Mechanisms of Surface Charge Modification of Carbonates in Aqueous Electrolyte Solutions

 ,
,  ,
, 

Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Sample Preparation
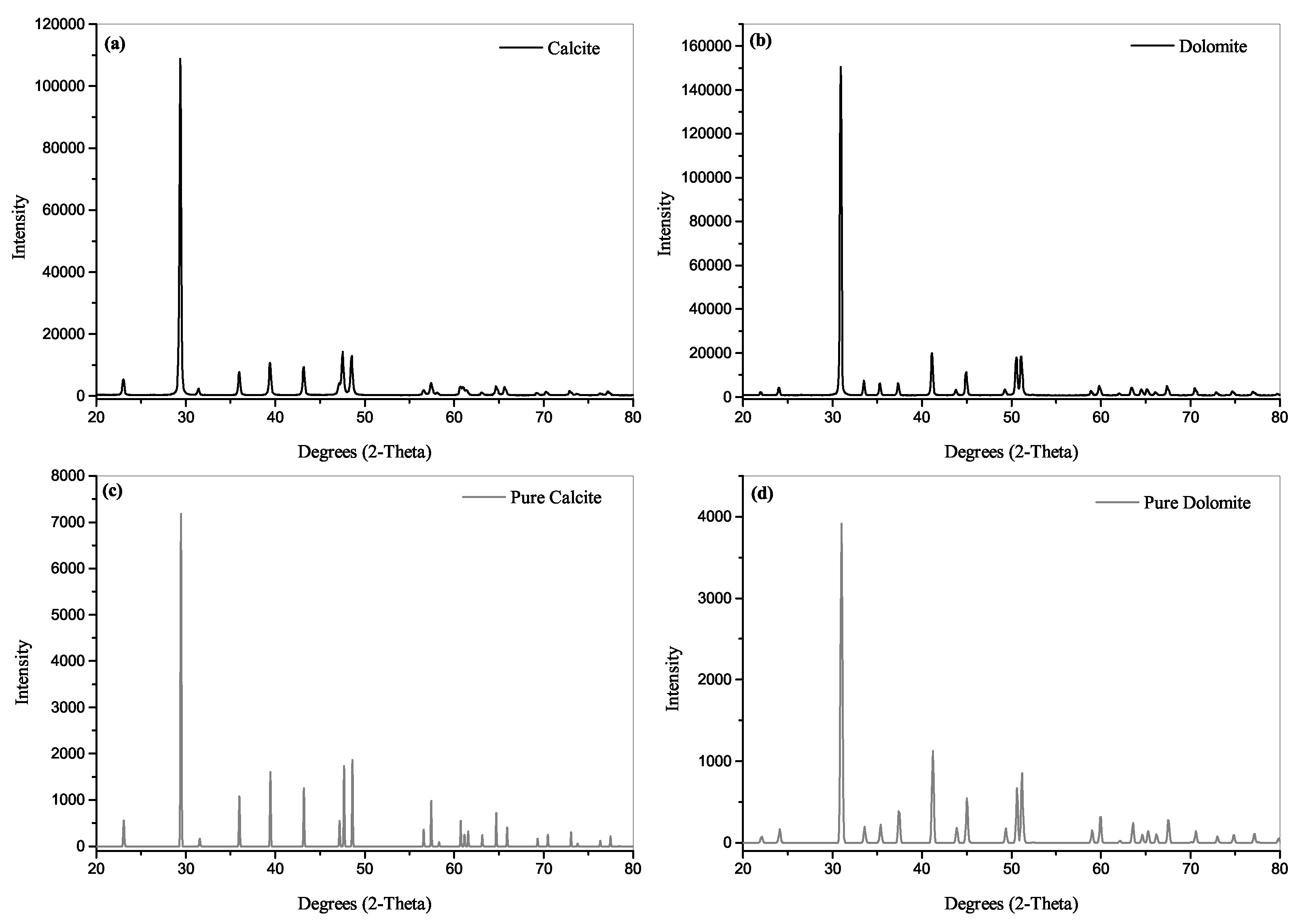
2.3. X-ray Diffraction
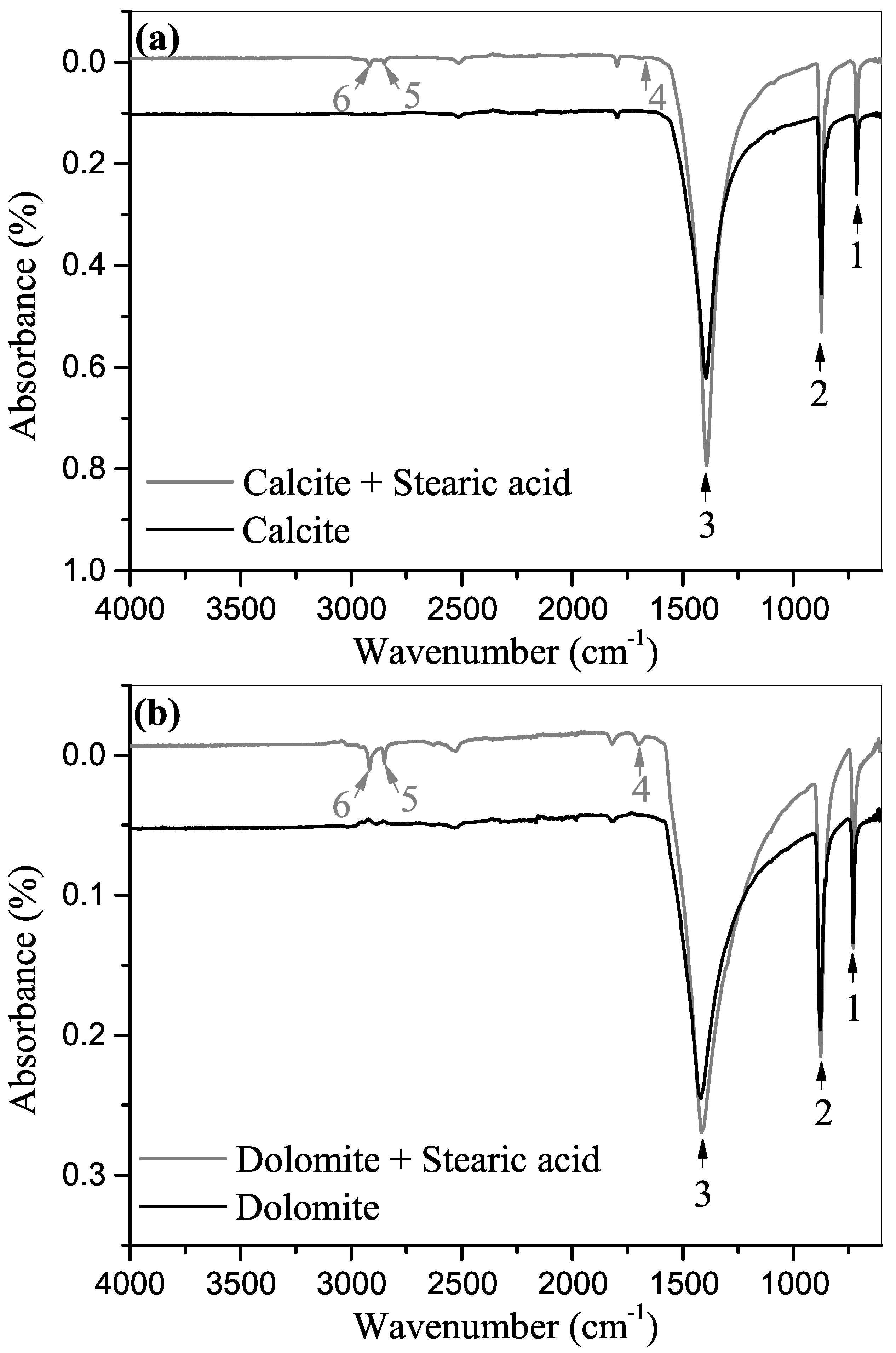
2.4. Fourier Transform Infrared Spectroscopy
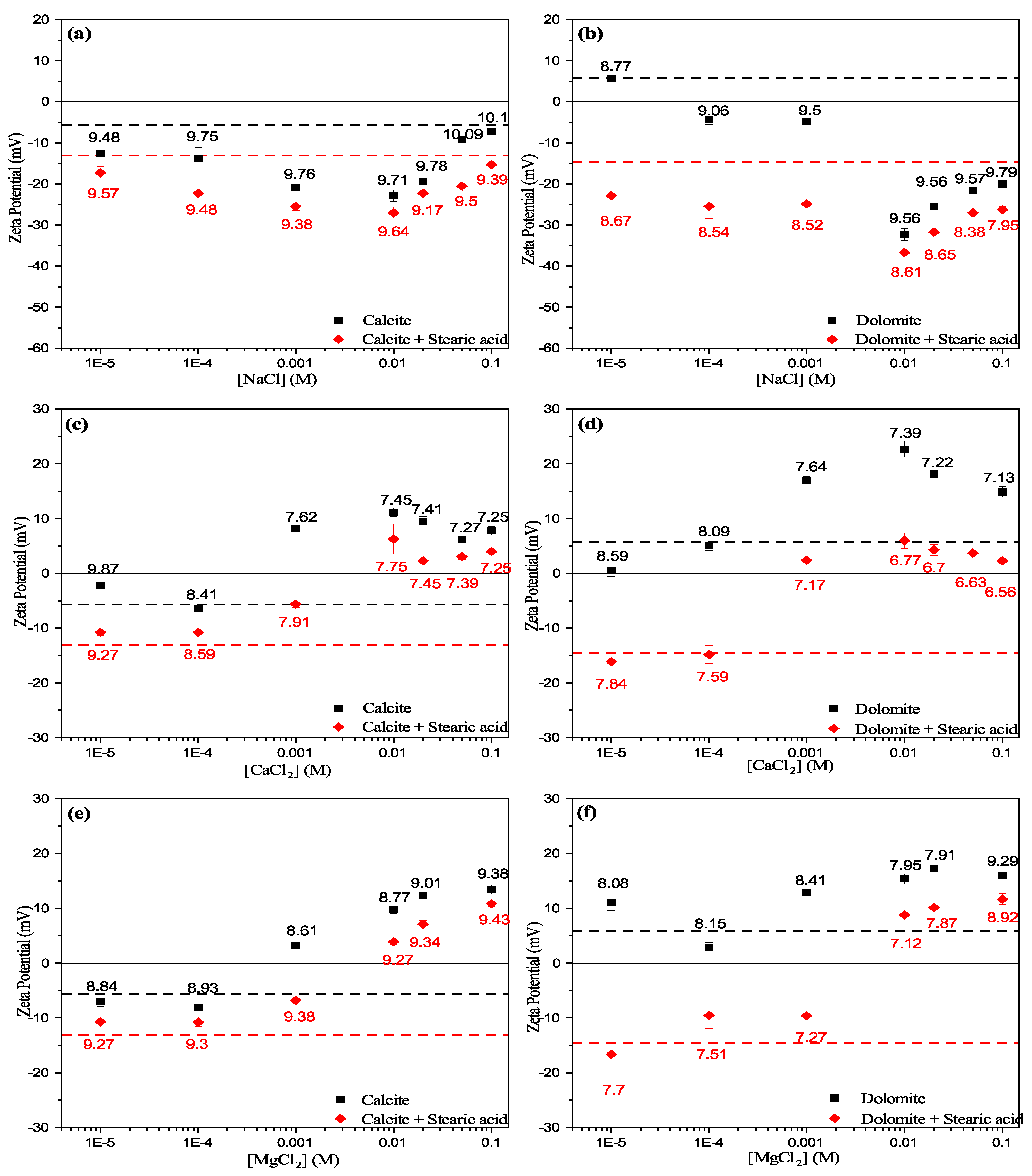
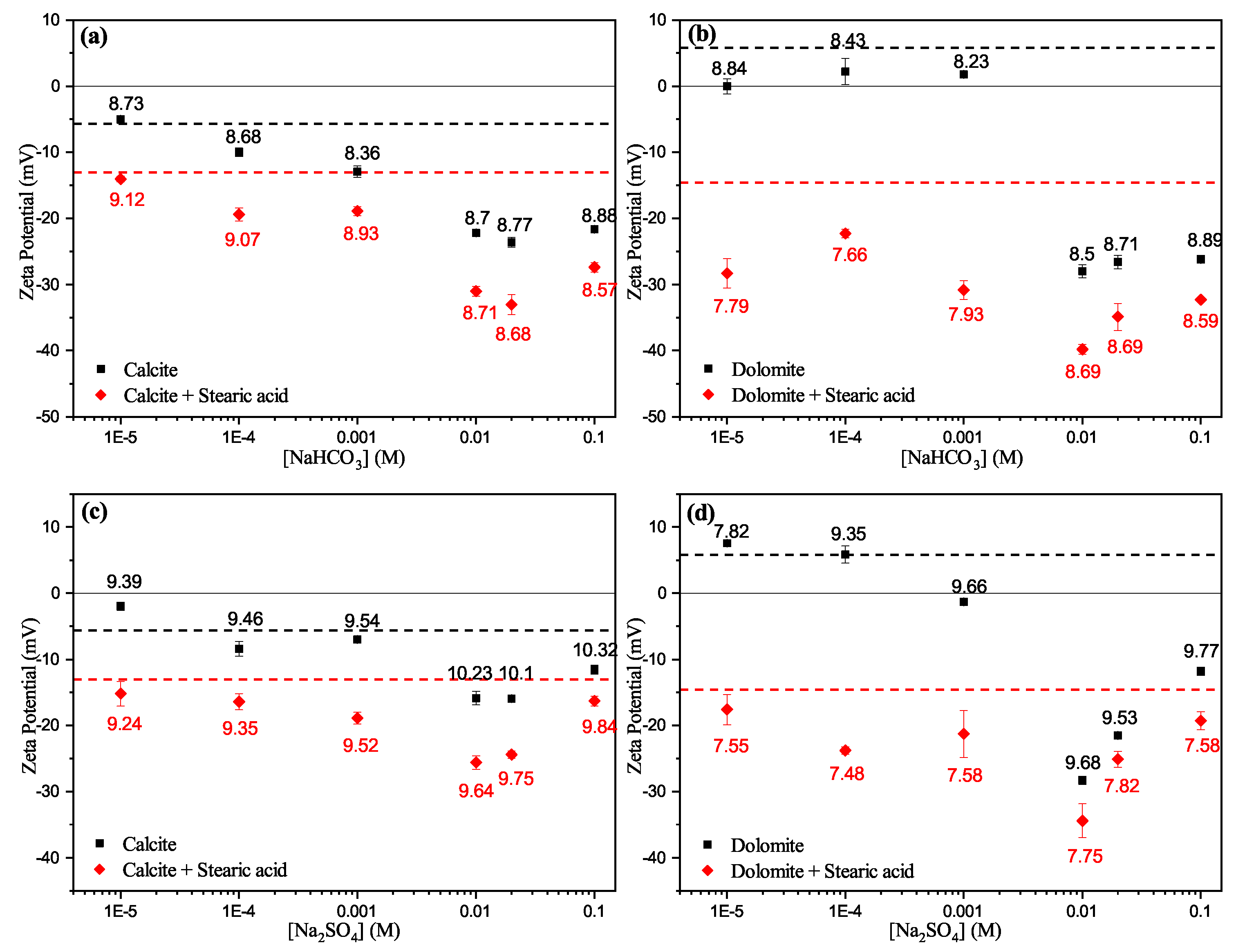
2.5. Zeta Potential Measurements
3. Results and Discussion
3.1. X-ray Diffraction
3.2. ATR-FTIR Spectroscopy
3.3. Zeta Potential
3.3.1. Effect of Cations
3.3.2. Effect of Anions
3.3.3. Effect of Stearic Acid
4. Conclusions
- Experimental results show that the unconditioned calcite particles are negatively charged in water, whereas dolomite particles showed positive surface charges. However, both calcite and dolomite particles showed similar trends of zeta potential by increasing the salt concentration.
- Zeta potential results show that the surface electrostatic charge of carbonate can be altered to strongly negative in the presence of HCO and SO ions (Group B), while higher concentrations of Ca and Mg ions (Group A) resulted in charge reversal from negative to positive.
- The current results shows that Ca and Mg cations act as potential-determining ions for calcite surfaces, while HCO and SO anions can be strong potential-determining ions toward dolomite surfaces.
- Experimental results show that pre-adsorbed stearic acid carbonate surfaces were more negatively charged compared to the unconditioned surface for the same electrolyte. This could be due to dissociative chemisorption (deprotonation) of stearic acid molecules on the solid particles.
- It is argued that divalent cations, e.g., Ca and Mg, would result in positive and neutral complexes with stearic acid molecules, which may result in strongly bound stearic acid film. In contrast, ions resulting in negative mineral surface charges (SO and HCO) will result in loosely bound stearic acid film to the carbonate mineral surface.
- The suggested mechanism in this study for surface charge modification of carbonates, in the presence of different ions, is changes in diffuse layer structure (expansion of electric double layer), due to adsorption of ions when changing electrolyte concentration.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Matthiesen, J.; Bovet, N.; Hilner, E.; Andersson, M.P.; Schmidt, D.; Webb, K.; Dalby, K.N.; Hassenkam, T.; Crouch, J.; Collins, I. How naturally adsorbed material on minerals affects low salinity enhanced oil recovery. Energy Fuels 2014, 28, 4849–4858. [Google Scholar] [CrossRef]
- Abdallah, W.; Gmira, A. Wettability assessment and surface compositional analysis of aged calcite treated with dynamic water. Energy Fuels 2013, 28, 1652–1663. [Google Scholar] [CrossRef]
- Valori, A.; Ali, F.; Abdallah, W. In-Situ Wettability Evaluation of Dynamic Water Flooding of Carbonate Rocks Based on NMR-T2 Distribution. In Proceedings of the SPE Middle East Oil & Gas Show and Conference, Manama, Kingdom of Bahrain, 6–9 March 2017; Society of Petroleum Engineers: Houston, TX, USA, 2017. [Google Scholar]
- Kazankapov, N. Enhanced Oil Recovery in Caspian Carbonates with “Smart Water”. In Proceedings of the SPE Russian Oil and Gas Exploration & Production Technical Conference and Exhibition, Moscow, Russia, 14–16 October 2014; Society of Petroleum Engineers: Houston, TX, USA, 2014. [Google Scholar]
- Derkani, M.; Fletcher, A.; Abdallah, W.; Sauerer, B.; Anderson, J.; Zhang, Z. Low salinity waterflooding in carbonate reservoirs: Review of interfacial mechanisms. Colloids Interfaces 2018, 2, 20. [Google Scholar] [CrossRef]
- Gomari, K.R.; Hamouda, A.; Davidian, T.; Fargland, D. Study of the effect of acidic species on wettability alteration of calcite surfaces by measuring partitioning coefficients, IFT and contact angles. Contact Angle Wettability Adhes. 2006, 4, 351–367. [Google Scholar]
- Gomari, K.R.; Hamouda, A. Effect of fatty acids, water composition and pH on the wettability alteration of calcite surface. J. Pet. Sci. Eng. 2006, 50, 140–150. [Google Scholar] [CrossRef]
- Marathe, R.; Turner, M.L.; Fogden, A. Pore-scale distribution of crude oil wettability in carbonate rocks. Energy Fuels 2012, 26, 6268–6281. [Google Scholar] [CrossRef]
- Sauerer, B.; Stukan, M.; Abdallah, W.; Derkani, M.H.; Fedorov, M.; Buiting, J.; Zhang, Z.J. Quantifying mineral surface energy by scanning force microscopy. J. Colloid Interface Sci. 2016, 472, 237–246. [Google Scholar] [CrossRef]
- Karimi, M.; Al-Maamari, R.S.; Ayatollahi, S.; Mehranbod, N. Mechanistic study of wettability alteration of oil-wet calcite: The effect of magnesium ions in the presence and absence of cationic surfactant. Colloids Surf. A Physicochem. Eng. Asp. 2015, 482, 403–415. [Google Scholar] [CrossRef]
- Gomari, K.R.; Denoyel, R.; Hamouda, A. Wettability of calcite and mica modified by different long-chain fatty acids (C 18 acids). J. Colloid Interface Sci. 2006, 297, 470–479. [Google Scholar] [CrossRef]
- Everett, D.H. Basic Principles of Colloid Science; Royal Society of Chemistry: Cambridge, UK, 2007. [Google Scholar]
- Alroudhan, A.; Vinogradov, J.; Jackson, M. Zeta potential of intact natural limestone: Impact of potential-determining ions Ca, Mg and SO 4. Colloids Surf. A Physicochem. Eng. Asp. 2016, 493, 83–98. [Google Scholar] [CrossRef]
- Hunter, R.J. Zeta Potential in Colloid Science: Principles and Applications; Academic Press: New York, NY, USA, 2013; Volume 2, pp. 1–386. [Google Scholar]
- Moulin, P.; Roques, H. Zeta potential measurement of calcium carbonate. J. Colloid Interface Sci. 2003, 261, 115–126. [Google Scholar] [CrossRef]
- Hanaor, D.; Michelazzi, M.; Leonelli, C.; Sorrell, C.C. The effects of carboxylic acids on the aqueous dispersion and electrophoretic deposition of ZrO2. J. Eur. Ceram. Soc. 2012, 32, 235–244. [Google Scholar] [CrossRef]
- Strand, S.; Høgnesen, E.J.; Austad, T. Wettability alteration of carbonates Effects of potential-determining ions (Ca2+ and SO) and temperature. Colloids Surf. A Physicochem. Eng. Asp. 2006, 275, 1–10. [Google Scholar] [CrossRef]
- Zhang, P.; Austad, T. Wettability and oil recovery from carbonates: Effects of temperature and potential-determining ions. Colloids Surf. A Physicochem. Eng. Asp. 2006, 279, 179–187. [Google Scholar] [CrossRef]
- Zhang, P.; Tweheyo, M.T.; Austad, T. Wettability alteration and improved oil recovery in chalk: The effect of calcium in the presence of sulfate. Energy Fuels 2006, 20, 2056–2062. [Google Scholar] [CrossRef]
- Zhang, P.; Tweheyo, M.T.; Austad, T. Wettability alteration and improved oil recovery by spontaneous imbibition of seawater into chalk: Impact of the potential-determining ions Ca2+, Mg2+, and SO. Colloids Surf. A Physicochem. Eng. Asp. 2007, 301, 199–208. [Google Scholar] [CrossRef]
- Gomari, K.R.; Hamouda, A.; Denoyel, R. Influence of sulfate ions on the interaction between fatty acids and calcite surface. Colloids Surf. A Physicochem. Eng. Asp. 2006, 287, 29–35. [Google Scholar] [CrossRef]
- Rezaei Gomari, K.A.; Karoussi, O.; Hamouda, A.A.a. Mechanistic study of interaction between water and carbonate rocks for enhancing oil recovery. In Proceedings of the SPE Europec/EAGE Annual Conference and Exhibition, Vienna, Austria, 12–15 June 2006; Society of Petroleum Engineers: Houston, TX, USA, 2006. [Google Scholar]
- Mahani, H.; Keya, A.L.; Berg, S.; Bartels, W.B.; Nasralla, R.; Rossen, W.R. Insights into the mechanism of wettability alteration by low-salinity flooding (LSF) in carbonates. Energy Fuels 2015, 29, 1352–1367. [Google Scholar] [CrossRef]
- Mahani, H.; Keya, A.L.; Berg, S.; Nasralla, R. Electrokinetics of carbonate/brine interface in low-salinity waterflooding: Effect of brine salinity, composition, rock type, and pH on ζ-potential and a surface-complexation model. SPE J. 2017, 22, 53–68. [Google Scholar] [CrossRef]
- Mahani, H.; Menezes, R.; Berg, S.; Fadili, A.; Nasralla, R.; Voskov, D.; Joekar-Niasar, V. Insights into the impact of temperature on the wettability alteration by low salinity in carbonate rocks. Energy Fuels 2017, 31, 7839–7853. [Google Scholar] [CrossRef]
- Alotaibi, M.B.; Nasr-El-Din, H.A.; Fletcher, J.J. Electrokinetics of limestone and dolomite rock particles. SPE Reserv. Eval. Eng. 2011, 14, 594–603. [Google Scholar] [CrossRef]
- Kasha, A.; Al-Hashim, H.; Abdallah, W.; Taherian, R.; Sauerer, B. Effect of Ca2+, Mg2+ and SO ions on the zeta potential of calcite and dolomite particles aged with stearic acid. Colloids Surf. A Physicochem. Eng. Asp. 2015, 482, 290–299. [Google Scholar] [CrossRef]
- Yousef, A.A.; Al-Saleh, S.; Al-Jawfi, M.S. The impact of the injection water chemistry on oil recovery from carbonate reservoirs. In Proceedings of the SPE EOR Conference at Oil and Gas West Asia, Muscat, Oman, 16–18 April 2012; Society of Petroleum Engineers: Houston, TX, USA, 2012. [Google Scholar]
- Yousef, A.A.; Al-Saleh, S.; Al-Jawfi, M.S. Improved/enhanced oil recovery from carbonate reservoirs by tuning injection water salinity and ionic content. In Proceedings of the SPE Improved Oil Recovery Symposium, Ulsa, OK, USA, 14–18 April 2012; Society of Petroleum Engineers: Houston, TX, USA, 2012. [Google Scholar]
- Al-Hashim, H.; Kasha, A.; Abdallah, W.; Sauerer, B. Impact of modified seawater on zeta potential and morphology of calcite and dolomite aged with stearic acid. Energy Fuels 2018, 32, 1644–1656. [Google Scholar] [CrossRef]
- Jackson, M.D.; Al-Mahrouqi, D.; Vinogradov, J. Zeta potential in oil–water–carbonate systems and its impact on oil recovery during controlled salinity water-flooding. Sci. Rep. 2016, 6, 37363. [Google Scholar] [CrossRef] [PubMed]
- Nyström, R.; Lindén, M.; Rosenholm, J.B. The Influence of Na+, Ca2+, Ba2+, and La3+ on the ζ Potential and the yield stress of calcite dispersions. J. Colloid Interface Sci. 2001, 242, 259–263. [Google Scholar] [CrossRef]
- Al Mahrouqi, D.; Vinogradov, J.; Jackson, M.D. Zeta potential of artificial and natural calcite in aqueous solution. Adv. Colloid Interface Sci. 2017, 240, 60–76. [Google Scholar] [CrossRef]
- Alotaibi, M.B.; Cha, D.; Alsofi, A.M.; Yousef, A.A. Dynamic interactions of inorganic species at carbonate/brine interfaces: An electrokinetic study. Colloids Surf. A Physicochem. Eng. Asp. 2018, 550, 222–235. [Google Scholar] [CrossRef]
- Merrill, L.; Bassett, W.A. The crystal structure of CaCO 3 (II), a high-pressure metastable phase of calcium carbonate. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1975, 31, 343–349. [Google Scholar] [CrossRef]
- Alruopn, P.L. Structural refinements of dolomite and a magnesian calcite and implications for dolomite formation in the marine environment. Am. Mineral. 1977, 62, 772–783. [Google Scholar]
- Rodriguez-Blanco, J.D.; Shaw, S.; Benning, L.G. The kinetics and mechanisms of amorphous calcium carbonate (ACC) crystallization to calcite, via vaterite. Nanoscale 2011, 3, 265–271. [Google Scholar] [CrossRef]
- Gunasekaran, S.; Anbalagan, G.; Pandi, S. Raman and infrared spectra of carbonates of calcite structure. J. Raman Spectrosc. 2006, 37, 892–899. [Google Scholar] [CrossRef]
- Jarrahian, K.; Seiedi, O.; Sheykhan, M.; Sefti, M.V.; Ayatollahi, S. Wettability alteration of carbonate rocks by surfactants: A mechanistic study. Colloids Surf. A Physicochem. Eng. Asp. 2012, 410, 1–10. [Google Scholar] [CrossRef]
- Lim, M.S.; Feng, K.; Chen, X.; Wu, N.; Raman, A.; Nightingale, J.; Gawalt, E.S.; Korakakis, D.; Hornak, L.A.; Timperman, A.T. Adsorption and desorption of stearic acid self-assembled monolayers on aluminum oxide. Langmuir 2007, 23, 2444–2452. [Google Scholar] [CrossRef] [PubMed]
- Pierre, A.; Lamarche, J.; Mercier, R.; Foissy, A.; Persello, J. Calcium as potential-determining ion in aqueous calcite suspensions. J. Dispers. Sci. Technol. 1990, 11, 611–635. [Google Scholar] [CrossRef]
- Monfared, A.D.; Ghazanfari, M.; Jamialahmadi, M.; Helalizadeh, A. Adsorption of silica nanoparticles onto calcite: Equilibrium, kinetic, thermodynamic and DLVO analysis. Chem. Eng. J. 2015, 281, 334–344. [Google Scholar] [CrossRef]
- Chibowski, E.; Hołysz, L.; Wójcik, W. Changes in zeta potential and surface free energy of calcium carbonate due to exposure to radiofrequency electric field. Colloids Surf. A Physicochem. Eng. Asp. 1994, 92, 79–85. [Google Scholar] [CrossRef]
- Farooq, U.; Tweheyo, M.T.; Sjøblom, J.; Øye, G. Surface characterization of model, outcrop, and reservoir samples in low salinity aqueous solutions. J. Dispers. Sci. Technol. 2011, 32, 519–531. [Google Scholar] [CrossRef]
- Ishido, T.; Mizutani, H. Experimental and theoretical basis of electrokinetic phenomena in rock–water systems and its applications to geophysics. J. Geophys. Res. Solid Earth 1981, 86, 1763–1775. [Google Scholar] [CrossRef]
- Huang, Y.C.; Fowkes, F.M.; Lloyd, T.B.; Sanders, N.D. Adsorption of calcium ions from calcium chloride solutions onto calcium carbonate particles. Langmuir 1991, 7, 1742–1748. [Google Scholar] [CrossRef]
- Andersen, J.; El-Mofty, S.; Somasundaran, P. Using electrophoresis for determining the mechanism of amine, sulfate and oleate adsorption on calcite. Colloids Surf. 1991, 55, 365–368. [Google Scholar] [CrossRef]
- Somasundaran, P.; Agar, G. The zero point of charge of calcite. J. Colloid Interface Sci. 1967, 24, 433–440. [Google Scholar] [CrossRef]
- Pavlovic, M.; Huber, R.; Adok-Sipiczki, M.; Nardin, C.; Szilagyi, I. Ion specific effects on the stability of layered double hydroxide colloids. Soft Matter 2016, 12, 4024–4033. [Google Scholar] [CrossRef] [PubMed]
- Awolayo, A.; Sarma, H.; AlSumaiti, A.M. A laboratory study of ionic effect of smart water for enhancing oil recovery in carbonate reservoirs. In Proceedings of the SPE EOR Conference at Oil and Gas West Asia, Muscat, Oman, 31 March–2 April 2014; Society of Petroleum Engineers: Houston, TX, USA, 2014. [Google Scholar]
- Foxall, T.; Peterson, G.C.; Rendall, H.M.; Smith, A.L. Charge determination at calcium salt/aqueous solution interface. JOurnal Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1979, 75, 1034–1039. [Google Scholar] [CrossRef]
- Cicerone, D.S.; Regazzoni, A.E.; Blesa, M.A. Electrokinetic properties of the calcite/water interface in the presence of magnesium and organic matter. J. Colloid Interface Sci. 1992, 154, 423–433. [Google Scholar] [CrossRef]
- Thompson, D.W.; Pownall, P.G. Surface electrical properties of calcite. J. Colloid Interface Sci. 1989, 131, 74–82. [Google Scholar] [CrossRef]
- Guichet, X.; Jouniaux, L.; Catel, N. Modification of streaming potential by precipitation of calcite in a sand–water system: Laboratory measurements in the pH range from 4 to 12. Geophys. J. Int. 2006, 166, 445–460. [Google Scholar] [CrossRef]
- Heberling, F.; Trainor, T.P.; Lützenkirchen, J.; Eng, P.; Denecke, M.A.; Bosbach, D. Structure and reactivity of the calcite–water interface. J. Colloid Interface Sci. 2011, 354, 843–857. [Google Scholar] [CrossRef]
- Sohal, M.A.; Thyne, G.; Søgaard, E.G. Review of recovery mechanisms of ionically modified waterflood in carbonate reservoirs. Energy Fuels 2016, 30, 1904–1914. [Google Scholar] [CrossRef]
- Fathi, S.J.; Austad, T.; Strand, S. Water-Based Enhanced Oil recovery (EOR) by “Smart Water” in Carbonate Reservoirs. In Proceedings of the SPE EOR Conference at Oil and Gas West Asia, Muscat, Oman, 16–18 April 2012; Society of Petroleum Engineers: Houston, TX, USA, 2012. [Google Scholar]
- Myint, P.C.; Firoozabadi, A. Thin liquid films in improved oil recovery from low-salinity brine. Curr. Opin. Colloid Interface Sci. 2015, 20, 105–114. [Google Scholar] [CrossRef]
- Eriksson, R.; Merta, J.; Rosenholm, J.B. The calcite/water interface: I. Surface charge in indifferent electrolyte media and the influence of low-molecular-weight polyelectrolyte. J. Colloid Interface Sci. 2007, 313, 184–193. [Google Scholar] [CrossRef]
- Pourchet, S.; Pochard, I.; Brunel, F.; Perrey, D. Chemistry of the calcite/water interface: Influence of sulfate ions and consequences in terms of cohesion forces. Cem. Concr. Res. 2013, 52, 22–30. [Google Scholar] [CrossRef]
- Douglas, H.; Walker, R. The electrokinetic behaviour of Iceland Spar against aqueous electrolyte solutions. Trans. Faraday Soc. 1950, 46, 559–568. [Google Scholar] [CrossRef]
- Fenter, P.; Geissbühler, P.; DiMasi, E.; Srajer, G.; Sorensen, L.; Sturchio, N. Surface speciation of calcite observed in situ by high-resolution X-ray reflectivity. Geochim. Cosmochim. Acta 2000, 64, 1221–1228. [Google Scholar] [CrossRef]
- Prédali, J.J.; Cases, J.M. Zeta potential of magnesian carbonates in inorganic electrolytes. J. Colloid Interface Sci. 1973, 45, 449–458. [Google Scholar] [CrossRef]
- Marouf, R.; Marouf-Khelifa, K.; Schott, J.; Khelifa, A. Zeta potential study of thermally treated dolomite samples in electrolyte solutions. Microporous Mesoporous Mater. 2009, 122, 99–104. [Google Scholar] [CrossRef]
- Schramm, L.L.; Mannhardt, K.; Novosad, J.J. Electrokinetic properties of reservoir rock particles. Colloids Surf. 1991, 55, 309–331. [Google Scholar] [CrossRef]
- Pokrovsky, O.S.; Schott, J.; Thomas, F. Dolomite surface speciation and reactivity in aquatic systems. Geochim. Cosmochim. Acta 1999, 63, 3133–3143. [Google Scholar] [CrossRef]
- Chen, G.; Tao, D. Effect of solution chemistry on flotability of magnesite and dolomite. Int. J. Miner. Process. 2004, 74, 343–357. [Google Scholar] [CrossRef]
- Mihajlović, S.R.; Vučinić, D.R.; Sekulić, Ž.T.; Milićević, S.Z.; Kolonja, B.M. Mechanism of stearic acid adsorption to calcite. Powder Technol. 2013, 245, 208–216. [Google Scholar] [CrossRef]
- Nalbach, M.; Raiteri, P.; Klassen, S.; Schafer, S.; Gale, J.D.; Bechstein, R.; Kuhnle, A. Where Is the Most Hydrophobic Region? Benzopurpurine Self-Assembly at the Calcite–Water Interface. J. Phys. Chem. C 2017, 121, 24144–24151. [Google Scholar] [CrossRef]
- Kanicky, J.R.; Shah, D.O. Effect of degree, type, and position of unsaturation on the pKa of long-chain fatty acids. J. Colloid Interface Sci. 2002, 256, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Wang, L.; Siretanu, I.; Duits, M.; Mugele, F. Salt dependent stability of stearic acid Langmuir–Blodgett films exposed to aqueous electrolytes. Langmuir 2013, 29, 5150–5159. [Google Scholar] [CrossRef] [PubMed]
- Haagh, M.E.; Siretanu, I.; Duits, M.H.; Mugele, F. Salinity-dependent contact angle alteration in oil/brine/silicate systems: The critical role of divalent cations. Langmuir 2017, 33, 3349–3357. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, D.K. Langmuir–Blodgett film structure. Surf. Sci. Rep. 1997, 27, 245–334. [Google Scholar] [CrossRef]
- Bloch, J.M.; Yun, W. Condensation of monovalent and divalent metal ions on a Langmuir monolayer. Phys. Rev. A 1990, 41, 844. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Salt | Conc. (g/L) | Conc. (ppm) | Conc. (mM) |
|---|---|---|---|
| NaCl | 40.30 | 40,323 | 690 |
| CaCl | 1.79 | 1799 | 16.20 |
| MgCl | 8.46 | 8461 | 88.80 |
| NaSO | 6.58 | 6580 | 46.30 |
| NaHCO | 0.16 | 165 | 1.96 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Derkani, M.H.; Fletcher, A.J.; Fedorov, M.; Abdallah, W.; Sauerer, B.; Anderson, J.; Zhang, Z.J. Mechanisms of Surface Charge Modification of Carbonates in Aqueous Electrolyte Solutions. Colloids Interfaces 2019, 3, 62. https://doi.org/10.3390/colloids3040062
Derkani MH, Fletcher AJ, Fedorov M, Abdallah W, Sauerer B, Anderson J, Zhang ZJ. Mechanisms of Surface Charge Modification of Carbonates in Aqueous Electrolyte Solutions. Colloids and Interfaces. 2019; 3(4):62. https://doi.org/10.3390/colloids3040062
Chicago/Turabian StyleDerkani, Maryam H., Ashleigh J. Fletcher, Maxim Fedorov, Wael Abdallah, Bastian Sauerer, James Anderson, and Zhenyu J. Zhang. 2019. "Mechanisms of Surface Charge Modification of Carbonates in Aqueous Electrolyte Solutions" Colloids and Interfaces 3, no. 4: 62. https://doi.org/10.3390/colloids3040062
APA StyleDerkani, M. H., Fletcher, A. J., Fedorov, M., Abdallah, W., Sauerer, B., Anderson, J., & Zhang, Z. J. (2019). Mechanisms of Surface Charge Modification of Carbonates in Aqueous Electrolyte Solutions. Colloids and Interfaces, 3(4), 62. https://doi.org/10.3390/colloids3040062