Rapid Hydrolysis of Organophosphates Induced by U(IV) Nanoparticles: A Kinetic and Mechanistic Study using Spectroscopic Analysis


Abstract

1. Introduction
2. Materials and Methods
2.1. Synthesis and Identification of U(IV) Species
2.1.1. U(IV) Nanoparticles (U(IV)NPs)
2.1.2. Amorphous U(IV)-Hydroxides (U(IV)–OH(am))
2.2. Particle Size and Zeta-Potential Measurements
2.3. Spectrophotometric Reaction Kinetic Analysis
2.4. ATR-FTIR Measurements
2.5. Reaction Modeling Analysis
3. Results and Discussion
3.1. Characterization of U(IV)NPs
3.2. Comparison of NPP Hydrolysis Rates by Different U(IV) Species
3.3. Mechanistic Studies on the NPP Hydrolysis
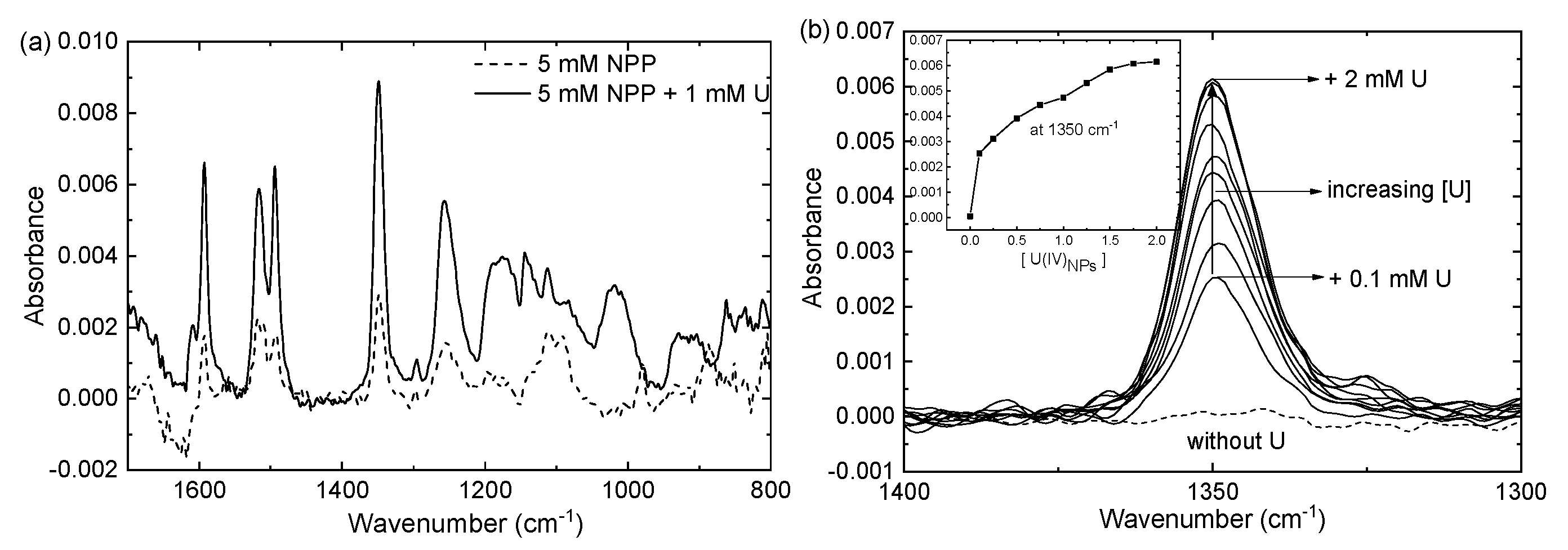
3.3.1. ATR-FTIR Studies and SEIRA
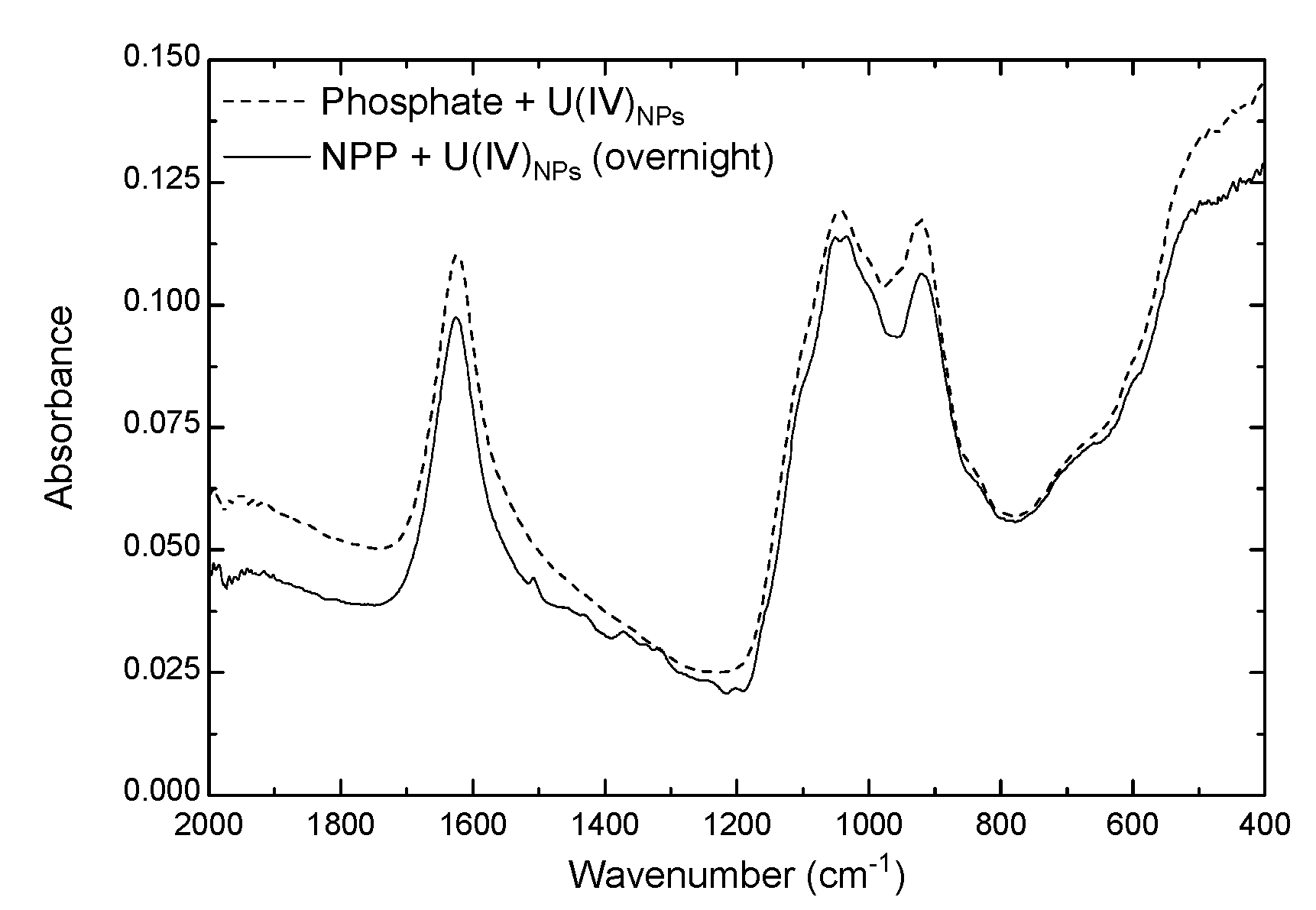
3.3.2. ATR-FTIR Spectra of Solid Phase U(IV)NPs
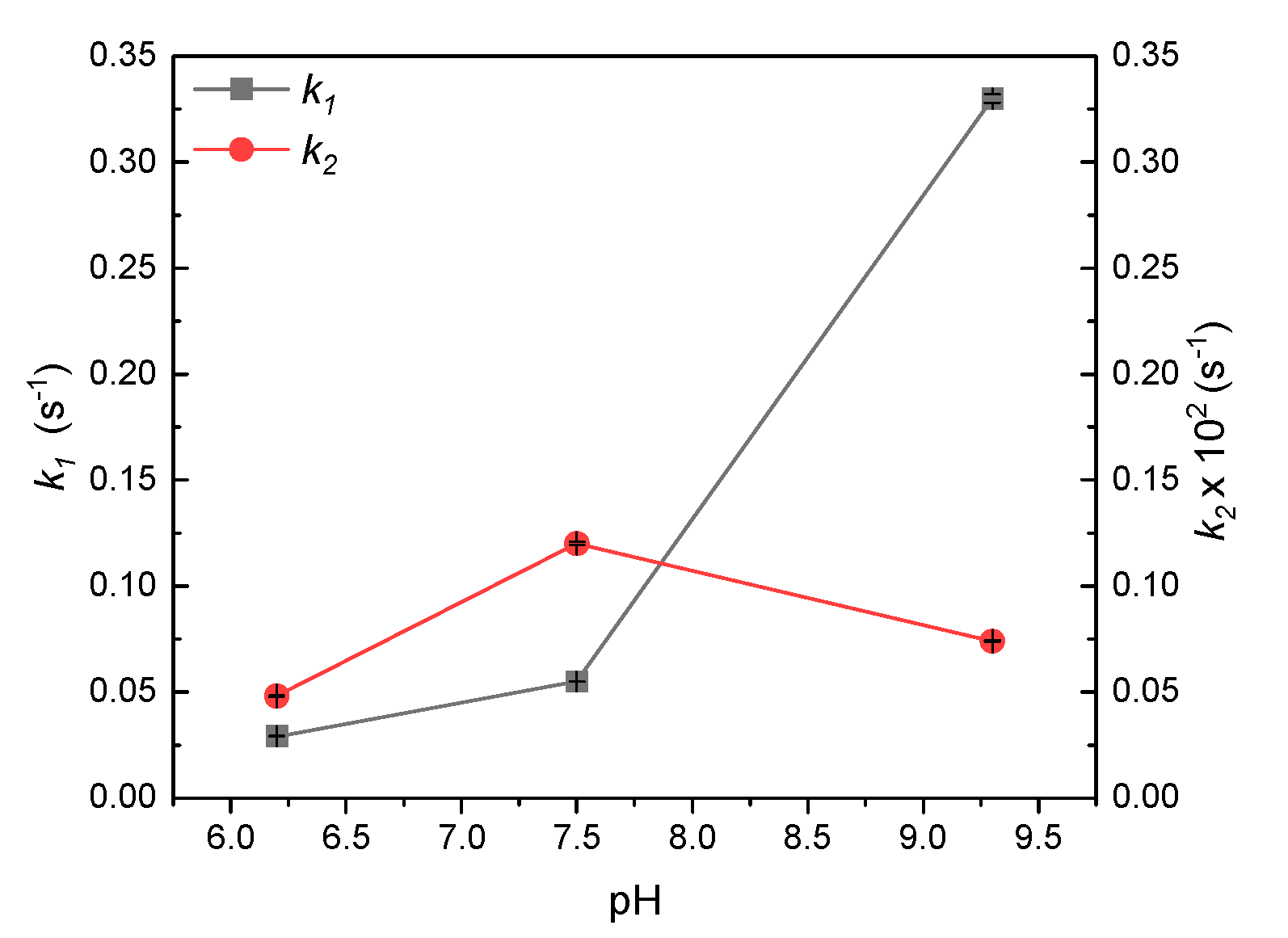
3.3.3. Reaction Modeling Analysis
3.4. Implication of U(IV)NPs Reactivity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kim, J.-I. Significance of actinide chemistry for the long-term safety of waste disposal. Nucl. Eng. Technol. 2006, 38, 459–482. [Google Scholar]
- Opel, K.; Weiss, S.; Hübener, S.; Zänker, H.; Bernhard, G. Study of the solubility of amorphous and crystalline uranium dioxide by combined spectroscopic methods. Radiochim. Acta 2007, 95, 143–149. [Google Scholar] [CrossRef]
- Kim, J.I. Actinide colloid generation in groundwater. Radiochim. Acta 1991, 52, 71–82. [Google Scholar] [CrossRef]
- Maher, K.; Bargar, J.R.; Brown, G.E., Jr. Environmental speciation of actinides. Inorg. Chem. 2012, 52, 3510–3532. [Google Scholar] [CrossRef] [PubMed]
- Geckeis, H.; Rabung, T.; Schäfer, T. Actinide-nanoparticle interaction: Generation, stability and mobility. In Actinide Nanoparticle Research; Springer: Berlin/Heidelberg, Germany, 2011; pp. 1–30. [Google Scholar] [CrossRef]
- Fuller, C.; Bargar, J.; Davis, J.; Piana, M. Mechanisms of uranium interactions with hydroxyapatite: Implications for groundwater remediation. Environ. Sci. Technol. 2002, 36, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Murray, F.; Brown, J.; Fyfe, W.; Kronberg, B. Immobilization of U-Th-Ra in mine wastes by phosphate mineralization. Can. Mineral. 1983, 21, 607–610. [Google Scholar]
- Arey, J.S.; Seaman, J.C.; Bertsch, P.M. Immobilization of uranium in contaminated sediments by hydroxyapatite addition. Environ. Sci. Technol. 1998, 33, 337–342. [Google Scholar] [CrossRef]
- Jerden, J.L., Jr.; Sinha, A. Phosphate based immobilization of uranium in an oxidizing bedrock aquifer. Appl. Geochem. 2003, 18, 823–843. [Google Scholar] [CrossRef]
- Langmuir, D. Aqueous Environmental Geochemistry; Prentice Hall: Upper Saddle River, NJ, USA, 1997. [Google Scholar]
- Martinez, R.J.; Wu, C.H.; Beazley, M.J.; Andersen, G.L.; Conrad, M.E.; Hazen, T.C.; Taillefert, M.; Sobecky, P.A. Microbial community responses to organophosphate substrate additions in contaminated subsurface sediments. PLoS ONE 2014, 9, e100383. [Google Scholar] [CrossRef]
- Newsome, L.; Morris, K.; Trivedi, D.; Bewsher, A.; Lloyd, J.R. Biostimulation by glycerol phosphate to precipitate recalcitrant uranium (IV) phosphate. Environ. Sci. Technol. 2015, 49, 11070–11078. [Google Scholar] [CrossRef]
- Newsome, L.; Morris, K.; Lloyd, J.R. The biogeochemistry and bioremediation of uranium and other priority radionuclides. Chem. Geol. 2014, 363, 164–184. [Google Scholar] [CrossRef]
- Stetten, L.; Blanchart, P.; Mangeret, A.; Lefebvre, P.; Le Pape, P.; Brest, J.; Merrot, P.; Julien, A.; Proux, O.; Webb, S.M. Redox fluctuations and organic complexation govern uranium redistribution from U (IV)-phosphate minerals in a mining-polluted wetland soil, Brittany, France. Environ. Sci. Technol. 2018, 52, 13099–13109. [Google Scholar] [CrossRef] [PubMed]
- Morin, G.; Mangeret, A.; Othmane, G.; Stetten, L.; Seder-Colomina, M.; Brest, J.; Ona-Nguema, G.; Bassot, S.; Courbet, C.; Guillevic, J. Mononuclear U (IV) complexes and ningyoite as major uranium species in lake sediments. Geochem. Perspect. Lett. 2016, 2, 95–105. [Google Scholar] [CrossRef]
- Ingall, E.D.; Schroeder, P.A.; Berner, R.A. The nature of organic phosphorus in marine sediments: New insights from 31P NMR. Geochim. Cosmochim. Acta 1990, 54, 2617–2620. [Google Scholar] [CrossRef]
- Espinosa, M.; Turner, B.L.; Haygarth, P.M. Preconcentration and separation of trace phosphorus compounds in soil leachate. J. Environ. Qual. 1999, 28, 1497–1504. [Google Scholar] [CrossRef]
- Pant, H.; Warman, P.; Nowak, J. Identification of soil organic phosphorus by 31P nuclear magnetic resonance spectroscopy. Commun. Soil Sci. Plant Anal. 1999, 30, 757–772. [Google Scholar] [CrossRef]
- Benitez-Nelson, C.R. The biogeochemical cycling of phosphorus in marine systems. Earth-Science Reviews 2000, 51, 109–135. [Google Scholar] [CrossRef]
- Turner, B.L.; McKelvie, I.D.; Haygarth, P.M. Characterisation of water-extractable soil organic phosphorus by phosphatase hydrolysis. Soil Biol. Biochem. 2002, 34, 27–35. [Google Scholar] [CrossRef]
- Paytan, A.; McLaughlin, K. The oceanic phosphorus cycle. Chem. Rev. 2007, 107, 563–576. [Google Scholar] [CrossRef]
- Balistrieri, L.S.; Chao, T. Adsorption of selenium by amorphous iron oxyhydroxide and manganese dioxide. Geochim. Cosmochim. Acta 1990, 54, 739–751. [Google Scholar] [CrossRef]
- Baldwin, D.S.; Beattie, J.K.; Coleman, L.M.; Jones, D.R. Phosphate ester hydrolysis facilitated by mineral phases. Environ. Sci. Technol. 1995, 29, 1706–1709. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, D.S.; Beattie, J.K.; Coleman, L.M.; Jones, D.R. Hydrolysis of an organophosphate ester by manganese dioxide. Environ. Sci. Technol. 2001, 35, 713–716. [Google Scholar] [CrossRef] [PubMed]
- Olsson, R.; Giesler, R.; Loring, J.S.; Persson, P. Adsorption, desorption, and surface-promoted hydrolysis of glucose-1-phosphate in aqueous goethite (α-FeOOH) suspensions. Langmuir 2010, 26, 18760–18770. [Google Scholar] [CrossRef] [PubMed]
- Morales-Rojas, H.; Moss, R.A. Phosphorolytic reactivity of o-iodosylcarboxylates and related nucleophiles. Chem. Rev. 2002, 102, 2497–2522. [Google Scholar] [CrossRef] [PubMed]
- Sigel, H. Metal Ions in Biological Systems. In The Lanthanides and Their Interrelations with Biosystems; Sigel, H., Sigel, A., Eds.; CRC Press: Basel, Switzerland, 2003; Volume 40, pp. 369–440. [Google Scholar]
- Liu, C.; Wang, M.; Zhang, T.; Sun, H. DNA hydrolysis promoted by di-and multi-nuclear metal complexes. Coord. Chem. Rev. 2004, 248, 147–168. [Google Scholar] [CrossRef]
- Morrow, J.R.; Iranzo, O. Synthetic metallonucleases for RNA cleavage. Curr. Opin. Chem. Biol. 2004, 8, 192–200. [Google Scholar] [CrossRef]
- Williams, N.H. Models for biological phosphoryl transfer. Biochim. Biophys. Acta 2004, 1697, 279–287. [Google Scholar] [CrossRef]
- Yatsimirsky, A.K. Metal ion catalysis in acyl and phosphoryl transfer: Transition states as ligands. Coord. Chem. Rev. 2005, 249, 1997–2011. [Google Scholar] [CrossRef]
- Ott, R.; Krämer, R. Rapid Phosphodiester Hydrolysis by Zirconium(IV). Angew. Chem. Int. Ed. 1998, 37, 1957–1960. [Google Scholar] [CrossRef]
- Blaskó, A.; Bruice, T.C. Recent Studies of Nucleophilic, General-Acid, and Metal Ion Catalysis of Phosphate Diester Hydrolysis. Acc. Chem. Res. 1999, 32, 475–484. [Google Scholar] [CrossRef]
- Lim, S.; Franklin, S.J. Lanthanide-binding peptides and the enzymes that Might Have Been. Cell. Mol. Life Sci. 2004, 61, 2184–2188. [Google Scholar] [CrossRef] [PubMed]
- Luong, T.K.N.; Shestakova, P.; Parac-Vogt, T.N. Kinetic studies of phosphoester hydrolysis promoted by a dimeric tetrazirconium(IV) Wells–Dawson polyoxometalate. Dalton Trans. 2016, 45, 12174–12180. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.; Carnall, W.T. Absorption spectra of Uranium(III) and Uranium(IV) in DClO4 solution 1. J. Phys. Chem. 1960, 64, 1933–1936. [Google Scholar] [CrossRef]
- Jolivet, J.P.; Tronc, E.; Chanéac, C. Iron oxides: From molecular clusters to solid. A nice example of chemical versatility. Geomaterials (Mineralogy) 2006, 338, 488–497. [Google Scholar] [CrossRef]
- Takasaki, B.K.; Chin, J. Synergistic effect between lanthanum (III) and hydrogen peroxide in phosphate diester cleavage. J. Am. Chem. Soc. 1993, 115, 9337–9338. [Google Scholar] [CrossRef]
- Moss, R.A.; Bracken, K.; Zhang, J. Actinide (uranyl) hydrolysis of phosphodiesters. Chem. Commun. 1997, 563–564. [Google Scholar] [CrossRef]
- Moss, R.A.; Zhang, J.; Bracken, K. Extraordinary acceleration of phosphodiester hydrolyses by thorium cations. Chem. Commun. 1997, 1639–1640. [Google Scholar] [CrossRef]
- Schneider, H.J.; Rammo, J.; Hettich, R. Catalysis of the hydrolysis of phosphoric acid diesters by lanthanide ions and the influence of ligands. Angew. Chem. Int. Ed. 1993, 32, 1716–1719. [Google Scholar] [CrossRef]
- Bracken, K.; Moss, R.A.; Ragunathan, K.G. Remarkably rapid cleavage of a model phosphodiester by complexed ceric ions in aqueous micellar solutions. J. Am. Chem. Soc. 1997, 119, 9323–9324. [Google Scholar] [CrossRef]
- Breslow, R.; Huang, D.-L. Effects of metal ions, including Mg2+ and lanthanides, on the cleavage of ribonucleotides and RNA model compounds. Proc. Natl. Acad. Sci. USA 1991, 88, 4080–4083. [Google Scholar] [CrossRef]
- Griffiths, P.R. Surface-enhanced infrared absorption spectroscopy: Principles and applications. Spectrosc. Prop. Inorg. Organomet. Compd. 2013, 44, 95–122. [Google Scholar] [CrossRef]
- Osawa, M.; Ataka, K.-I.; Yoshii, K.; Nishikawa, Y. Surface-enhanced infrared spectroscopy: The origin of the absorption enhancement and band selection rule in the infrared spectra of molecules adsorbed on fine metal particles. Appl. Spectrosc. 1993, 47, 1497–1502. [Google Scholar] [CrossRef]
- Osawa, M.; Ikeda, M. Surface-enhanced infrared absorption of p-nitrobenzoic acid deposited on silver island films: Contributions of electromagnetic and chemical mechanisms. J. Phys. Chem. 1991, 95, 9914–9919. [Google Scholar] [CrossRef]
- Ahmad, I.; Dines, T.J.; Rochester, C.H.; Anderson, J.A. IR study of nitrotoluene adsorption on oxide surfaces. J. Chem. Soc., Faraday Trans. 1996, 92, 3225–3231. [Google Scholar] [CrossRef]
- Osawa, M. Surface-enhanced infrared absorption spectroscopy. In Handbook of Vibrational Spectroscopy; Chalmers, J., Griffiths, P., Eds.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2006; pp. 785–799. [Google Scholar] [CrossRef]
- Li, B.; Raff, J.; Barkleit, A.; Bernhard, G.; Foerstendorf, H. Complexation of U (VI) with highly phosphorylated protein, phosvitin: A vibrational spectroscopic approach. J. Inorg. Biochem. 2010, 104, 718–725. [Google Scholar] [CrossRef]
- Klein, A.R.; Bone, S.E.; Bakker, E.; Chang, Z.; Aristilde, L. Abiotic phosphorus recycling from adsorbed ribonucleotides on a ferrihydrite-type mineral: Probing solution and surface species. J. Colloid Interface Sci. 2019, 547, 171–182. [Google Scholar] [CrossRef]
- Arai, Y.; Sparks, D.L. ATR–FTIR spectroscopic investigation on phosphate adsorption mechanisms at the ferrihydrite–water interface. J. Colloid Interface Sci. 2001, 241, 317–326. [Google Scholar] [CrossRef]
- Maeder, M.; Neuhold, Y.-M. Practical data analysis in chemistry. In Data Handling in Science and Technology; Rutan, S., Walczak, B., Eds.; Elsevier: Oxford, UK, 2007. [Google Scholar]
- Maeder, M.; King, P. ReactLab KINETICS; Jplus Consulting Pty Ltd.: East Freemantle, WA, Australia, 2009. [Google Scholar]
- Salome, K.R.; Green, S.J.; Beazley, M.J.; Webb, S.M.; Kostka, J.E.; Taillefert, M. The role of anaerobic respiration in the immobilization of uranium through biomineralization of phosphate minerals. Geochim. Cosmochim. Acta 2013, 106, 344–363. [Google Scholar] [CrossRef]
- Turner, B.L.; Papházy, M.J.; Haygarth, P.M.; McKelvie, I.D. Inositol phosphates in the environment. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2002, 357, 449–469. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Metal Species | T (°C) | kobs/10−4 s−1 | |||||
|---|---|---|---|---|---|---|---|
| pH 3.3 ± 0.2 | pH 5.2 ± 0.2 | pH 6.1 ± 0.2 | pH 7.2 ± 0.3 | pH 9.3 ± 0.2 | |||
| U(IV)NPs | 25 | 0.90 | 1.1 | 1.2 | 1.4 | 0.21 | this work [a] |
| 50 | 15 | 16 | 19 | 20 | 2.2 | ||
| U(IV)–OH(am) | 25 | 0.24 | 0.23 | 0.21 | 0.17 | 0.080 | |
| 50 | 2.4 | 2.3 | 2.2 | 1.7 | 0.82 | ||
| U(VI), Th(IV), La(III) | 37 | 0.095(UO22+) [b] | 280(Th4+) [c] | 0.0014(La3+) [d] | ref. [37,38,39] [f] | ||
| Ce(IV) [e] | 50 | 180 | 260 | 190 | ref. [40,41] [f] | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, H.; Cha, W. Rapid Hydrolysis of Organophosphates Induced by U(IV) Nanoparticles: A Kinetic and Mechanistic Study using Spectroscopic Analysis. Colloids Interfaces 2019, 3, 63. https://doi.org/10.3390/colloids3040063
Cho H, Cha W. Rapid Hydrolysis of Organophosphates Induced by U(IV) Nanoparticles: A Kinetic and Mechanistic Study using Spectroscopic Analysis. Colloids and Interfaces. 2019; 3(4):63. https://doi.org/10.3390/colloids3040063
Chicago/Turabian StyleCho, Hyejin, and Wansik Cha. 2019. "Rapid Hydrolysis of Organophosphates Induced by U(IV) Nanoparticles: A Kinetic and Mechanistic Study using Spectroscopic Analysis" Colloids and Interfaces 3, no. 4: 63. https://doi.org/10.3390/colloids3040063
APA StyleCho, H., & Cha, W. (2019). Rapid Hydrolysis of Organophosphates Induced by U(IV) Nanoparticles: A Kinetic and Mechanistic Study using Spectroscopic Analysis. Colloids and Interfaces, 3(4), 63. https://doi.org/10.3390/colloids3040063