Abstract
The characteristics of the wetting process of the porous surface of silica gel when penetrated by base liquids (water and n-octane), ethanol and stable drug systems (naproxen, ibuprofen and cyclosporine A), as biologically active substances in two ethanol concentrations, were determined by the wetting rate vs. time. The tests were performed for contacted and non-contacted plates with the vapours of the wetting liquid. Thin-layer liquid chromatography was used to determine the penetration rate of the SiO2-coated plates, taking into account the linear dependence consistent with the Washburn equation. Additionally, the changes in the adhesive tension ΔG were determined for the tested drugs. Drug stability tests were conducted using the dynamic light scattering technique and microelectrophoresis. The penetration time of the plate depends on the properties and structure of the wetting liquid droplets. The types of interactions (dispersive, electrostatic and hydrogen bonding) formed between the silanol surface groups of the silica gel and the groups contained in the adsorbate particles are also very important factors. The greater the impact force, the slower the wetting process due to the strong penetration of the liquid into the pores of the substrate. The characteristics of the drug wetting/stability process may contribute to the development of their new forms, creating delivery systems with greater efficiency and lower side effects.
1. Introduction
Drugs play an important role in human functioning, improving the patient’s condition, but their incorrect intake may lead to undesirable side effects. Each pharmaceutical product consists of a biologically active substance causing a specific effect, and auxiliary substances. The main agents used to treat pain are non-steroidal anti-inflammatory drugs (NLPs). They have a comprehensive anti-inflammatory, analgesic and antipyretic effect [1,2,3,4,5,6]. Modern drug delivery systems are developing particularly intensively nowadays due to the development of technology and digitization, allowing the creation of new drug carriers with much greater therapeutic effectiveness. Research into designing drug delivery systems capable of encapsulating and releasing drugs with different characteristics and wettability is of great interest. Accordingly, it is important to carefully control the hydrophobic/hydrophilic drug release kinetics, in terms of wettability, to avoid the undesirable effect of so-called burst release. Burst release denotes an unpredictable and uncontrolled release of encapsulated drugs from nanocarriers when introduced to a release medium. This phenomenon leads to higher initial drug delivery and also reduces the effective lifetime of the drug. Burst release can be used to deliver drugs at high release rates as one of the different drug administration strategies when immediate effect is desired [4,5,6,7,8,9].
1.1. Non-Steroidal Anti-Inflammatory Drugs
Non-steroidal anti-inflammatory drugs (NSAID) are a very wide group with a heterogeneous chemical structure and a range of applications. However, they all have at least three things in common: identical pharmacological properties, the same basic mechanism of action and similar side effects. NSAIDs include anti-inflammatory drugs, analgesics and antipyretics. The name of non-steroids is due to their structure, which is different from corticosteroids—also drugs with anti-inflammatory properties. Two main properties, i.e., versatile action as well as easy availability, contributed significantly to the prevalence of NSAIDs, most of which are available over the counter. Such drugs have found applications in the treatment of many diseases, and are the first agents used to treat pain, ranging from minor pain to the pain associated with osteoarthritis and cancer [4]. NSAIDs are also administered to patients struggling with inflammatory and non-inflammatory rheumatic disease, in the treatment of severe menstrual pains, hepatic and renal colic, migraines, neuralgia, root syndromes, muscle pains, post-traumatic pains, after-surgery pains and discopathies. NSAIDs increase the production of glycosaminoglycans as well as hyaluronan, so they constitute a protective substance for the articular cartilage. The most commonly used drugs in this group are ibuprofen and naproxen [4,5,6,7,8,9,10,11,12].
Ibuprofen is a safe drug belonging to the phenylpropionic acid derivatives (Figure 1). Its main advantage is the ability to control inflammation, fever and pain, as well as the side effects of another drug. It has a completely different structure, differing significantly from its predecessors, and is more readily used than paracetamol due to its anti-inflammatory effect. Like all drugs, ibuprofen can also cause side effects after exceeding the appropriate dose, but the side effects are minor, due to the good tolerance of the drug by the body [4,5,6,7,8,9,10,11,12].
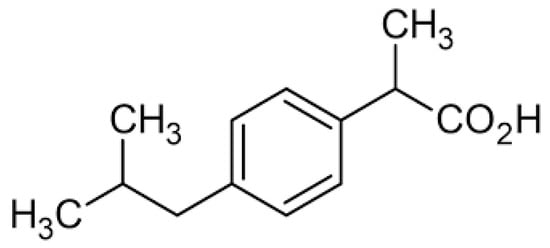
Figure 1.
(±)-Ibuprofen structure.
Naproxen, like ibuprofen, is a drug from the same NSAID group, belonging to the phenylpropionic acid derivatives (Figure 2). A common feature between ibuprofen and naproxen is their carboxylic acid group, which is not present in cyclosporine. This functional group contributes to their higher polarity and clarifies the low solubility. Taking naproxen, apart from the typical side effects of NSAIDs, may be associated with more serious side effects, such as angioedema, nephrotic syndrome or interstitial nephritis [4].
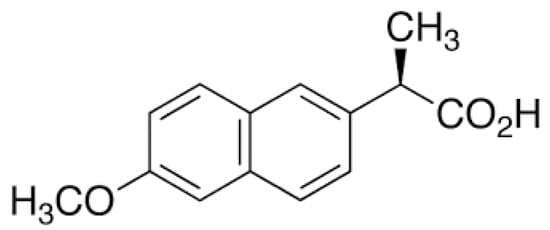
Figure 2.
(+)-Naproxen structure.
The naproxen molecule is relatively large if we relate its dimension to the pore diameter of typical silica material. The length of a naproxen molecule estimated from model calculations is ~1.17 nm, and its width is ~0.5 nm. However, with consideration of the dimension with the van der Waals spheres of extreme hydrogen atoms, the size increases to ~1.41 nm in length and ~0.74 nm in width [6]. The pKa of the NSAIDs’ carboxylic acids varies within 3.9 to 5.3. Therefore, at the acidic pH, the COOH is mostly protonated, decreasing the solubility of those drugs. On the contrary, at a neutral pH, the COOH group is deprotonated, increasing the drugs’ solubility in those media. Possible differences in behavior between ibuprofen and naproxen could be caused by additional methoxy group, which changes naproxen’s polarity [10].
1.2. Immunosuppressive Drugs
Immunosuppressive drugs (immunosuppressants) are known as drugs which reduce the activity of the immune system. They lead to the suppression or weakening of the immune system by reducing the number of immune cells produced and maturing. Immunosuppressants are mainly used to prevent the rejection of organ transplants, such as the heart, liver, kidney, lungs, marrow and cornea of the eye. They are also used in patients who have problems with their autoimmune system. They help in the treatment of diseases such as multiple sclerosis and rheumatoid arthritis. One of the immunosuppressants is cyclosporine A. Initially, cyclosporine was used as an antifungal drug, but later in the course of research it turned out to be a drug with strong immunosuppressive properties. Cyclosporine is a polypeptide consisting of 11 amino acids (Figure 3), and this compound is produced by the Tolypocladium inflatum fungus [13,14,15,16,17].
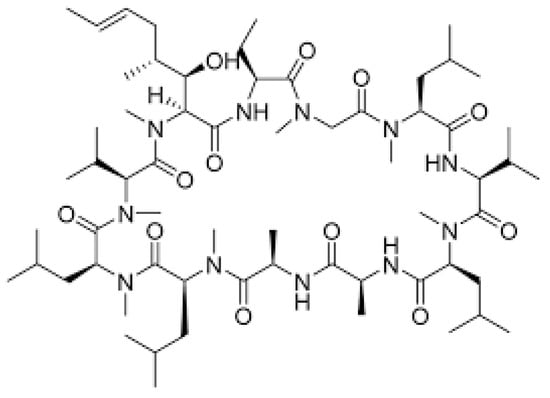
Figure 3.
Cyclosporine A (CsA) structure.
Taking cyclosporine is also associated with serious side effects, such as an increased risk of cancer or infections, increased levels of creatinine and urea, structural changes in the kidneys, hypertension, hyperkalemia, hypomagnesaemia, lymphadenopathy and many others. Therapy with cyclosporine, as a drug with strong effects, desirable and undesirable, should be closely monitored by a physician [13,14,15,16,17].
1.3. Stability of Drugs
Usually, drugs have a rather complicated structure, which is related to their reactivity, causing the degradation and consumption of the biologically active substance. The decomposition processes start at the time of synthesis, or with the formulation of a given pharmaceutical, and may lead to the creation of a drug with a lower activity than intended (i.e., with low efficacy), as well as with an increased activity of the active substance, causing toxic effects on the patient’s body. Then, the drug removal process is essential, and the drug wettability/stability is directly related to this. For example, the process of removing cyclosporine from the body, the drug used in our studies, is a first-order kinetic process. Cyclosporine is a drug whose metabolism takes place in the liver. Only a small amount of the drug, about 1% unchanged, is eliminated along with bile, which is transported to the gastrointestinal tract. It can also be eliminated from the body via the kidneys [13,14,15].
The absorption of CsA takes approximately 30 min and varies with various factors. The preparation should be administered before a meal, because then its concentration in the serum is higher than in the case of administration after a meal. Then, 1–2 h after taking the drug in the form of a microemulsion, its maximum concentration in the serum occurs, while in the case of the administration of the drug in the form of capsules, this concentration is achieved only after 1–6 h. In children, the rate of excretion is almost four times faster than in people over 40 years of age. It follows that with the same dose of the drug taken, its concentration is lower. About 50–70% of CsA is found in the blood in red blood cells. Over 80% of it is present in the plasma in a form bound to proteins, mainly to lipoproteins [13,14,15,16,17].
There are many ways to break down drugs, but most instabilities are due to the oxidation and hydrolysis processes. In the broadest sense, hydrolysis means the formation of new chemical compounds by breaking the chemical bond of another compound through a reaction with water. Other forms of drug decomposition, much less common, are hydration, polymerization and dimerization. Medicines contain a large number of functional groups exposed to hydrolysis during storage, most often ester and amide groups. Hydrolysis is a fairly slow reaction, but in the presence of an acid or a base, the reaction rate increases, and the drug can decompose significantly faster [15,16,17]. The wettability and stability studies conducted in this paper may be helpful when considering the susceptibility of drugs to hydrolysis.
1.4. The Routes of Drug Delivery
Different forms of the drug (tablets, capsules, powders, aerosols, etc.) contribute to the release control the active substance depending on the route of delivery of the pharmaceutical to the body (Table 1), and are closely related to the particle size of the preparation as well as the general physicochemical characteristics. Controlled release drugs allow the active substance to be delivered to the desired section of the gastrointestinal tract. The advantage of this method is the low burden on the stomach. In the case of modified release drugs, the active ingredient produces the desired therapeutic effect when the appropriate pH or temperature is maintained. Drugs that are salts of weak bases and strong acids maintain a low pH as a result of hydrolysis, and the hydrogen ion formed during this process can lead to the hydrolysis of the drug itself (acid catalyzed hydrolysis). Similarly, base-catalyzed hydrolysis applies to drugs that are the salts of strong bases and weak acids; here, a hydroxyl ion is formed, which can also hydrolyze the drug, becoming a catalyst [1,2,3,4,15,16,17].

Table 1.
The routes of drug delivery.
Drugs with modified actions are aimed at delivering the active substance at a given concentration to the appropriate place in the body, and they can be divided into: XL (extra-long), with a very slow release; SR (slow release), with a slow release; CR (controlled release), with a controlled release; MR (modified release), with a modified release [1,2,3,4,15,16,17].
1.4.1. Passive Diffusion
Passive diffusion is usually the most important mechanism for the transport of small drugs or particles, such as ions and metabolites, and cargo smaller than ~40 kDa and/or with a diameter of up to 10 nm. Thus, it is impossible to allow macromolecules, e.g., proteins (polyelectrolytes), to travel to their destination. Larger molecules require active transport-mediated and signal-dependent mechanisms to pass through. An important case is the drugs strongly bound to proteins in the bloodstream, which results in a lack of access to cells and no flow through them. Access to the body takes place only when the drug molecule is in a solution and is broken down on the biological membrane. The pharmaceutical molecule then diffuses through the cell, where it breaks down outside the cell and ends up in the water space inside the cell. Drugs that are very lipid soluble (insoluble in water, e.g., antifungal drugs) penetrate the biological membrane without leaving it, and are trapped in its hydrophobic part. However, drugs that are highly soluble in water, and which do not dissolve in the lipid layer, remain in the intracellular aqueous solution. The absorption efficiency of a drug depends on an appropriate, intermediate degree of solubility; therefore, a chemical compound cannot be highly hydrophilic or highly hydrophobic, so that it can penetrate cell membranes. The organs in which the activation of the drug occurs most often are the intestine, therefore pharmaceuticals that are strongly acidic (Kpa < 2) or strongly alkaline (Kpa > 10) will not pass through the membrane, as they will be almost completely ionized at the values of the pH in these organs [1,2,3,4,15,16,17]. In this aspect, studies into the wettability and the hydrophilic–hydrophobic nature of the active substances tested in drugs, or in modified drug forms, may be helpful.
1.4.2. Active Transport
In situations where a chemical substance is very important for the proper functioning of the body, special mechanisms are created that allow an essential molecule or ion to cross cell membranes. The basic particles that must move inside the cell are glucose, sodium and chloride ions. However, they are too polar to diffuse passively (1.4.1) across cell membranes, and therefore active transport occurs, thanks to globular proteins. They act as carriers, because they contain hydrophilic ion channels responsible for the transport of the required substances. This transport takes place against the concentration gradient, and requires metabolic energy in the form of ATP (adenosine triphosphate) and its hydrolysis to ADP (adenosine diphosphate). A good example of active transport is the transport of amino acids that are too hydrophilic to use passive diffusion. The carrier is specific for a given molecule, and recognizes the desired compound that it must form with to create a complex on the surface of the cell. The resulting protein complex diffuses through the phospholipid membrane and, using energy, arrives at the intracellular space, where it dissociates, thus releasing the active substance. The protein carrier returns freely to the outside of the cell membrane to pick up another molecule for transport. Drugs that are similar in structure to an essential natural compound use an active transport mechanism. What makes active transport an important and useful is its chiral nature, meaning, therefore, that it can easily distinguish enantiomers [1,2,3,4,15,16,17].
Particle size is a very critical attribute of passive diffusion and active transport in drug delivery systems, because it affects stability, encapsulation efficiency, the drug release profile, the effectiveness of the drug, bio-distribution, mucoadhesion and cellular uptake. On the other hand, the size is strictly dependent on the route of administration. One dimension would be ideal for skin applications, another one for inhalation, and another one for drugs administered orally and directly into the bloodstream. Nanocarrier formulations with a constant and narrow size distribution are necessary to achieve optimum clinical outcomes. Moreover, particle size and size distribution (low polydispersity, PDI) are important factors for evaluating the stability of a colloidal dosage form upon storage. In drug delivery applications using lipid-based carriers, such as liposome and nanoliposome formulations, a PDI of 0.3 and below is considered to be acceptable, and indicates a homogenous population of phospholipid vesicles [1,2,3,4,5,6,18].
For example, nanocarriers in the size range of 100–150 nm display eight to nine times more internalization into lung tumor cells compared to microparticles with a size range of 3–5 μm. Consequently, the precise tailoring of aerosol particle size is required to achieve deep lung deposition and the best internalization into the tumor cells. For comparison, regarding skin applications, other parameters are optimal. Vesicles with a diameter of 600 nm or above are not able to deliver the encapsulated material into deeper layers of the skin. These vesicles are inclined to stay in or on the stratum corneum, and may form a lipid layer on the skin after drying. Nanovesicles with a diameter of 300 nm or below are able to deliver their contents to some extent into the deeper skin layers. However, nanovesicles with a diameter of 70 nm or below have shown a maximum deposition of contents in both viable dermal and epidermal layers. Nanoparticles below 6–7 nm in size can be absorbed through the lipidic transepidermal routes, while those with a particle size of below 36 nm can be absorbed through the aqueous pores. Taking into account all these conditions, approximate particle size ranges for drug deposition in various body organs via different dosage forms and routes of administration have been collected on the basis of the literature data, and they are as follows: lymphatic (10–50 nm); long-circulating carriers, e.g., brain, tumors (50–200 nm); transdermal (10–600 nm); intravenous/intramuscular (200–2000 nm); ocular (100–3000 nm); aerosol (1–10 μm); nasal (8–20 µm) [1,2,3,4,5,6,18].
1.5. Wettability of Drugs
The aim of this study was to investigate the effect of ethyl alcohol concentration and drug type on the electrokinetic properties of active substance systems in a silica matrix. In this study, preparations from the NSAID group, belonging to phenylpropionic acid derivatives, i.e., ibuprofen and naproxen, were used. Most NSAID drugs require the intake of higher amounts than the therapeutic dose due to their generally low solubility/wettability in the physiological media. The next drug tested was cyclosporine. It is an essential immunosuppressive substance in the treatment of transplant patients, but it can cause much more serious effects than ibuprofen or naproxen, so it should be taken under close medical supervision. As a nanocarrier, silica gel was chosen, because it has a several advantages, such as a unique ordered structure of a high specific area and pore volume, and consequently high sorption capacity. Moreover, the production of these materials is relatively simple, inexpensive and easily controllable. Finally, they are nontoxic and biocompatible, which are the key requirements for pharmaceutical applications. A detailed description of this carrier, supported by additional research, has been presented in our previous manuscripts [5]. The decrease/increase in the degree of drug release is strictly dependent on the ionic interactions between the amine/carboxylic groups of drugs, and the total amount of drug loaded into silica material, and this in turn is closely related to the wettability and the polar/apolar nature of the drug. For example, it is well known that the better adsorption of anionic drugs is possible on a modified silica material (MCM-41) with a cationic character [6].
Measurements for all obtained systems were performed applaying dynamic light scattering techniques and microelectrophoresis. Additionally, wettability tests and assessments of the hydrophilic–hydrophobic character of the active substances were carried out using the thin-layer wicking method. This method gives the possibility to compare, in an easy manner, the drug solubility/behavior in different solutions, e.g., in conventional solvents (water, ethanol, glycerol), buffer, physiological fluid or simulated body fluid, and to check the effectiveness of charging/releasing drugs in a silica material through, e.g., electrostatic interactions between the cationic functional groups of the polymer and the anionic ones in drug groups, and vice versa. We hope that the obtained data on the effective particle diameter, electrophoretic mobility (zeta potential), polydispersity and wicking time (polar and apolar test liquid) values make it possible to determine the factors that have the greatest impact on the stability of the systems and the wettability process. The physico-chemical characteristic enables a more complete understanding of the correlation and dependence between the determined parameters and the composition of the tested pharmacologically active substance systems with respect to transport and efficiency.
For porous materials, apart from the particle size and surface size, the type and size of the pores are also important. Usually, the delivery rate of a drug increases with the growing pore size of the silica material. A very useful feature is also the regularity of the pore shape and the uniformity of its dimension. If the pore diameters are too small during adsorption from the gas and liquid phase, the sieve effect may be expected. The adsorption of drugs in porous material can be, for example, difficult, not because of the chemical character, but for geometrical reasons. When the geometrical restrictions are comparable with the dimensions of silica, their presence may be the reason for the irreversibility of adsorption. Additionally, the shape of pores mainly influences the accessibility of interior pores by drug molecules, as well as the diffusion into and out of pores. Pores with constricts, or those that are partially closed, may be only partially filled with drug molecules [19,20,21,22,23,24,25,26,27].
The surface of the silica gel contains the silanol groups with an acidic character. On the other hand, the presence of hydroxyl groups enhances the interactions with polar compounds, such as water, alcohols and other species containing heteroatoms. The wetting process can also be affected by the siloxane bridges of a hydrophobic nature that are formed during the synthesis of this type of silica material. The use of ethanol in our research, to increase the solubility of the drug, may cause competitive adsorption in the pores of the gel, which will be discussed in more detail in the following sections. The polarity of a solvent should be one of the parameters considered when evaluating its capacity to dissolve a drug, through the principle ‘like dissolves like’ [5,6].
This study has thus reported, for the first time, a thorough investigation, at both the nano-scale (DLS method, electrophoresis) and the macro-scale (wettability test), of the interactions occurring between silica-based materials and drugs differing in wettability from various chemical groups. For the first time, a simplified technique of draining moisture from a thin layer was used to compare drugs with a completely different structure, mechanism of action and purpose. To the best of our knowledge, studies using these methods for this set of drugs (in particular the immunosuppressive drug (cyclosporine A), have not yet been published.
2. Materials and Methods
2.1. Materials
The main materials and substances used during research are as follows: ibuprofen (70280, Cayman Chemical Company, Ann Arbor, MI, USA, 206.28 g/mol), naproxen (70290, Cayman Chemical Company, Ann Arbor, MI, USA, 230.3 g/mol), cyclosporine A (Alfa Aesar, Kandel, Germany, 1202.64 g/mol)), 96% ethyl alcohol p.a. (POCH S.A., Gliwice, Poland), n-octane (p.a. ≥ 99.5%, Fluka, Buchs, Switzerland), silica gel 60 (Merck Sp. z o.o., Poznań, Poland) and deionized water from the Milli-Q plus 185 Lab Water System.
2.2. Silica-Based Drug Delivery
The route of nanocarriers to the destination is usually abundant in many barriers due to the low number of nanoparticles delivered to the target. The manipulation of nanocarrier properties is based on the selection of the appropriate size, material composition and surface properties, surface chemistry and target ligand conjugation in order to overcome sequential physiological barriers and precisely deliver the drug. The size of the nanocarriers should be carefully controlled, as larger nanoparticles (>500 nm) may clog capillaries due to their small diameters, while smaller nanocarriers (<10 nm) may be trapped by macrophages [19,20,21,22,23,24,25,26,27].
One such carrier used in drug delivery is silica gel or silica gel-based systems. Studies of monodisperse silica-based carriers in the range of 20–200 nm show the highest efficiency of accumulation and penetration for the value of 50 nm. The upper and lower values are not as efficient anymore. The preferred range focuses on drug delivery to the tumor is the range of 10–200 nm, from which nanoparticles smaller than 100 nm are most often selected [19,20,21,22,23,24,25,26,27]. In this respect, the study of particle size using the dynamic light scattering method, discussed in detail in the next section, and their changes over time are necessary.
2.3. Methods
2.3.1. Dynamic Light Scattering
The mean, effective diameters and multimodal size distributions were determined by the Zeta Plus/Bi-Mass Brookhaven (Holtsville, NY, USA) apparatus using the dynamic lights scattering method, with DLS as a function of time [27]. A monochromatic laser beam falls on the cuvette and, due to the different size and number of particles present in the path of the light beam, it can be scattered. In this method, a close relationship is observed between particle size and light scattering. Any changes are submitted and analyzed by a photo detector. The effective diameter is the hydrodynamic diameter (including the compact part of the electrical double layer) that a sphere would need to have in order to diffuse at the same rate as the particle being measured. The prepared solutions for the dynamic light scattering method are strictly defined [28,29,30]. Although the instrument can measure solutions up to 40%, the measured value in these cases will be incorrect. It is not a DLS measurement if the sample is not pure and diluted, because the Stokes–Einstein equation correctly applies to infinitely diluted solutions. Highly concentrated measurements can be conducted in other applications, such as diffusion wave spectroscopy (DWS). According to the assumptions of this methodology, the best results are obtained for samples with a concentration of up to 10%, or after the prior dilution of the stock solution. In our case, all tested samples fell within this range, and there was no need to dilute them before the actual measurement.
DLS measurements were conducted to investigate the effect of drug loading on the corresponding effective diameter of the system. If the system is polydisperse, the effective diameter weighed by intensity is a value calculated from the averaged intensity of the scattered light by each particle. Measurements are carried out in special polyacrylic cuvettes, into which the analyzed system is introduced, and then the whole is placed in the measuring cell. The measurements of the effective diameter were carried out vs. time (5, 15, 30, 60, 120 min after pouring the test systems into the measuring cuvettes) [28]. Drug release was determined not only by diffusion and swelling/relaxation phenomena, but also by the occurrence of other processes, such as the electrostatic interactions taking place between drug molecules and the silica material, which can be monitored by measuring effective diameter and zeta potential changes.
2.3.2. Microelectrophoresis
The same instrument (Zeta Plus/Bi-Mass Brookhaven (Holtsville, NY, USA)) was used during microelectrophoresis. These two measurements, i.e., particle size and zeta potential, are closely related, and this solution is not available in other devices of this type. The electrophoretic mobility is tested by inserting the electrode into the polyacrylic cuvette with the solution. This experimental part is based on the phenomenon of microelectrophoresis [31,32,33]. The electrophoretic mobility is measured and converted by the system into electrokinetic potential directly through the computer software. The zeta potentials of the suspensions at a natural pH were calculated from the Smoluchowski equation. The obtained results are counted and presented in specially adapted computer software. The measurement of electrophoretic mobility is taken directly after measuring the effective diameter, because this parameter is necessary when entering data in the electrophoretic technique procedure. A detailed mathematical description of this method is presented in our previous papers [28].
2.3.3. Thin-Layer Wicking Method
Wetting is the process of fluid interaction with a variety of solid surfaces. A drop of liquid on a solid can spread over its surface to a varying degree, depending on the intermolecular forces that occur between the studied phases. The shape of such a drop depends on the wetting ability of a given surface. Both the surface tension and the surface free energy for liquids remain in equilibrium. However, in the case of solids, these quantities do not have the same value. This is due to the low mobility of atoms, molecules or ions in the crystal lattice. When large surfaces, such as porous solids, are wetted, it is not practically possible to achieve equilibrium. In the case of the powdered solids used in the pharmaceutical industry, the degree of wetting is described not by contact angle measurements, but rather by the kinetics of the wetting process; therefore, often the thin-layer wicking method is used. This method relies on the penetration of a probe liquid into a thin, porous layer of the solid powder deposited, for example, on a metal, plastic or glass support. Such a process may be described by Washburn’s equation, based on Poiseuille’s law [34,35,36,37]:
where x2 is the square of penetration distance; t is the penetration time, η is the liquid viscosity, R is the effective radius of the intergranular capillaries that are formed in a porous layer or column of a powdered solid body, ∆G is the change of free energy (enthalpy) accompanying the replacement of the unit interface surface, i.e., the solid–gas and solid–liquid phases, during liquid movement (wetting) in a porous film.
x2 = ΔG R t/2 η
To describe the wetting process, it is necessary to determine the surface tension of the tested solid, 𝛾SV, and the solid–liquid interfacial tension, 𝛾SL. In practice, it is not possible to determine these parameters directly [34,35,36,37]. For solids in particulate form, the difference between 𝛾SV and 𝛾SL is usually determined by measuring the rate of liquid penetration into the porous layer of the moisturized body. Washburn was the first to investigate the relationship between penetration time and the wettability of a solid in the horizontally positioned capillary. Moreover, he determined that this equation is also true for the layer of powdered solid body [31,32,33,34].
There are three main types of wetting processes: flow wetting, adhesive wetting and immersion wetting. By definition, “drip wetting” is a process involving the spreading of a liquid over the surface of a solid, with the simultaneous displacement of gas molecules from it [25]. The driving force behind this process is the wetting factor (work of spreading, WS). It is defined as the difference between the work of adhesion of the tested liquid to the surface of a solid and its work of cohesion. When the wetting ratio reaches a value greater than or equal to zero, the liquid completely spreads over the surface of the solid. When WS becomes negative, the liquid does not spread over the surface of the solid, but forms droplets on the tested surface. In the case of the bare surface, providing that n-octane also completely wets the solid surface, but no film is present ahead of the front, the below equations are used [34,35,36,37]:
where expression WA means the work of adhesion of the liquid to the solid surface, WC is the work of cohesion of the liquid and expression WA − WC is the work of spreading. Additionally, is the Lifshitz van der Waals component for solid (s) or liquid (l), is the surface tension of the liquid and ∆G is the change of free energy (enthalpy) accompanying the replacement of the unit interface surface, i.e., the solid–gas and solid–liquid phases, during liquid movement (wetting) in a porous film.
The powder film can be represented as a collection of circular capillaries of a certain mean radius. It is the radius of the equivalent capillary that represents the pores in the powdered layer of a solid, called the effective radius of intergranular capillaries, in a porous layer (R). According to this assumption, all dependencies derived for the singular capillary are also right for the powder film [34,35].
The measurements of the wettability of silica gel-coated plastic plates were conducted two ways: (i) without contact with liquid vapours and (ii) in contact with liquid vapours. In measurement (i), plastic plates coated with SiO2 and a centimeter scale at one edge was placed in the thin-layer chromatography chamber. The liquid to be tested (n-octane, water and the drug solution) was poured in by means of the infusion of the mobile phase located on the side of the smaller glass plate. Then, a plastic plate coated with SiO2 was slid over the protrusion of the chamber, which started the process of soaking the test liquid. At the same time, a timer was started, and the chamber was covered with a glass plate, closing the access to air and guaranteeing the course of the process under constant conditions were present inside the chamber. At the moment when the front of the tested liquid reached another, distances (from 1 cm to 8 cm) were read, and the time was recorded.
Measurements (ii), in contact with liquid vapours, were conducted as follows: a few drops of the given liquid were poured into the chromatography chamber, and plastic plates covered with silica gel were placed in it without any contact with the liquid. The chamber was then tightly closed with a cover pane, and the prepared plates were left in the vapours of the liquid for a week. After one week, measurements of the wetting rate were performed using the same liquid that had previously been introduced into the chamber, and in the same manner as measurements (i), without contact with liquid vapours. The wetting times of the given liquid for successive distances (from 1 cm to 8 cm) were read and recorded. For each tested liquid, the measurements were repeated 3–4 times.
Understandably, the measurements for the contacted and non-contacted plates give different results, to a greater or lesser extent speaking to the interactions between the carrier surface and the tested liquid. The selection of liquids of different characters (strongly polar, less polar or apolar) gives an overview of the various types of interactions, and enables a full description of the phenomenon of the wetting process affecting the drugs of different natures and structures (including those with functional groups).
3. Results
3.1. Physico-Chemical Characteristic of Nanocarriers
Generally, drug delivery systems are built of two main components: a nanocarrier and a biological active substance. The nanocarrier matrix plays a crucial role in the proper functioning of such systems, so should be biocompatible, non-toxic and thermally/chemically stable. As nanocarrier the silica gel was chosen because it has several advantages, such as a uniquely ordered structure with a high specific area and pore volume and, consequently, a high sorption capacity. A detailed description of this carrier, supported by additional research, has been presented in our previous manuscripts [5,7].
Biological membranes are negatively charged; therefore, positively charged nanocarriers often show higher in vitro delivery efficiency compared to negatively charged or neutral nanocarriers. Membrane potential (also transmembrane potential or membrane voltage) is the difference in electric potential between the interior and the exterior of a biological cell. For the exterior of the cell, typical values of membrane potential normally range from −80 mV to −40 mV. All animal cells are surrounded by a membrane composed of a lipid bilayer with proteins embedded in it. Transmembrane proteins (ion pumps) and ion channels are electrically equivalent to a set of batteries and resistors inserted in the membrane, and therefore create a voltage between the two sides of the membrane. The negative charge within the cell is created by the cell membrane being more permeable to potassium ion movement than sodium ion movement. As more cations are expelled from the cell than taken in, the inside of the cell remains negatively charged relative to the extracellular fluid. However, when the drug should remain in systemic circulation for a long time, carriers with a negative or neutral charge are more often chosen. Nanocarriers are made of positively charged cores and negatively charged shells, so they can change their charge depending on the environment [1,2,3,4,5,6]. When the microenvironment shows a low pH, negatively charged coatings are discharged, which causes a change in the charge of the carrier.
The surface of the nanocarriers is the interface that allows the contact of the carrier with cells and their interactions. Silica materials are frequently used as carriers of active substances because they show high biocompatibility and chemical stability, and are also non-toxic. These carriers most often have the form of xerogels, mesoporous materials and spherically mesoporous silica materials with absorption properties [18,19,20,21,22,23]. The hydrophobicity and hydrophilicity of the surface have a significant impact here, and the presence or absence of target ligands on the surface of nanocarriers may affect their adhesion and penetration into cells [18,19,20,21,22,23,24,25,26]. A detailed description of the process of wetting the solid surface with drugs can be helpful in this regard.
3.2. Wettability Process
As a carrier, solid powdered silica gel 60, which is widely used, was applied to plastic plates. Silica hydrogels are most often in the form of ointments, and are frequently applied to release the active substance on the mucous membranes of the nose, mouth and eye. Such materials usually have an amorphous and porous structure, thanks to which the active substance can be released relatively quickly from surface as a result of the diffusion, erosion or degradation of the matrix. In addition, a better diffusion of the active substance can be ensured by adding a significant amount of water during the production; as a consequence, the obtained material has a larger specific surface area and larger pores. The extended duration of action for this carrier results from the longer path that the active substance has to travel upon its introduction and release from the silicate matrix. The big advantages of spherical materials are their biodegradability and the possibility of modification with various functional groups. Moreover, the active substance can be added directly during the gel formation [5,19,20,21,22,23,24,25,26,27].
Under constant pressure and temperature conditions with a series of plates for which the R parameter was experimentally determined, the plate wetting time t as a function of the square of the distance should be a straight line with the slope defined by ∆G. Additionally, it is assumed that ∆G does not change along the plate in the wetting process. The effective radius of the intergranular capillaries of the tested SiO2 substrate on plastic plates was determined by averaging the radius obtained for three successive alkanes (n-octane, nonane and decane). It is assumed that the mentioned apolar liquids, practically, do not interact with the tested porous surface, comparably to a set of intergranular circular capillaries, and thus their radius can be indirectly determined. The mean determined effective radius of the intergranular capillaries R for this silica layer applied on plastic plates is 5.63 ± 0.36 × 10–5 cm. This value was used for all calculations. Detailed considerations regarding this methodology have been presented in previous works [5,28]. The naproxen solution penetrates the plate surface with 81 cm2 of silica gel faster than ibuprofen, in both the cases of contacted and non-contacted plates with drug vapours, which may be due to interactions between the charged silica surface and groups with the opposite sign in the naproxen molecule (Figure 4a,b). The time discrepancy exceeds 1015 s and is almost four times faster than the penetration rate of a solution with a higher alcohol concentration (Figure 5a,b). The rate of penetration of the non-contacted plate by the naproxen solution differs slightly for both solvent (ethanol) concentrations. Penetration with the ibuprofen solution is definitely more favorable for a lower alcohol concentration, which may be related to the competitive adsorption process.
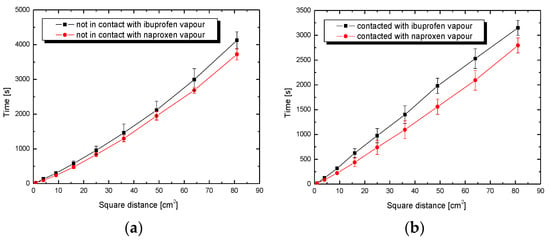
Figure 4.
Penetration time of plates coated with SiO2 by solutions of drugs in ethanol (5 mL) as a function of the square of the distance x2 for (a) non-contacted and (b) contacted plates with drug vapours.
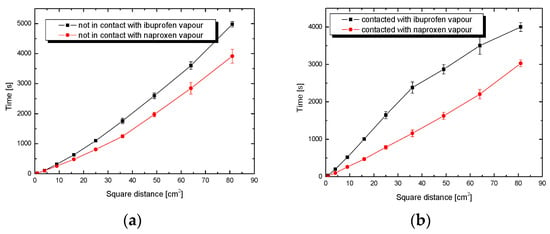
Figure 5.
Penetration time of plates coated with SiO2 by solutions of drugs in ethanol (10 mL) as a function of the square of the distance x2 for (a) non-contacted and (b) contacted plates with drug vapours.
Regarding dependence in Figure 5a,b, evidently, the naproxen solution wets the silica surface faster than ibuprofen, despite the comparable molecular weight of both drugs. The discrepancy in the wetting rate between the drugs is considerable, and amounts to about a minimum of 1050 s for the distance, which equals 9 cm of plate (square distance 81 cm2). This is probably due to the polar groups contained in the naproxen molecule interacting with the polar silanol groups of the silica. The presence of ethanol and dipoles in the tested system does not exclude the formation of hydrogen bonds, which can be responsible for the movement of liquid on the surface of the SiO2 plate. In the case of neutral ibuprofen, the interaction with the polar carrier is slowed down.
Competition, being the characteristic phenomenon in adsorption from solutions, plays an important role during the impregnation of a porous solid with a drug solution. In the case of multi-component systems (several components in a solution) in the adsorption process, one of the components is adsorbed faster (takes up free adsorption sites), and then the remaining ones adsorb slower or not at all. Thus, it is very important to match appropriately the solvent (in our case ethanol with the dipole structure) to the drug character. The solvent should be minimally active for the silica surface, biocompatible and easily removable at lower temperatures. Ethanol seems to be a suitable solvent for this type of application, but its effect is noticeable, especially at higher concentrations.
In the case of wetting the contact plates with the ibuprofen solution, the increase in alcohol concentration causes a much slower liquid adsorption process. Comparing this process to the infiltration of the naproxen solution, the time difference, depending on the ethanol concentration, is visible. Comparing all the curves showing the penetration rate of ibuprofen and naproxen solutions, regardless of the parameters of a given process, it was concluded that the naproxen solution penetrates the silica gel plates faster. Changing the ethanol concentration of the ibuprofen solution has a direct impact on the wetting process. The higher the ethanol concentration, the more hydroxyl groups in the solution, and the stronger the adsorption on the gel surface.
It can also mean that naproxen is located mainly on the external surface of particles, or around the openings of silica pores. It is also possible that, during the wetting process into the pore interior, it penetrates mainly alcohol, and a large part of naproxen remains out of the pores; hence, these shorter penetration times result (Figure 5). In turn, large changes in the course of the wetting process with a higher concentration of alcohol may arise from strong competition between the drug and ethanol for the internal surface. This is especially observable for neutral ibuprofen (Figure 5b). When designing drug delivery systems, it is important to remember that, in such cases, the drug release is determined mainly by the dissolution process.
Additionally, the wetting process is influenced by the surface properties of all three phases in contact with each other and, therefore, it can take place according to various mechanisms. There is a high probability of adsorption on the wetted surface of the tested solution, as a consequence of which its character changes, e.g., from hydrophobic to hydrophilic, or vice versa. In order to determine the amount of adsorbed solution or test liquid at the interface, depending on the contact angle, changes in the adhesive tension (delta G, Equations (1) and (2)) are determined as a function of the surface tension of the solution.
3.3. Adhesion Tension
The changes in the adhesive tension determined as a function of the surface tension of the solution give more information about wettability process, especially in complicated multicomponent solutions. The obtained relationships of delta G vs. time are presented in Figure 6 and Figure 7. The effect of the influence of ethanol concentration on the behavior of the tested drug formulations is immediately visible. Comparing the changes in the adhesive tension for two ibuprofen systems (Figure 6a), clear fluctuations can be noticed, especially in the case of the curve obtained for a drug solution with a lower ethanol concentration. The curve shows a sigmoid character with a noticeable minimum (at a distance of 2 cm) and maximum (at a distance of 5 cm). The adhesive tension values for these extremes are, respectively, 24.63 mN/m and 40.94 mN/m. In the case of the curve obtained for a solution of ibuprofen in a higher concentration of ethanol, its course is milder, without clearly marked extremes. This may be evidence of much greater homogeneity of the adhesion process as an effect of the greater number of hydroxyl groups derived from the ethanol.
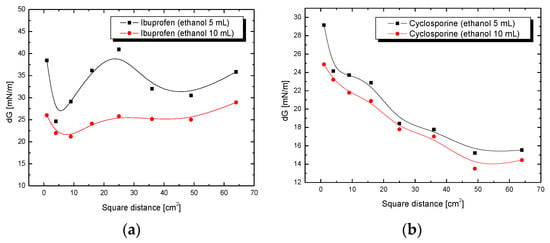
Figure 6.
Adhesion tension of (a) ibuprofen or (b) cyclosporine drug solution to plates coated with SiO2 as a function of the square of the distance x2.
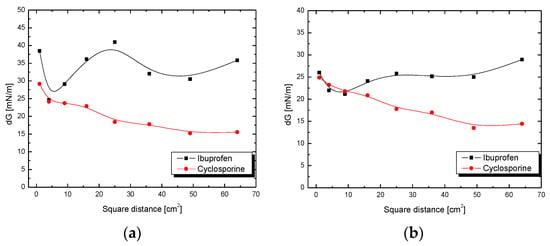
Figure 7.
Adhesion tension of ibuprofen or cyclosporine solution to plates coated with SiO2 (a) in ethanol (5 mL) or (b) in ethanol (10 mL) as a function of the square of the distance x2.
The higher wetting rate of non-contacted plates than that of the contacted plates with vapours may be a direct result of the large particle size of drug, especially cyclosporine (not presented). During contact with the plates and adsorption on their surface, these particles slow down the penetration of the drug solution through the porous structure of the gel. The curves showing changes in the adhesive tension for ibuprofen and cyclosporine solutions show clear differences in their course (Figure 6 and Figure 7).
The more variable adhesion process is demonstrated by ibuprofen in ethanol solutions, while the adhesion process for the cyclosporine solution is more homogeneous. The reason for this is probably the differences in the structure of the studied drugs and the difference in particle size, as well as the type of interaction taking place in the drug–silica gel system. A more homogeneous process means a better fit to a linear function according to the Washburn equation. Any deviation from this trend, regardless of the direction in which the process is accelerated or delayed, indirectly indicates non-homogeneity or additional impacts. Figure 6a,b shows completely different scales; when one takes a closer look at them, one can see that for cyclosporine, the fluctuations are much smaller.
After some time, for a distance of about 4–5 cm, the values for cyclosporine fluctuate around a constant value (average 14 mV), and for ibuprofen, they still fluctuate. The analogous curves for cyclosporine (Figure 6b) do not show large fluctuations, and the extremes of these functions are not observed. Changes in the adhesive tension along the entire test section for a solution of cyclosporine in 5 mL of ethanol are in the range from 29.16 mN/m to 15.21 mN/m, while for a cyclosporine in 10 mL of ethanol, it is from 24.87 mN/m to 13.50 mN/m. Slight differences in the changes to the adhesive tension for drug solutions with different ethanol concentrations may result from the fact that additional hydroxyl groups present in the ethanol molecule or ethanol dipoles present in the cyclosporine solution do not have such a significant importance for the adhesion process, as was observed in the case of the ibuprofen solution. It could have a connection with the structure of cyclosporine, which, in its structure, has more polar functional groups interacting with the surface of the silica gel compared to neutral ibuprofen.
When comparing the solutions of drugs, for ibuprofen and cyclosporine in 10 mL of ethanol, on the surface of plates not in contact with vapours (Figure 7b), slight fluctuations and non-homogeneous course of the curves for both solutions over the tested length can be observed. This may indicate changes in the adhesion process that took place throughout the duration of the test. In addition, it is worth noting that the adhesive tension values for the first three centimeters in the case of the ibuprofen solution and cyclosporine are at a similar level; for ibuprofen they are, successively, 25.99 mN/m (square distance 1 cm2), 21.97 mN/m (4 cm2) and 21.17 mN/m (9 cm2), and for cyclosporine, 24.87 mN/m (1 cm2), 23.21 mN/m (4 cm2) and 21.78 mN/m (9 cm2).
The amount of drug deposited in pores depends on the equilibrium of the adsorption process. In the case of highly porous solids containing pores of small dimensions, the relation of pore dimension to dimensions of adsorbed molecules is very important. If the dimensions of drug molecules are comparable with the pore diameter of a silica material, then the competition between drug and ethanol molecules for silica surface plays the main role in drug loading process. While discussing the results obtained by us for three different drugs, this substantial relationship can be found. The longer release of the drug from the silica system is more preferable because it takes place without burst effect [8,9,19,20,21,22,23,24,25,26,27].
By comparing the results obtained for two tested drug solutions from two different groups, ibuprofen and cyclosporine, at the same concentration of ethanol (5 mL) on plates coated with silica gel not in contact with drug vapours (Figure 7a), much larger fluctuations and a more heterogeneous course of the curve for ibuprofen than that for cyclosporine can be observed. These sharp differences in the course of the curves for ibuprofen solutions and cyclosporine suggest that the course of the adhesion process is more variable in ibuprofen in ethanol solution. The reason for this is probably the differences in the structure of the studied drugs and the significant difference in the size of the molecules. As previously mentioned, ibuprofen contains only one polar carboxyl group, while cyclosporine contains a hydroxyl group, cyclically repeating secondary and tertiary amines and carbonyl groups. Although these are less polar groups than the one found in ibuprofen, their predominance and amount may make the adhesion process of this drug on the SiO2 surface more homogeneous.
3.4. The Influence of the Drug Type on the Effective Diameter
As was mentioned, effective diameter measurements were made using the ZetaPlus meter. With this device, not only the effective diameter can be measured, but also the mean diameter, polydispersity, quality and electrophoretic mobility (zeta potential). The aim of this experiment was to determine the effect of the type of drug on the effective diameter and the zeta potential of silica gel solutions and suspensions in the aqueous solutions of active substances. For this study, aqueous solutions of naproxen, ibuprofen and cyclosporine, with the addition of two different concentrations of ethanol, were used, as well as silica gel suspensions in analogous drug solutions. Table 2 presents the obtained data of the effective diameter depending on the type of drug in an aqueous solution with lower concentration of alcohol. Knowledge of these parameters and their stability or variability as a function of time directly translates into drug stability and indirectly informs us about the degree of drug loading in the silica matrix. In the case of a solution with naproxen and ibuprofen, the effective diameter remains similar in both, and it increases at the beginning of the measurements.
Table 2.
Dependence of the effective diameter on the type of drug in the aqueous solution with the addition of ethanol (5 mL).
It is well known that the encapsulation of hydrophobic molecules within the silica material ensured prolonged drug release over time, thus avoiding their common burst release from hydrophilic networks. Furthermore, the different drug molecule arrangement within the gel network, as a consequence of their hydrophilic/hydrophobic nature and loading protocol, resulted in a different drug releasing mechanism and timing [6,7,9]. The obtained values of the effective diameter for an aqueous solution of the drugs with different concentrations of ethanol allow us to state that the increase in the concentration of ethanol contributes to the stabilization of the tested solution; it is possible due to the electrostatic interactions of alcohol dipoles. These results confirmed the earlier conclusion obtained from the wettability tests.
For the naproxen and ibuprofen solution, the lowest diameter value was reached within 5 min, and both solutions achieved the highest values of the tested parameter at the same time, i.e., 2 h. In contrast to the system with the NSAID drugs, the effective diameter for the cyclosporine solution decreases as a function of time. The highest value of the tested parameter was obtained at the beginning of the measurements, and the lowest within 120 min from the moment of pouring the solution into the measuring cuvette. This is probable due to the highest drug mass and its lowest stability.
3.5. The Influence of the Drug Type on the Zeta Potential
The table below (Table 3) shows the zeta potential values depending on the type of drug in an aqueous solution with the addition of ethanol. The largest fluctuations of the examined parameter can be noticed for the ibuprofen solution. In the case of the naproxen and cyclosporine solutions, there are also significant deviations from the mean zeta potential, but they do not change as rapidly as for ibuprofen solution. For the naproxen system, the highest value of the potential was recorded within 15 min, and then the parameter decreased and fluctuated until 2 h. For a solution with cyclosporine, the zeta potential reaches the highest value within 5 min, then decreases to 0.49 mV, and next continues to increase as a function of time.
Table 3.
Dependence of the zeta potential on the type of drug in the aqueous solution with the addition of ethanol (5 mL).
Naproxen, with positively charged functional groups, gives positive potential values. In the case of ibuprofen, which, in its structure, has both positively and negatively charged groups, which are visible in the obtained zeta potential values as a function of time, so both positive and negative values appear. However, it does not translate into the stability of the entire system. On the other hand, values for cyclosporine are proportionally higher, and positive values dominate. These conclusions can also confirm the polydispersity values discussed in detail in the next section.
3.6. The Influence of the Drug Type on pH and Polydispersity
All drug solutions and suspensions with silica gel have a slightly acidic or acidic pH in the range of 3.84–6.52. Most often, an increase in the ethanol concentration causes a slight decrease in pH; in turn, the addition of silica gel to the test solutions increases the pH of the systems. The smallest pH changes were observed for the systems with cyclosporine. On the basis of the obtained polydispersity values, the homogeneity of the tested systems can be arranged in the following order: naproxen systems, ibuprofen systems and cyclosporine systems. It can be concluded that the lower the polydispersity value, the more homogeneous each of the tested systems was. Taking into account the principle that optimal pharmaceutical formulations exhibit a polydispersity of around 0.3 and below, only the naproxen solution meets this criterion (Table 4) [18].
Table 4.
Dependence of the polydispersity on the type of drug in the aqueous solution with the addition of ethanol.
Referring to the polydispersity parameter changes, it can be stated that, for the naproxen and/or cyclosporine solution and their suspensions of silica gel, the increase in the ethanol concentration stabilizes this parameter, which can confirm the greater homogeneity of these systems and, indirectly, also their long-term stability. In the case of the ibuprofen solution, an increase in the ethanol concentration causes the appearance of polydispersity fluctuations and higher values of the tested parameter, which may be related to the next conclusions. The greatest deviations of the zeta potential were also obtained for the ibuprofen solution and the greatest changes in pH.
4. Conclusions
The use of modern drug delivery systems (DDS) allows for the creation of innovative methods of treating diseases, proposing further development of the already known methods, as well as an unconventional approach to a pharmaceutical at every stage of its presence in human body. DDS, using targeted therapy, create greater possibilities of delivering a drug to its destination in a specific way, even in the case of drug resistance. Drug delivery systems can also provide a solution to previously incurable diseases. In this regard, the most important conclusions resulting from the conducted research into the stability and wettability of such systems for three selected drugs with different structures and properties are as follows.
For an aqueous solution of drugs and their silica-based suspension, it can be concluded that the increase in alcohol concentration significantly contributes to the stabilization of the measuring system, because alcohol dipoles stabilize the solution with forces of an electrostatic nature. In the drug solution, destabilization processes can take place; however, this process is usually faster in the case of a solution with a lower ethanol concentration. Similarly, for the suspension of silica gel in a drug solution with an additive of ethanol, the effective diameter changes as a function of time; however, it usually shows lower fluctuations for the system with a higher concentration of ethanol, which may indicate a greater stability of the system. The optimal size is strictly dependent on the route of administration, one value for skin applications, one for inhalation, and another one for drugs administered orally and directly into the bloodstream. However, in all cases, systems with a constant and narrow size distribution (low polydispersity), optimal zeta potentials are necessary to achieve the stability of the dosage form upon storage, and especially to obtain optimum clinical outcomes.
Changing the ethanol concentration also has a direct effect on the wetting process, especially for the naproxen and ibuprofen systems. The higher the ethanol concentration, the more hydroxyl groups in the solution, and the stronger the adsorption on the gel surface. Greater homogeneity of the adhesion mechanism was achieved for the contacted plates with drug vapours versus non-contacted plates. The curves of adhesive tension for the ibuprofen or cyclosporine solutions show marked differences in their course. The more variable adhesion process is demonstrated by ibuprofen in an ethanol solution, while the adhesion process for the cyclosporine solution is more homogeneous. The reason for this is probably the differences in the structure of the studied drugs and the difference in particle size, as well as the type of interaction taking place in the drug–silica gel system.
On the other hand, varying the ethanol concentration in the cyclosporine solution does not cause drastic changes to the wetting rate. With a large number of cyclosporine functional groups, the additional amount of ethanol hydroxyl groups does not play a significant role. The better linear dependence of the wetting time as a function of the square of the distance for cyclosporine compared to ibuprofen (or naproxen) is due to the structural differences of the drug molecules.
Determining the influence of carriers on the physicochemical characteristics of the tested drugs and the environment in which biologically active substances are found is an important aspect of the development of the pharmacological industry. Thanks to this, it is possible to improve various forms of the drug in order to maximize its bioavailability and indirectly regulate the method of drug delivery in the most optimal way for a specific place of the organism. The conducted experiment shows the possibilities of transporting various types of pharmaceuticals with different structures (understood to be the content of specific positively or negatively charged functional groups that determine the nature of interactions) and applications, regulating their stability and/or wettability through various modifications, using available and relatively simple physicochemical methods. In our opinion, the here-developed methodological platform could open the possibility to design silica-based materials able to encapsulate/release different biological active substances, thus eventually increasing therapeutic efficacy through their target delivery into a tissue/organ.
Author Contributions
Conceptualization, A.E.W.; methodology, A.E.W. and K.P.; validation, A.E.W. and K.P.; formal analysis, A.E.W. and K.P.; investigation, A.E.W. and K.P.; writing—original draft preparation, A.E.W. and K.P.; writing—review and editing, A.E.W. and K.P.; visualization, A.E.W. and K.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rainsford, K.D. Ibuprofen: Pharmacology, efficacy and safety. Inflammopharmacology 2009, 17, 275–342. [Google Scholar] [CrossRef] [PubMed]
- Cairns, D. Essentials of Pharmaceutical Chemistry, 4th ed.; Pharmaceutical Press: Aberdeen, UK, 2012. [Google Scholar]
- Sun, W.; Hu, Q.; Ji, W.; Wright, G.; Gu, Z. Leveraging Physiology for Precision Drug Delivery. Physiol. Rev. 2017, 97, 189–225. [Google Scholar] [CrossRef]
- Ghlichloo, I.; Gerriets, V. Nonsteroidal Anti-inflammatory Drugs (NSAIDs); Stat Pearls Publishing: Treasure Islands, FL, USA, 2020. [Google Scholar]
- Wiącek, A.E. Changes in wetting properties of silica surface treated with DPPC in the presence of phospholipase A2 enzyme. Appl. Surf. Sci. 2010, 256, 7672–7677. [Google Scholar] [CrossRef]
- Krasucka, P.; Goworek, J. MCM-41 silica as a host material for controlled drug delivery systems. Ann. Univ. Maria Curie-Skłodowska 2015, 70, 45–66. [Google Scholar]
- Wiącek, A.E. Investigation of DPPC adsorption on SiO2 particles from aqueous solution and in the presence of enzymes by the zeta potential and effective diameter measurements. Powder Technol. 2011, 213, 141–147. [Google Scholar] [CrossRef]
- Yoo, J.; Won, Y.-Y. Phenomenology of the Initial Burst Release of Drugs from PLGA Microparticles. ACS Biomater. Sci. Eng. 2020, 6, 6053–6062. [Google Scholar] [CrossRef]
- Han, G.; Ceilley, R. Chronic Wound Healing: A Review of Current Management and Treatments. Adv. Ther. 2017, 34, 599–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roda, A.; Paiva, A.; Duarte, A.R.C. Therapeutic Liquid Formulations Based on Low Transition Temperature Mixtures for the Incorporation of Anti-Inflammatory Drugs. Pharmaceutics 2021, 13, 1620. [Google Scholar] [CrossRef] [PubMed]
- Allen, L.V., Jr.; Ansel, H.C. Ansel’s Pharmaceutical Dosage Forms and Drug Delivery Systems, 4th ed.; Wolter Kluwer Health: Philadelphia, PA, USA, 2014; pp. 34–66. [Google Scholar]
- Laurano, R.; Boffito, M. Thermosensitive Micellar Hydrogels as Vehicles to Deliver Drugs with Different Wettability. Front. Bioeng. Biotechnol. 2020, 8, 708. [Google Scholar] [CrossRef]
- Colombo, D.; Poggi, S. Clinical profile of cyclosporine in dermatology. Drug Dev. Res. 2011, 72, 634–646. [Google Scholar] [CrossRef]
- Žid, L.; Zeleňák, V.; Almáši, M.; Zeleňáková, A.; Szücsová, J.; Bednarčík, J.; Šuleková, M.; Hudák, A.; Váhovská, L. Mesoporous Silica as a Drug Delivery System for Naproxen: Influence of Surface Functionalization. Molecules 2020, 25, 4722. [Google Scholar] [CrossRef] [PubMed]
- Przykaza, K.; Woźniak, K.; Jurak, M.; Wiącek, A.E.; Mroczka, R. Properties of the Langmuir and Langmuir–Blodgett mono-layers of cholesterol-cyclosporine A on water and polymer support. Adsorption 2019, 25, 923–936. [Google Scholar] [CrossRef] [Green Version]
- Kaaz, K.; Reich, A. Treatment of atopic dermatitis with cyclosporine A: A case report. Dermatol. Rev. 2017, 104, 433. [Google Scholar]
- Wiącek, A.E.; Jurak, M.; Ładniak, A.; Przykaza, K.; Szafran, K. Cyclosporine CsA—The Physicochemical Characterization of Liposomal and Colloidal Systems. Colloids Interfaces 2020, 4, 46. [Google Scholar] [CrossRef]
- Danaei, M.; Dehghankhold, M.; Ataei, S.; Hasanzadeh Davarani, F.; Javanmard, R.; Dokhani, A.; Khorasani, S.; Mozafari, M.R. Impact of Particle Size and Polydispersity Index on the Clinical Applications of Lipidic Nanocarrier Systems. Pharmaceutics 2018, 10, 57. [Google Scholar] [CrossRef] [Green Version]
- Kwon, S.; Singh, R.K.; Perez, R.; Neel, E.A.A.; Kim, H.-W.; Chrzanowski, W. Silica-based mesoporous nanoparticles for controlled drug delivery. J. Tissue Eng. 2013, 4, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Iturrioz-Rodríguez, N.; Correa-Duarte, M.A.; Fanarraga, M.L. Controlled drug delivery systems for cancer based on meso-porous silica nanoparticles. Int. J. Nanomed. 2019, 14, 3389–3401. [Google Scholar] [CrossRef] [Green Version]
- Ma, P.; Xiao, H.; Li, C.; Dai, Y.; Cheng, Z.; Hou, Z.; Lin, J. Inorganic nanocarriers for platinum drug delivery. Mater. Today 2015, 18, 554–564. [Google Scholar] [CrossRef]
- Salazar-Hernández, C.; Salazar-Hernández, M.; Lona-Ramos, R.; Elorza-Rodríguez, E.; Rocha-Ramírez, A.H.; Carmen, S.-H.; Mercedes, S.-H.; Rocío, L.R.; Enrique, E.-R.; Hilario, R.-R.A. Silica from Rice as New Drug Delivery Systems; IntechOpen Book Series: London, UK, 2017; p. 5. [Google Scholar] [CrossRef] [Green Version]
- Selvarajan, V.; Obuobi, S.; Lai, P.; Ee, R. Silica Nanoparticles—A Versatile Tool for the Treatment of Bacterial Infections. Front. Chem. 2020, 5, 1–16. [Google Scholar] [CrossRef]
- Chen, J.-F.; Ding, H.-M.; Wang, J.-X.; Shao, L. Preparation and characterization of porous hollow silica nanoparticles for drug delivery application. Biomaterials 2003, 25, 723–727. [Google Scholar] [CrossRef]
- Liberman, A.; Mendez, N.; Trogler, W.C.; Kummel, A.C. Synthesis and surface functionalization of silica nanoparticles for nanomedicine. Surf. Sci. Rep. 2014, 69, 132–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giret, S.; Chi Man, M.W.; Carcel, C. Mesoporous-Silica-Functionalized Nanoparticles for Drug Delivery. Chemistry 2015, 21, 13850–13865. [Google Scholar] [CrossRef] [PubMed]
- Bharti, C.; Nagaich, U.; Pal, A.K.; Gulati, N. Mesoporous silica nanoparticles in target drug delivery system: A review. Int. J. Pharm. Investig. 2015, 5, 124–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiącek, A.E. Investigations of DPPC effect on Al2O3 particles in the presence of (phospho)lipases by the zeta potential and effective diameter measurements. Appl. Surf. Sci. 2011, 257, 4495–4504. [Google Scholar] [CrossRef]
- Kralchevsky, P.A.; Danov, K.D.; Denkov, N.D. Chemical physics of colloid systems and interfaces. In Handbook of Surface and Colloid Chemistry, 3rd ed.; Birdi, K.S., Ed.; CRC Press: Boca Raton, FL, USA, 2008; Volume 7, pp. 197–377. [Google Scholar]
- Otsuki, A.; de Campo, L.; Garvey, C.; Rehm, C. H2O/D2O Contrast Variation for Ultra-Small-Angle Neutron Scattering to Minimize Multiple Scattering Effects of Colloidal Particle Suspensions. Colloids Interfaces 2018, 2, 37. [Google Scholar] [CrossRef] [Green Version]
- Guide for Dynamic Light Scattering (DLS) Sample Preparation. Available online: https://www.brookhaveninstruments.com/guide-for-dls-sample-preparation/08.11.2019 (accessed on 27 December 2021).
- Otsuki, A.; Hayagan, N.L. Zeta potential of inorganic fine particle-Na-bentonite binder mixture systems. Electrophoresis 2020, 41, 1405–1412. [Google Scholar] [CrossRef]
- Vergouw, J.; Difeo, A.; Xu, Z.; Finch, J. An agglomeration study of sulphide minerals using zeta-potential and settling rate. Part 1: Pyrite and galena. Miner. Eng. 1998, 11, 159–169. [Google Scholar] [CrossRef]
- Suchata, K.; Muenduen, P.; Zhang, N.B.-M. Applicability of Washburn capillary rise for determining contact angles of powders/porous materials. J. Colloid Interface Sci. 2013, 397, 169–176. [Google Scholar]
- Wiącek, A.E. Changes in wetting properties of alumina surface treated with DPPC in the presence of phospholipase A2 enzyme. Colloids Surf. B Biointerfaces 2011, 87, 54–60. [Google Scholar] [CrossRef]
- Chibowski, E. Some problems of characterization of a solid surface via the surface free energy changes. Adsorption Sci. Technol. 2017, 35, 647–659. [Google Scholar] [CrossRef] [Green Version]
- Terpiłowski, K.; Bielska, M.; Prochaska, K.; Chibowski, E. Wettability of ultrafiltration membranes. In Advances in Contact Angle, Wettability and Adhesion; Mittal, K.L., Ed.; Wiley Online Library: Hoboken, NJ, USA, 2019; Volume 4, pp. 57–72. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).