
The Structure of Liquid and Glassy Carbamazepine

 , , , ,
, , , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Nuclear Magnetic Resonance Spectroscopy
2.2. Vibrational Spectroscopy
2.3. Thermal Analysis
2.4. High-Energy X-ray Diffraction
2.5. Empirical Potential Structure Refinement
3. Results
3.1. Sample Characterization
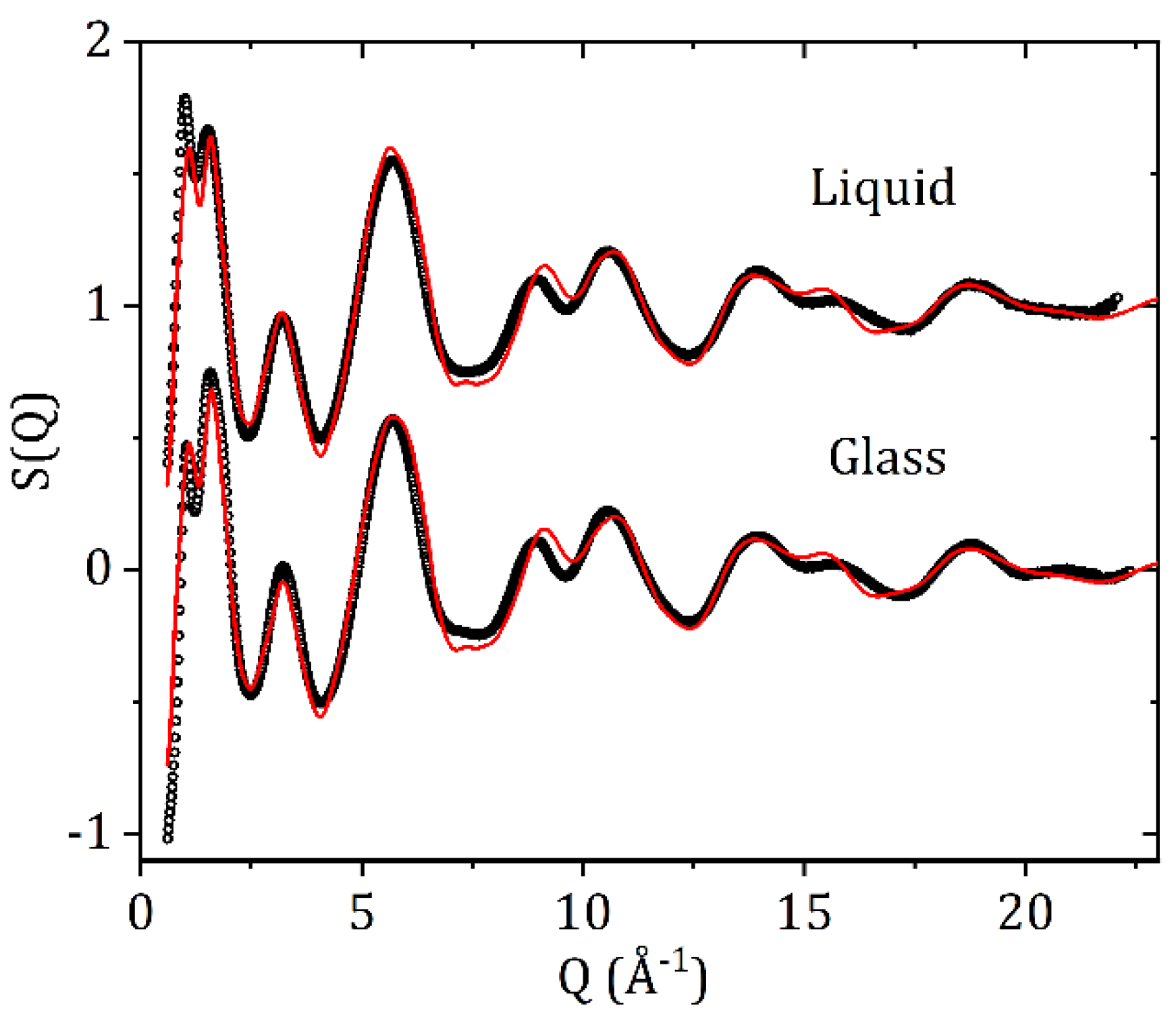
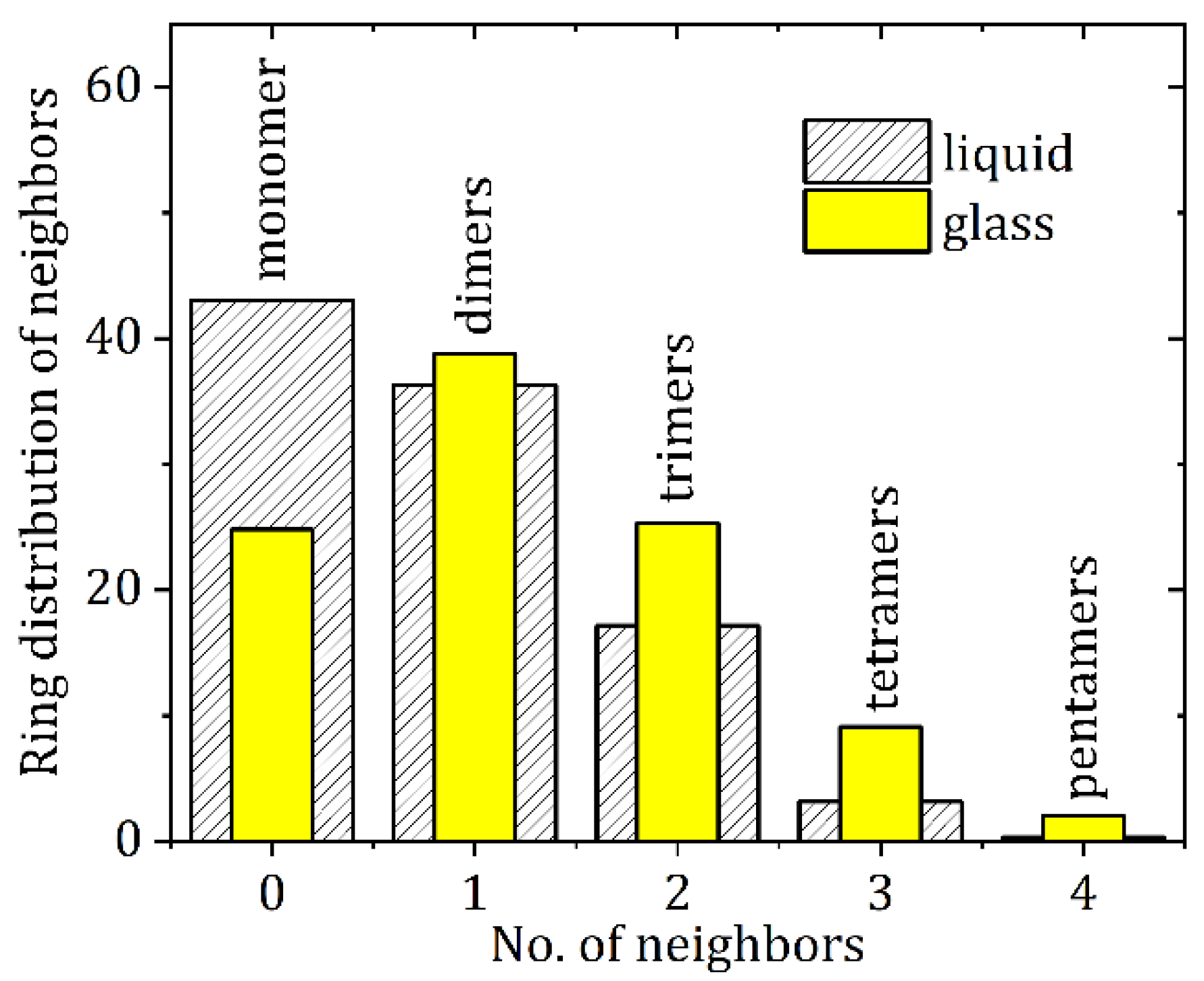
3.2. High-Energy X-ray Diffraction and EPSR Modeling Results
4. Discussion
Hydration of CBZ
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Yu, L. Amorphous pharmaceutical solids: Preparation, characterization and stabilization. Adv. Drug Deliv. Rev. 2001, 48, 27–42. [Google Scholar] [CrossRef]
- Willart, J.F.; Descamps, M. Solid State Amorphization of Pharmaceuticals. Mol. Pharm. 2008, 5, 905–920. [Google Scholar] [CrossRef] [PubMed]
- Byrn, S.R.; Zografi, G.; Chen, X.S. Solid State Properties of Pharmaceutical Material; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2017. [Google Scholar]
- Healy, A.M.; Worku, Z.A.; Kumar, D.; Madi, A.M. Pharmaceutical solvates, hydrates and amorphous forms: A special emphasis on cocrystals. Adv. Drug Deliv. Rev. 2017, 117, 25–46. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.K.; Ghi, P.Y.; Puschmann, H.; Apperley, D.C.; Griesser, U.J.; Hammond, R.B.; Ma, C.; Roberts, K.J.; Pearce, G.J.; Yates, J.R.; et al. Structural Studies of the Polymorphs of Carbamazepine, Its Dihydrate, and Two Solvates. Org. Process Res. Dev. 2005, 9, 902–910. [Google Scholar] [CrossRef]
- Billinge, S.; Dykhne, T.; Juhas, P.; Bozin, E.; Taylor, R.; Florence, A.; Shankland, K. Characterisation of amorphous and nanocrystalline molecular materials by total scattering. CrystEngComm 2010, 12, 1366–1368. [Google Scholar] [CrossRef]
- Petkov, V.; Ren, Y.; Suchomel, M. Molecular arrangement in water: Random but not quite. J. Phys. Condens. Matter 2012, 24, 155102. [Google Scholar] [CrossRef]
- Skinner, L.B.; Benmore, C.J.; Parise, J.B. Comment on ‘Molecular arrangement in water: Random but not quite’. J. Phys. Condens. Matter 2012, 24, 338001, Discussion 338002. [Google Scholar] [CrossRef]
- Soper, A.K. Recent water myths. Pure Appl. Chem. 2010, 82, 1855–1867. [Google Scholar] [CrossRef]
- Wright, A.C. The Great Crystallite Versus Random Network Controversy: A Personal Perspective. Int. J. Appl. Glass Sci. 2014, 5, 31–56. [Google Scholar] [CrossRef]
- Benmore, C.J.; Benmore, S.R.; Edwards, A.D.; Shrader, C.D.; Bhat, M.H.; Cherry, B.R.; Smith, P.; Gozzo, F.; Shi, C.; Smith, D.; et al. A High Energy X-ray Diffraction Study of Amorphous Indomethacin. J. Pharm. Sci. 2022, 111, 818–824. [Google Scholar] [CrossRef]
- Dołęga, A.; Juszyńska-Gałązka, E.; Osiecka-Drewniak, N.; Natkański, P.; Kuśtrowski, P.; Krupa, A.; Zieliński, P.M. Study on the thermal performance of carbamazepine at different temperatures, pressures and atmosphere conditions. Thermochim. Acta 2021, 703, 178990. [Google Scholar] [CrossRef]
- Benmore, C.J. A Review of High-Energy X-ray Diffraction from Glasses and Liquids. ISRN Mater. Sci. 2012, 2012, 852905. [Google Scholar] [CrossRef]
- Hammersley, A.P.; Svensson, S.O.; Hanfland, M.; Fitch, A.N.; Hausermann, D. Two-dimensional detector software: From real detector to idealised image or two-theta scan. High Press. Res. 1996, 14, 235–248. [Google Scholar] [CrossRef]
- Qiu, X.; Thompson, J.W.; Billinge, S.J.L. PDFgetX2: A GUI-driven program to obtain pair distribution function from X-ray powder diffraction data. J. Appl. Crystallogr. 2004, 37, 678. [Google Scholar] [CrossRef]
- Skinner, L.B.; Benmore, C.J.; Parise, J.B. Area detector corrections for high quality synchrotron X-ray structure factor measurements. Nucl. Instrum. Methods Phys. Res. Sect. A Accel. Spectrometers Detect. Assoc. Equip. 2012, 662, 61–70. [Google Scholar] [CrossRef]
- Soper, A.K. Partial structure factors from disordered materials diffraction data: An approach using empirical potential structure refinement. Phys. Rev. B 2005, 72, 104204. [Google Scholar] [CrossRef]
- Soper, A.K. Joint structure refinement of X-ray and neutron diffraction data on disordered materials: Application to liquid water. J. Phys. Condens. Matter 2007, 19, 335206. [Google Scholar] [CrossRef]
- Dołęga, A.; Juszyńska-Gałązka, E.; Deptuch, A.; Baran, S.; Zieliński, P.M. Cold-crystallization and physical stability of glassy carbamazepine. Thermochim. Acta 2022, 707, 179100. [Google Scholar] [CrossRef]
- Dołęga, A.; Zieliński, P.M.; Osiecka-Drewniak, N. New Insight Into Thermodynamical Stability of Carbamazepine. J. Pharm. Sci. 2019, 108, 2654–2660. [Google Scholar] [CrossRef]
- Dołęga, A.; Zieliński, P.M. Kinetics of non-isothermal cold-crystallization of carbamazepine in the glassy state studied by DSC. J. Non-Cryst. Solids 2022, 575, 121198. [Google Scholar] [CrossRef]
- Dołęga, A.; Krupa, A.; Zieliński, P.M. Enhanced thermal stability of carbamazepine obtained by fast heating, hydration and re-crystallization from organic solvent solutions: A DSC and HPLC study. Thermochim. Acta 2020, 690, 178691. [Google Scholar] [CrossRef]
- Naima, Z.; Siro, T.; Juan-Manuel, G.-D.; Chantal, C.; René, C.; Jerome, D. Interactions between carbamazepine and polyethylene glycol (PEG) 6000: Characterisations of the physical, solid dispersed and eutectic mixtures. Eur. J. Pharm. Sci. 2001, 12, 395–404. [Google Scholar] [CrossRef]
- Grzesiak, A.L.; Lang, M.; Kim, K.; Matzger, A.J. Comparison of the four anhydrous polymorphs of carbamazepine and the crystal structure of form I. J. Pharm. Sci. 2003, 92, 2260–2271. [Google Scholar] [CrossRef] [PubMed]
- Suhasini, M.; Sailatha, E.; Gunasekaran, S.; Ramkumaar, G.R. Molecular structure and spectroscopic characterization of Carbamazepine with experimental techniques and DFT quantum chemical calculations. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 141, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Khankari, R.K.; Grant, D.J.W. Pharmaceutical hydrates. Thermochim. Acta 1995, 248, 61–79. [Google Scholar] [CrossRef]
- Metz, G.; Ziliox, M.; Smith, S.O. Towards quantitative CP-MAS NMR. Solid State Nucl. Magn. Reson. 1996, 7, 155–160. [Google Scholar] [CrossRef]
- Johnson, R.L.; Schmidt-Rohr, K. Quantitative solid-state 13C NMR with signal enhancement by multiple cross polarization. J. Magn. Reson. 2014, 239, 44–49. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atom | Coulomb Charges (e) | ε (kJ/mole) | σ (Å) |
|---|---|---|---|
| H1 | +0.25 | 0.00 | 0.00 |
| N1 & N2 | 0.00 | 0.70 | 3.20 |
| O1 | −0.50 | 0.65 | 3.10 |
| NMR Labels for Carbamazepine | Carbon Number | CDCl3 Solution | CBZ III | Dihydrate | Glass |
|---|---|---|---|---|---|
 | 15 | 157.19 | 159.0 | 158.6 | 159.1 157.6 |
| 1, 14 | 140.05 | 140.3 137.2 | 140.9 140.1 | 141.9 140.2 | |
| 6, 9 | 135.02 | 134.7 | 135.3 134.7 | 135.0 | |
| 7, 8 | 130.48 | 133.2 | 131.2 | ||
| 2, 13 | 129.59 | 132.1 | 129.9 | ||
| 4, 11 | 129.47 | 130.9 | 129.5 | 129.3 | |
| 5, 10 | 128.74 | 129.2 | 128.9 | ||
| 3, 12 | 127.76 | 127.3 | 126.8 126.0 |
| Structure | Tg (°C) | ∆H (J/g) | Onset (°C) | Tm (°C) |
|---|---|---|---|---|
| CBZ I | ||||
| Form I fusion | 113.95 | 190.38 | 191.38 | |
| CBZ III | ||||
| CBZ III fusion | 20.856 | 173.35 | 174.95 | |
| CBZ I cold-crystallization | 5.217 | 175.44 | 175.95 | |
| CBZ I fusion | 110.91 | 191.10 | 191.26 | |
| CBZ·2H2O | ||||
| Dehydration | 362.85 | 73.03 | 84.49, 95.35 | |
| Mixed phase melting | 7.085 | 155.99 | 164.47 | |
| CBZ I fusion | 95.052 | 190.75 | 191.16 | |
| CBZ glass | ||||
| Glass transition, onset | ||||
| Glass transition, midpoint | 42.83 | |||
| CBZ I cold-crystallization | 46.03 | 57.083 | 81.98 | 97.62 |
| CBZ I fusion | 102.21 | 189.00 | 190.31 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benmore, C.J.; Edwards, A.; Alderman, O.L.G.; Cherry, B.R.; Smith, P.; Smith, D.; Byrn, S.; Weber, R.; Yarger, J.L. The Structure of Liquid and Glassy Carbamazepine. Quantum Beam Sci. 2022, 6, 31. https://doi.org/10.3390/qubs6040031
Benmore CJ, Edwards A, Alderman OLG, Cherry BR, Smith P, Smith D, Byrn S, Weber R, Yarger JL. The Structure of Liquid and Glassy Carbamazepine. Quantum Beam Science. 2022; 6(4):31. https://doi.org/10.3390/qubs6040031
Chicago/Turabian StyleBenmore, Chris J., Angela Edwards, Oliver L. G. Alderman, Brian R. Cherry, Pamela Smith, Daniel Smith, Stephen Byrn, Richard Weber, and Jeffery L. Yarger. 2022. "The Structure of Liquid and Glassy Carbamazepine" Quantum Beam Science 6, no. 4: 31. https://doi.org/10.3390/qubs6040031
APA StyleBenmore, C. J., Edwards, A., Alderman, O. L. G., Cherry, B. R., Smith, P., Smith, D., Byrn, S., Weber, R., & Yarger, J. L. (2022). The Structure of Liquid and Glassy Carbamazepine. Quantum Beam Science, 6(4), 31. https://doi.org/10.3390/qubs6040031