvre, G.; Rabung, T.; Altmaier, M.; Kalmykov, S.; Duro, L.; et al. Sorption of Eu(III) on quartz at high salt concentrations. Colloids Surf. A Physicochem. Eng. Asp. 2019, 578, 123610. [Google Scholar] [CrossRef] [Green Version]
- Morelová, N.; Finck, N.; Lützenkirchen, J.; Schild, D.; Dardenne, K.; Geckeis, H. Sorption of americium/europium onto magnetite under saline conditions: Batch experiments, surface complexation modelling and X-ray absorption spectroscopy study. J. Colloid Interface Sci. 2020, 561, 708–718. [Google Scholar] [CrossRef]
- Předota, M.; Machesky, M.L.; Wesolowski, D.J.; Cummings, P.T. Electric double layer at the rutile (110) surface. 4. Effect of temperature and pH on the adsorption and dynamics of ions. J. Phys. Chem. C 2013, 117, 22852–22866. [Google Scholar] [CrossRef]
- Schindler, P.W. A solution chemists view of surface chemistry. Pure Appl. Chem. 1991, 63, 1697–1704. [Google Scholar] [CrossRef]
- Westall, J.C. Mineql: A Computer Program for the Calculation of Chemical Equilibrium Composition of Aqueous Systems; Massachusetts Institute of Technology, Water Quality Laboratory: Cambridge, MA, USA, 1976. [Google Scholar]
- Westall, J.; Hohl, H. A comparison of electrostatic models for the oxide/solution interface. Adv. Colloid Interface Sci. 1980, 12, 265–294. [Google Scholar] [CrossRef]
- Hiemstra, T.; De Wit, J.C.M.; Van Riemsdijk, W.H. Multisite proton adsorption modeling at the solid/solution interface of (hydr)oxides: A new approach: II. Application to various important (hydr)oxides. J. Colloid Interface Sci. 1989, 133, 105–117. [Google Scholar] [CrossRef]
- Hiemstra, T.; Van Riemsdijk, W.H. A surface structural approach to ion adsorption: The charge distribution (CD) model. J. Colloid Interface Sci. 1996, 179, 488–508. [Google Scholar] [CrossRef]
- Hiemstra, T. Ferrihydrite interaction with silicate and competing oxyanions: Geometry and hydrogen bonding of surface species. Geochim. Cosmochim. Acta 2018, 238, 453–476. [Google Scholar] [CrossRef]
- Tasciotti, E.; Liu, X.; Bhavane, R.; Plant, K.; Leonard, A.D.; Price, B.K.; Cheng, M.C.-C.; Decuzzi, P.; Tour, J.; Roberta, F.M.; et al. Mesoporous silicon particles as a multistage delivery system for imaging and theraputic applications. Nat. Nanotechnol. 2008, 3, 151–157. [Google Scholar] [CrossRef]
- Iler, R.K. The Chemistry of Silica: Solubility, Polymerization, Colloid and Surface Properties and Biochemistry of Silica; Wiley: New York, NY, USA, 1979. [Google Scholar]
- Sahai, N. Is silica really an anomalous oxide? Surface acidity and aqueous hydrolysis revisited. Environ. Sci. Technol. 2002, 36, 445–452. [Google Scholar] [CrossRef]
- Brown, M.A.; Arrigoni, M.; Heroguel, F.; Beloqui Redondo, A.; Giordano, L.; van Bokhoven, J.A.; Pacchioni, G. pH dependent electronic and geometric structures at the water-silica nanoparticle interface. J. Phys. Chem. C 2014, 118, 29007–29016. [Google Scholar] [CrossRef]
- Kosmulski, M. Adsorption of cesium hydroxide on controlled porous glasses. J. Radioanal. Nucl. Chem. Artic. 1989, 129, 149–154. [Google Scholar] [CrossRef]
- Redhead, H.; Davis, S.; Illum, L. Drug Delivery in poly (lactide-co-glycolide) nanoparticles surface modified with poloxamer 407 and poloxamine 908: In vitro characterization and in vivo evaluation. J. Control. Release 2001, 70, 353–363. [Google Scholar] [CrossRef]
- Kosmulski, M.; Matijevic, E. Zeta-potentials of silica in water alcohol mixtures. Langmuir 1992, 8, 1060–1064. [Google Scholar] [CrossRef]
- Brown, M.A.; Goel, A.; Abbas, Z. Effect of electrolyte concentration on the stern layer thickness at a charged interface. Angew. Chem. Int. Ed. 2016, 55, 3790–3794. [Google Scholar] [CrossRef]
- Jena, K.C.; Covert, P.A.; Hore, D.K. The effect of salt on the water structure at a charged solid surface: Differentiating second- and third-order nonlinear contributions. J. Phys. Chem. Lett. 2011, 2, 1056–1061. [Google Scholar] [CrossRef]
- Ostroverkhov, V.; Waychunas, G.A.; Shen, Y.R. Vibrational spectra of water at water/A-quartz (0001) interface. Chem. Phys. Lett. 2004, 386, 144–148. [Google Scholar] [CrossRef]
- Ong, S.; Zhao, X.; Eisenthal, K.B. Polarization of water molecules at a charged interface: Second harmonic studies of the silica/water interface. Chem. Phys. Lett. 1992, 191, 327–335. [Google Scholar] [CrossRef]
- Keller, A.A.; Wang, H.T.; Zhou, D.X.; Lenihan, H.S.; Cherr, G.; Cardinale, B.J.; Miller, R.; Ji, Z.X. Stability and aggregation of metal oxide nanoparticles in natural aqueous matrices. Environ. Sci. Technol. 2010, 44, 1962–1967. [Google Scholar] [CrossRef]
- Iler, R.K. The Colloidal Chemistry of Silica and Silicates; Cornell University Press: Ithaca, NY, USA, 1955. [Google Scholar]
- Parks, G.A. The isoelectric points of solid oxide, solid hydroxides and aqueous hydro complex systems. Chem. Rev. 1965, 65, 177–198. [Google Scholar] [CrossRef]
- Duval, Y.; Mielczarski, J.A.; Pokrovsky, O.S.; Meilczarski, E.; Ehrhardt, J.J. Evidence of the existence of three types of species at the quartz-aqueous solution interface at pH 0-10: XPS surface group quantification and surface complexation. J. Phys. Chem. B 2002, 106, 2937–2945. [Google Scholar] [CrossRef]
- Dove, P.M.; Craven, C.M. Surface charge density on silica in alkali and alkaline earth chloride electrolyte solutions. Geochim. Cosmochim. Acta 2005, 69, 4963–4970. [Google Scholar] [CrossRef]
- Bolt, G.H. Determination of the charge density of silica sols. J. Phys. Chem. 1957, 61, 1166–1169. [Google Scholar] [CrossRef]
- Tadros, T.F.; Lyklema, J. Adsorption of potential-determining ions at the silica-aqueous electrolyte interface and the role of some cations. J. Electroanal. Chem. 1968, 17, 267–275. [Google Scholar] [CrossRef]
- Milonjic, S.K. Determination of surface ionization and complexation constants at colloidal silica/electrolyte interface. Colloids Surf. 1987, 23, 301–312. [Google Scholar] [CrossRef]
- Sonnefeld, J.; Göbel, A.; Vogelsberger, W. Surface charge density on spherical silica particles in aqueous alkali chloride solutions part 1. Experimental results. Colloid Polym. Sci. 1995, 273, 926–931. [Google Scholar] [CrossRef]
- Brown, M.A.; Abbas, Z.; Kleibert, A.; Goel, A.; Green, R.; May, S.; Squires, T.M. Determination of surface potential and electrical double layer structure at the aqueous electrolyte-nanoparticle interface. Phys. Rev. X 2016, 6, 011007. [Google Scholar] [CrossRef] [Green Version]
- Jalil, A.H.; Pyell, U. Quantification of zeta-potential and electrokinetic surface charge density for colloidal silica nanoparticles dependent on type and concentration of the counterion: Probing the outer Helmholtz plane. J. Phys. Chem. C 2018, 122, 4437–4453. [Google Scholar] [CrossRef]
- Boakye-Ansah, S.; Khan, M.A.; Haase, M.F. Controlling surfactant adsorption on highly charged nanoparticles to stabilize bijels. J. Phys. Chem. C 2020, 124, 12417–12423. [Google Scholar] [CrossRef] [PubMed]
- Hunt, J.D.; Ezzedine, S.M.; Bourcier, W.; Roberts, S. Kinetics of the Gelation of Colloidal Silica at Geothermal Conditions, and Implications for Reservoir Modification and Management. In Proceedings of the 38th Workshop on Geothermal Reservoir Engineering, Stanford, CA, USA, 11–13 February 2013. [Google Scholar]
- Trompette, J.-L.; Meireles, M. Ion-specific effect on the gelation kinetics of concentrated colloidal silica suspensions. J. Colloid Interface Sci. 2003, 263, 522–527. [Google Scholar] [CrossRef]
- Trompette, J.-L.; Clifton, M.J.; Bacchin, P. Ion-specific repulsive interactions in colloidal silica dispersions evidenced through osmotic compression measurements and implication in frontal ultrafiltration experiments. J. Colloid Interface Sci. 2005, 290, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Döpke, M.F.; Lützenkirchen, J.; Moultos, O.A.; Siboulet, B.; Dufrêche, J.-F.; Padding, J.T.; Hartkamp, R. Preferential adsorption in mixed electrolytes confined by charged amorphous silica. J. Phys. Chem. C 2019, 123, 16711–16720. [Google Scholar] [CrossRef] [Green Version]
- Vayssieres, L. On the thermodynamic stability of metal oxide nanoparticles in aqueous solutions. Int. J. Nanotechnol. 2005, 2, 411–439. [Google Scholar] [CrossRef]
- Zackrisson, A.S.; Martinelli, A.; Matic, A.; Bergenholtz, J. Concentration effects on irreversible colloid cluster aggregation and gelation of silica dispersions. J. Colloid Interface Sci. 2006, 301, 137–144. [Google Scholar] [CrossRef]
- Trompette, J.-L. Influence of Co-ion nature on the gelation kinetics of colloidal silica suspensions. J. Phys. Chem. B 2017, 121, 5654–5659. [Google Scholar] [CrossRef]
- Fossum, J.O.; Gudding, E.; Fonseca, D.d.M.; Meheust, Y.; DiMasi, E.; Gog, T.; Venkataraman, C. Observations of orientational ordering in aqueous suspensions of a nano-layered silicate. Energy 2005, 30, 873–883. [Google Scholar] [CrossRef]
- Philips, I.R.; Sheehan, K.J. Importance of surface charge characteristics when selecting soils for wastewater re-use. Aust. J. Soil Res. 2005, 43, 915–927. [Google Scholar] [CrossRef]
- Lützenkirchen, J.; Preočanin, T.; Kovačević, D.; Tomišić, V.; Lövgren, L.; Kallay, N. Potentiometric titrations as a tool for surface charge determination. Croat. Chem. Acta 2012, 85, 391–417. [Google Scholar] [CrossRef]
- Stumm, W.; Huang, C.P.; Jenkins, S.R. Specific chemical interaction affecting stability of dispersed systems. Croat. Chem. Acta 1970, 42, 223–245. [Google Scholar]
- Lützenkirchen, J. The constant capacitance model and variable ionic strength: An evaluation of possible applications and applicability. J. Colloid Interface Sci. 1999, 217, 8–18. [Google Scholar] [CrossRef]
- Lyklema, J. Fundamentals of Interface and Colloid Science; Academic Press: San Diego, CA, USA, 1995; Volume 2. [Google Scholar]
- Sverjensky, D.A. Interpretation and prediction of triple-layer model capacitances and the structure of the oxide-electrolyte-water interface. Geochim. Cosmochim. Acta 2001, 65, 3643–3655. [Google Scholar] [CrossRef]
- Westall, J. FITEQL: A Computer Program for Determination of Chemical Constants from Experimental Data. Version 2.0; Oregon State University: Corvallis, OR, USA, 1982. [Google Scholar]
- Zhuravlev, L.T. The surface chemistry of amorphous silica. Zhuravlev model. Colloids Surf. A Physicochem. Eng. Asp. 2000, 173, 1–38. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goel, A.; Lützenkirchen, J. Relevance of Colloid Inherent Salt Estimated by Surface Complexation Modeling of Surface Charge Densities for Different Silica Colloids. Colloids Interfaces 2022, 6, 23. https://doi.org/10.3390/colloids6020023
Goel A, Lützenkirchen J. Relevance of Colloid Inherent Salt Estimated by Surface Complexation Modeling of Surface Charge Densities for Different Silica Colloids. Colloids and Interfaces. 2022; 6(2):23. https://doi.org/10.3390/colloids6020023
Chicago/Turabian StyleGoel, Alok, and Johannes Lützenkirchen. 2022. "Relevance of Colloid Inherent Salt Estimated by Surface Complexation Modeling of Surface Charge Densities for Different Silica Colloids" Colloids and Interfaces 6, no. 2: 23. https://doi.org/10.3390/colloids6020023
APA StyleGoel, A., & Lützenkirchen, J. (2022). Relevance of Colloid Inherent Salt Estimated by Surface Complexation Modeling of Surface Charge Densities for Different Silica Colloids. Colloids and Interfaces, 6(2), 23. https://doi.org/10.3390/colloids6020023