
First Principles Calculations of the Optical Response of LiNiO2

 , , ,
, , ,  , ,
, ,  , , and
, , and
Abstract
1. Introduction
2. Computational Methods
3. Results and Discussions
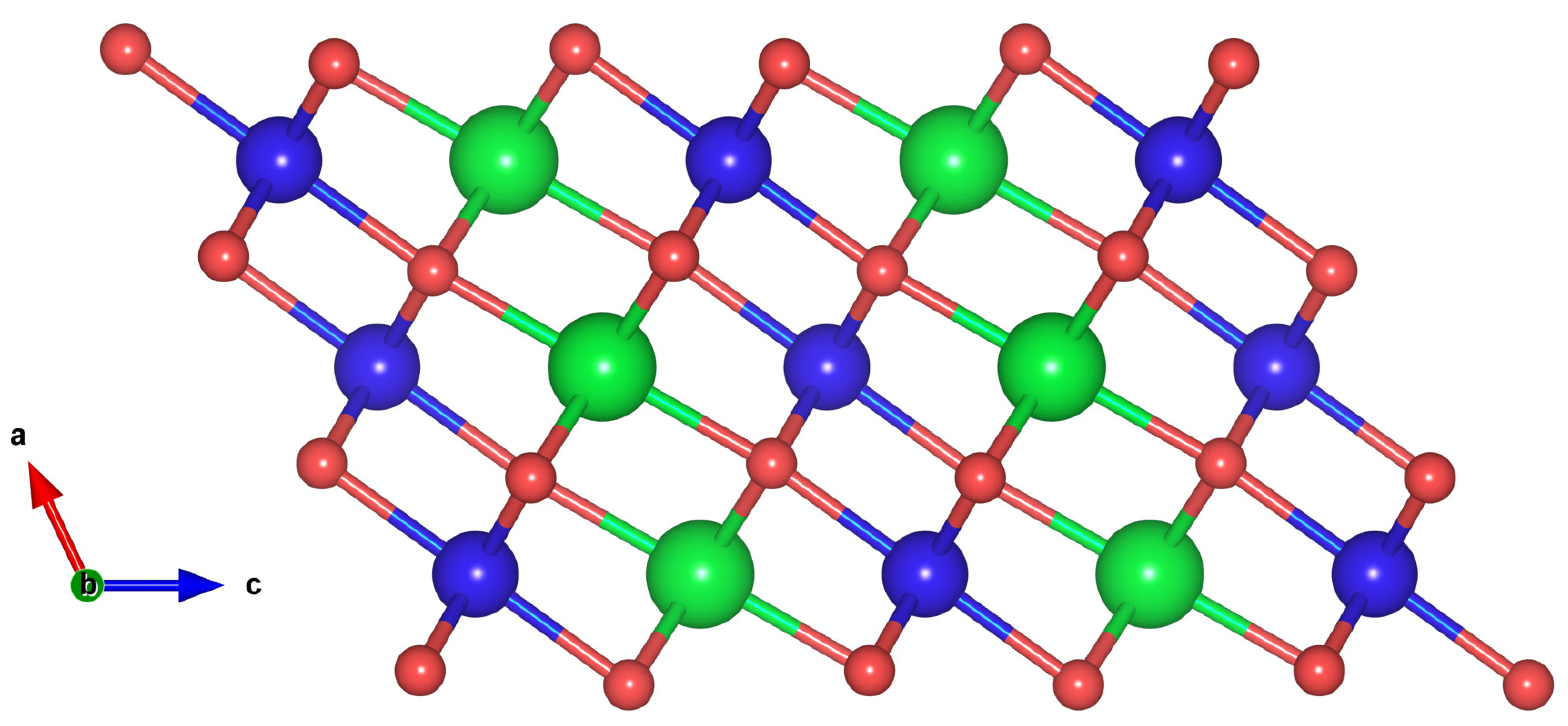
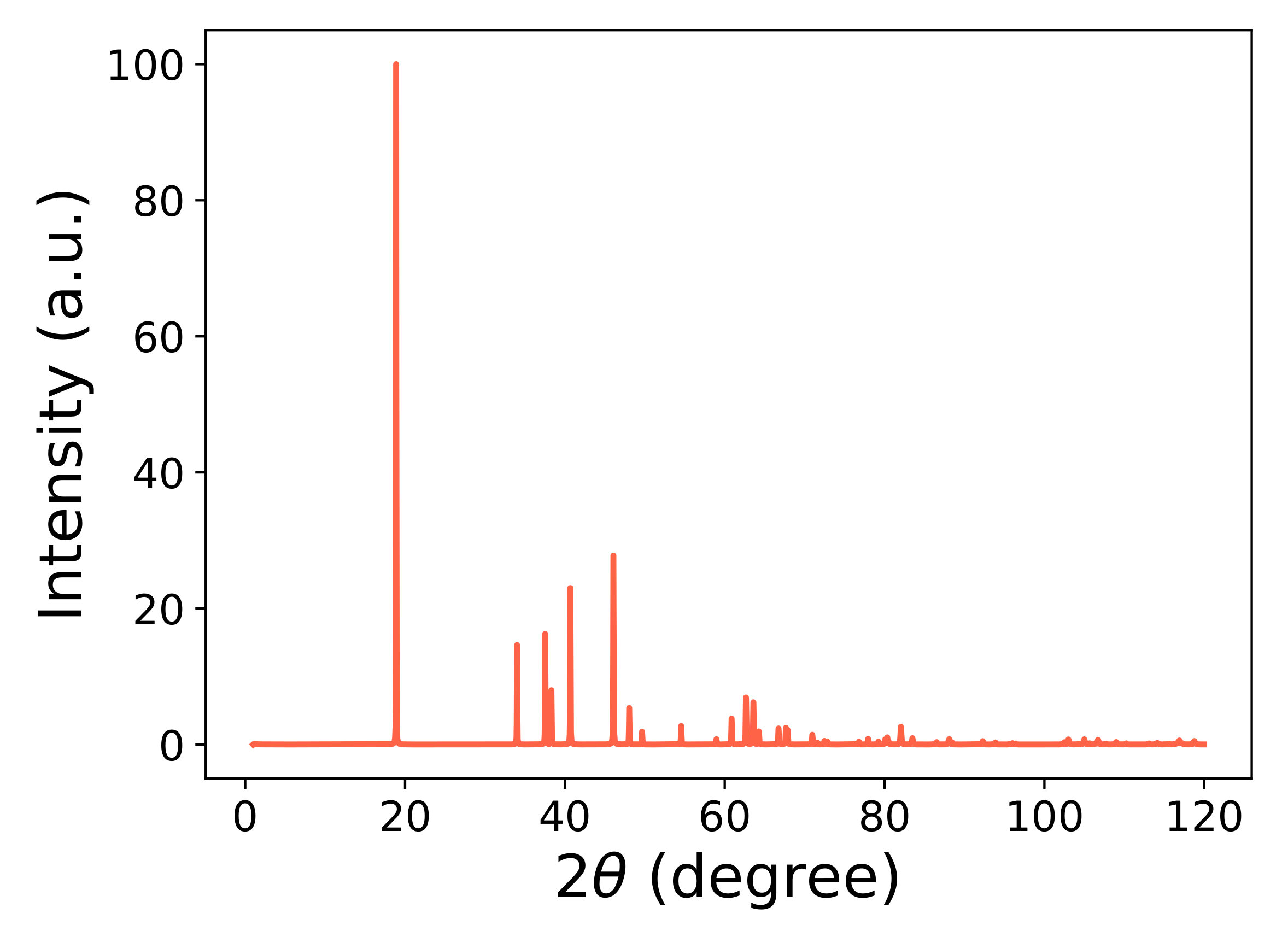
3.1. Structural Properties
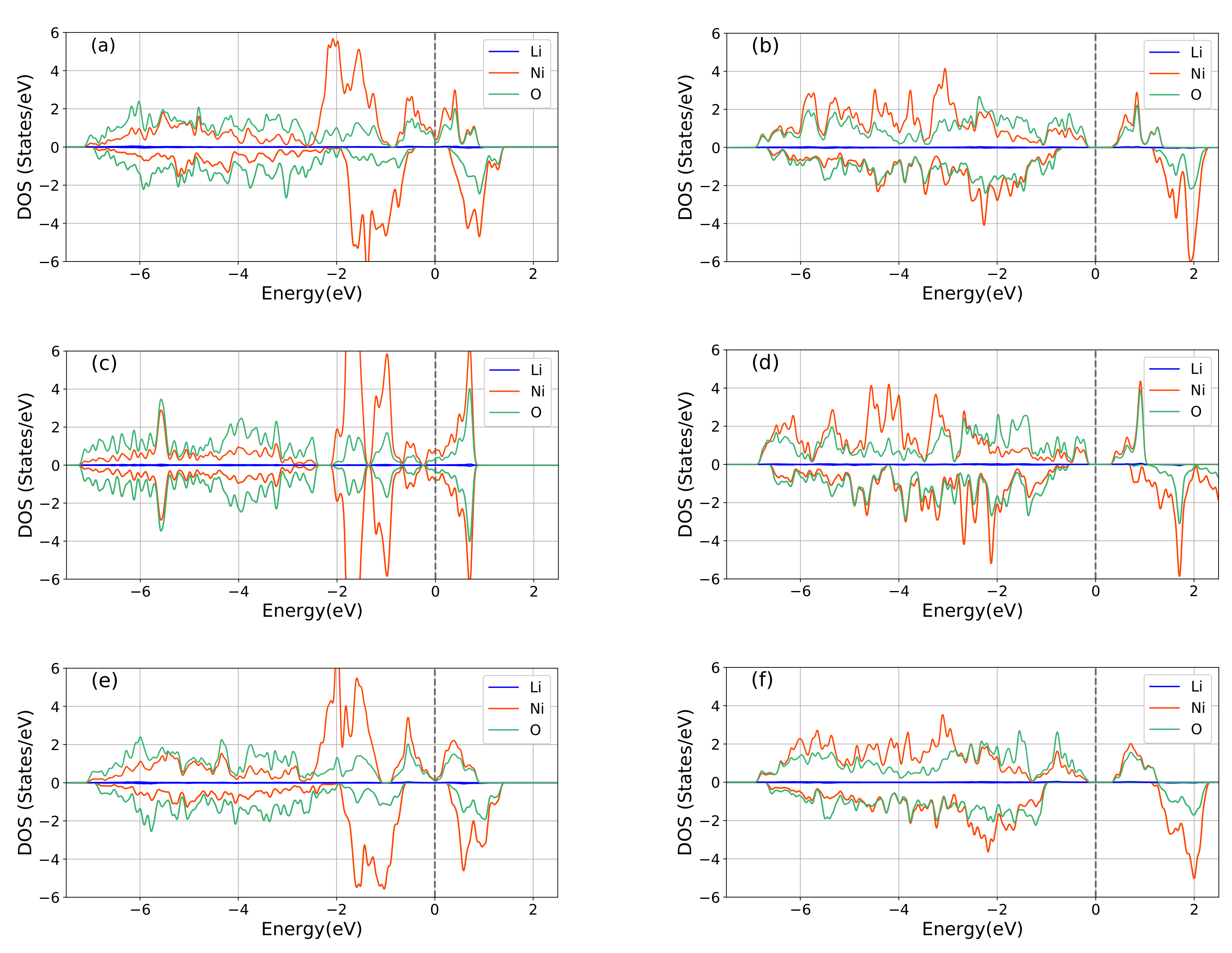
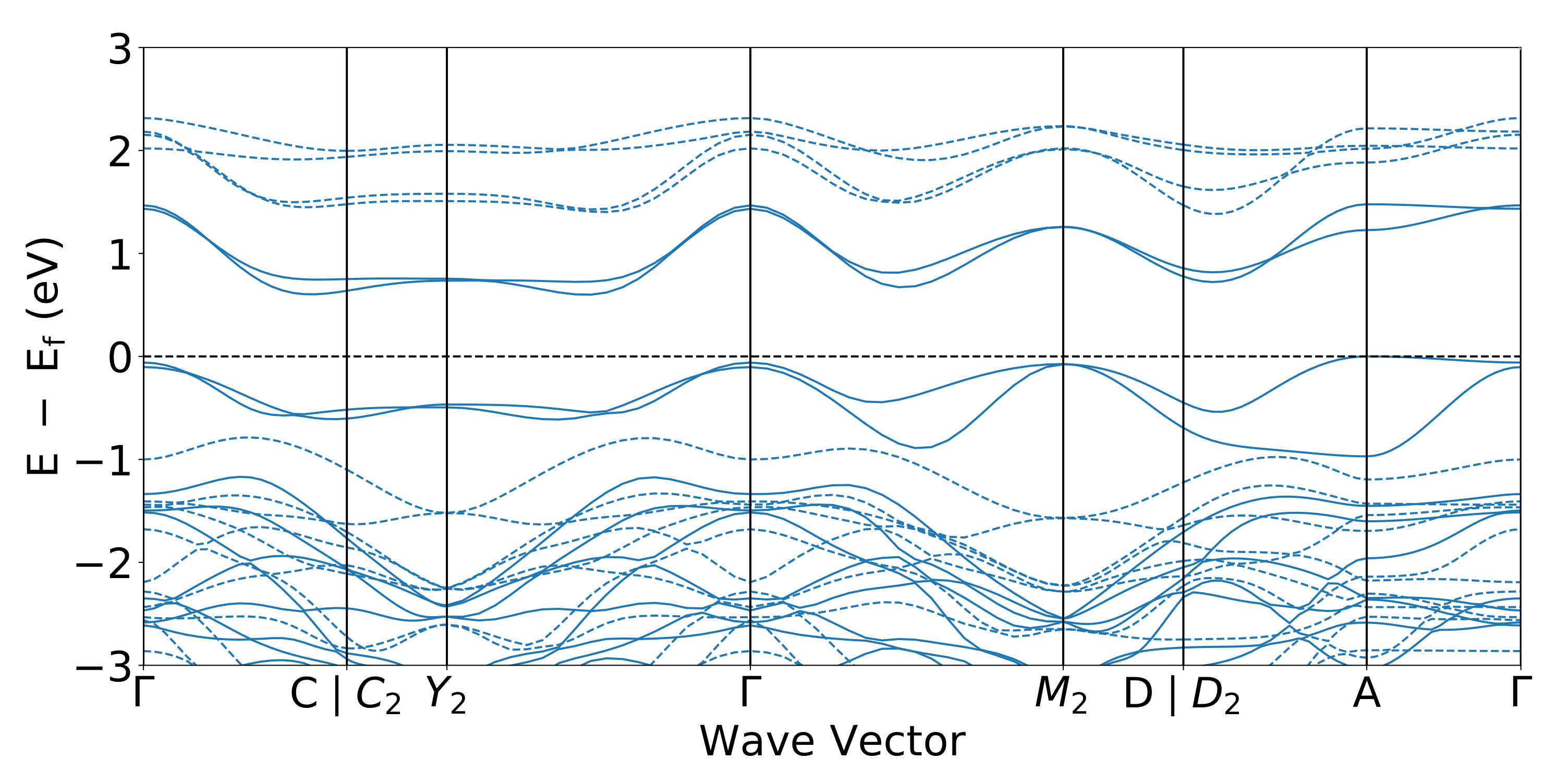
3.2. Electronic Structures
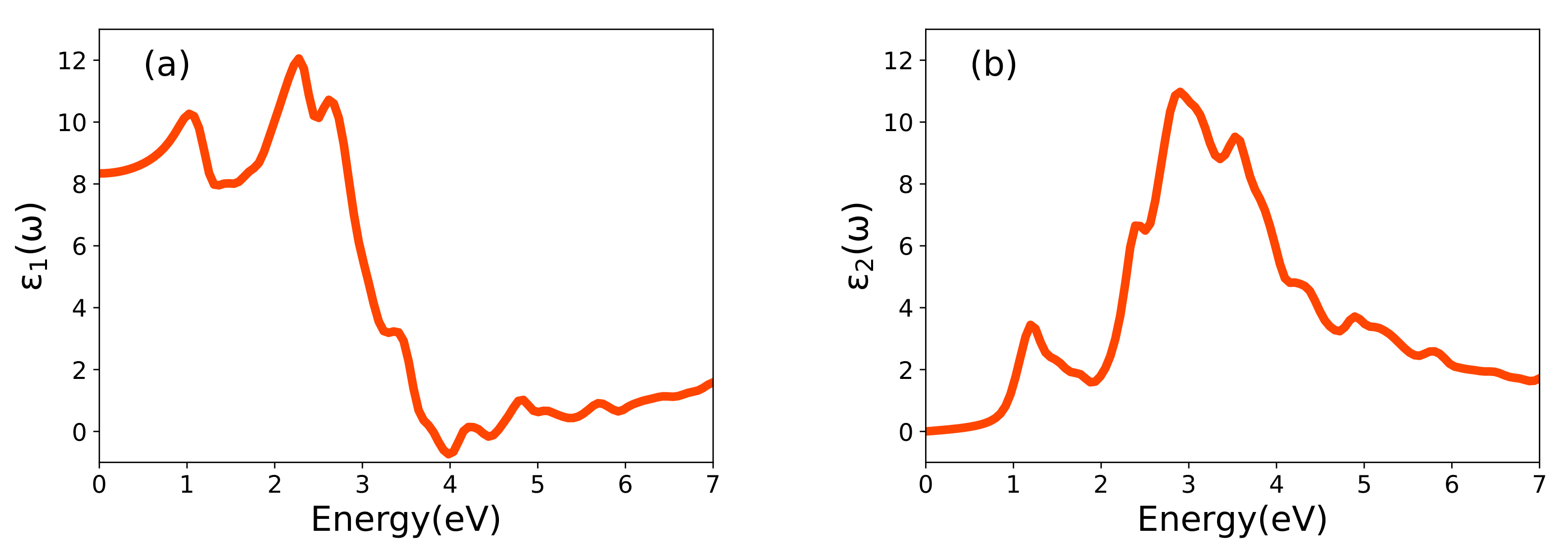
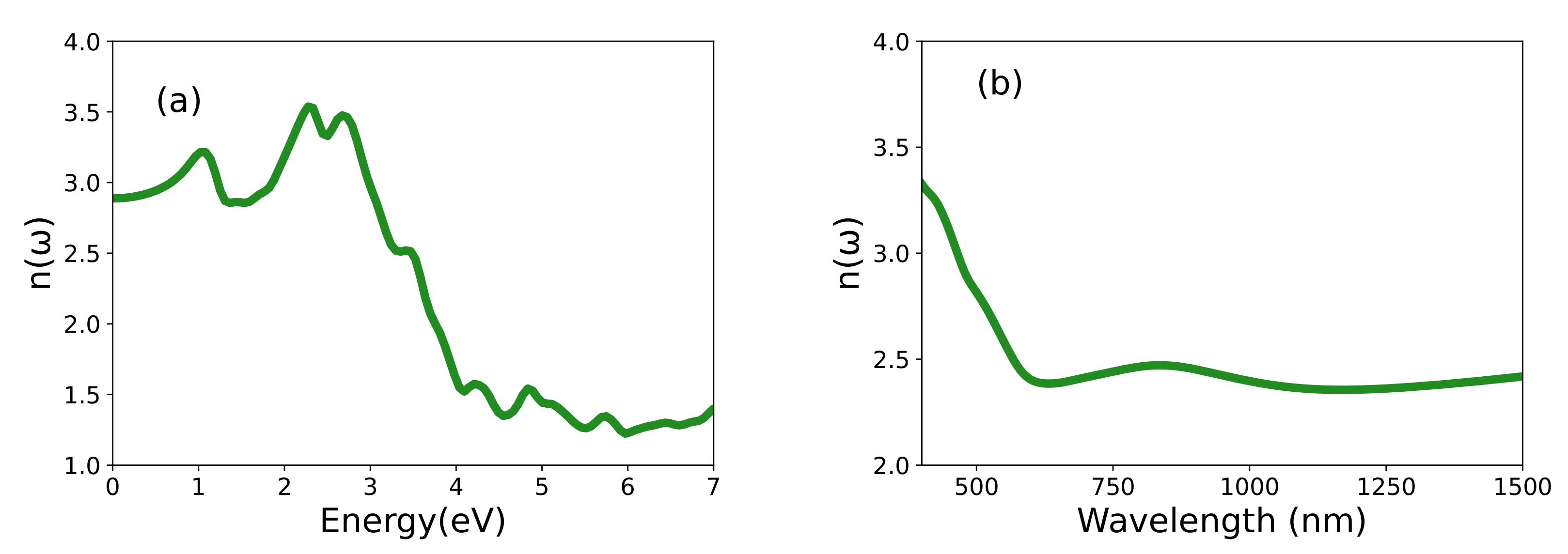
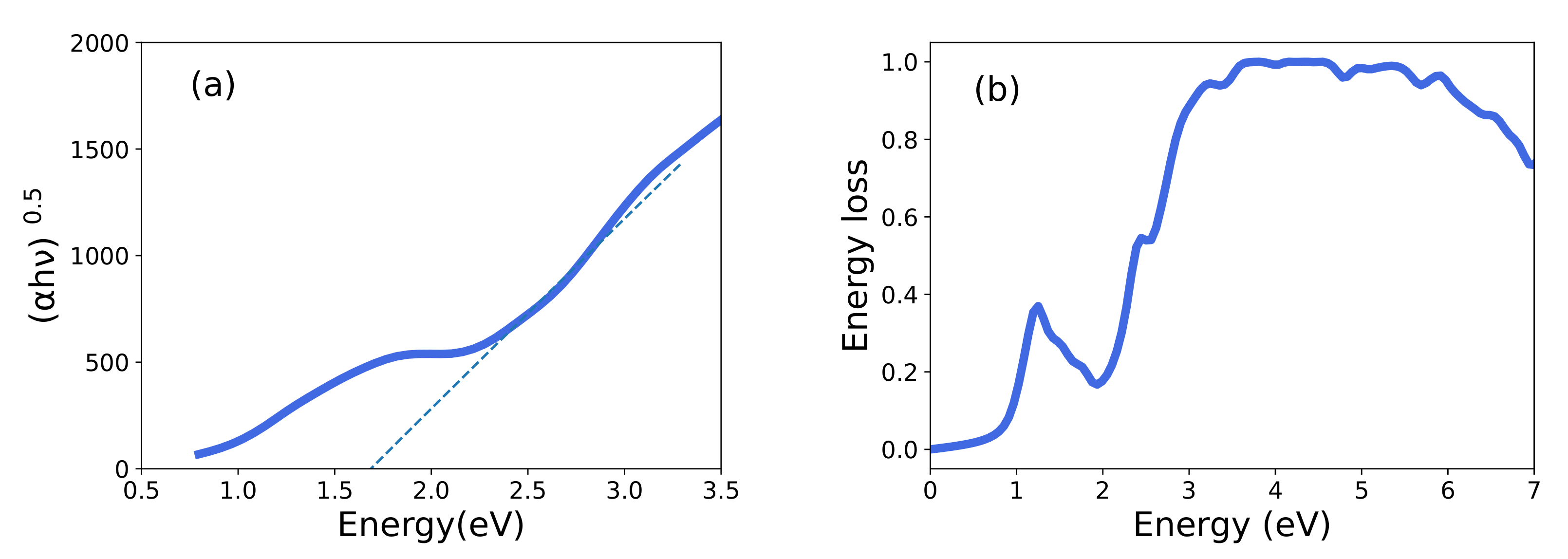
3.3. Optical Properties
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BZ | Brillouin Zone |
| DFT | Density Functional Theory |
| DOS | Density of states |
| GGA | Generalized Gradient approximation |
| LIB | Lithium-Ion Battery |
| LNO | Lithium Nickel Oxide |
| PAW | Projector-Augmented-Wave |
| PBE | Perdew–Burke–Ernzerhof |
| PDOS | Partial density of states |
| RIXS | Resonant Inelastic X-ray Scattering (RIXS) |
| SCAN | Strongly constrained and appropriately normed |
| XRD | X-ray diffraction |
References
- Larcher, D.; Tarascon, J. Towards greener and more sustainable batteries for electrical energy storage. Nat. Chem. 2015, 7, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, M.; James, C.; Seung-Taek, M.; Seung-Min, O.; Yang-Kook, S. Nickel-Rich and Lithium-Rich Layered Oxide Cathodes: Progress and Perspectives. Adv. Energy Mater. 2016, 6, 1501010. [Google Scholar] [CrossRef]
- Arumugam, M. A reflection on lithium-ion battery cathode chemistry. Nature 2020, 11, 1550. [Google Scholar] [CrossRef]
- Zhang, H.; Li, C.; Eshetu, G.; Laruelle, S.; Grugeon, S.; Zaghib, K.; Julien, C.; Mauger, A.; Guyomard, D.; Rojo, T.; et al. From Solid-Solution Electrodes and the Rocking-Chair Concept to Today’s Batteries. Angew. Chem. Int. Ed. 2020, 59, 534–538. [Google Scholar] [CrossRef] [PubMed]
- Du Pasquier, A.; Plitz, I.; Menocal, S.; Amatucci, G. A comparative study of Li-ion battery, supercapacitor and nonaqueous asymmetric hybrid devices for automotive applications. J. Power Sources 2003, 115, 171–178. [Google Scholar] [CrossRef]
- Kim, Y.; Seong, W.M.; Manthiram, A. Cobalt-free, high-nickel layered oxide cathodes for lithium-ion batteries: Progress, challenges, and perspectives. Energy Storage Mater. 2021, 34, 250–259. [Google Scholar] [CrossRef]
- Schipper, F.; Erickson, E.M.; Erk, C.; Shin, J.; Francois, F.C.; Aurbach, D. Review—Recent Advances and Remaining Challenges for Lithium Ion Battery Cathodes. J. Electrochem. Soc. 2016, 164, A6220–A6228. [Google Scholar] [CrossRef]
- Bianchini, M.; Roca-Ayats, M.; Hartmann, P.; Brezesinski, T.; Janek, J. There and Back Again—The Journey of LiNiO2 as a Cathode Active Material. Angew. Chem. Int. Ed. 2019, 58, 10434–10458. [Google Scholar] [CrossRef]
- Anisimov, V.I.; Zaanen, J.; Andersen, O.K. Band theory and Mott insulators: Hubbard U instead of Stoner I. Phys. Rev. B 1991, 44, 943–954. [Google Scholar] [CrossRef]
- Devi, A.A.S.; Nokelainen, J.; Barbiellini, B.; Devaraj, M.; Alatalo, M.; Bansil, A. Re-examining the giant magnetization density in α -Fe16N2 with the SCAN+U method. Phys. Chem. Chem. Phys. 2022, 24, 17879–17884. [Google Scholar] [CrossRef]
- El-Bana, M.; El Radaf, I.; Fouad, S.; Sakr, G. Structural and optoelectrical properties of nanostructured LiNiO2 thin films grown by spray pyrolysis technique. J. Alloys Compd. 2017, 705, 333–339. [Google Scholar] [CrossRef]
- Wang, Y.J.; Barbiellini, B.; Lin, H.; Das, T.; Basak, S.; Mijnarends, P.E.; Kaprzyk, S.; Markiewicz, R.S.; Bansil, A. Lindhard and RPA susceptibility computations in extended momentum space in electron-doped cuprates. Phys. Rev. B 2012, 85, 224529. [Google Scholar] [CrossRef]
- Barbiellini, B.; Hancock, J.N.; Monney, C.; Joly, Y.; Ghiringhelli, G.; Braicovich, L.; Schmitt, T. Inelastic x-ray scattering from valence electrons near absorption edges of FeTe and TiSe2. Phys. Rev. B 2014, 89, 235138. [Google Scholar] [CrossRef]
- Li, N.; Sallis, S.; Papp, J.K.; Wei, J.; McCloskey, B.D.; Yang, W.; Tong, W. Unraveling the cationic and anionic redox reactions in a conventional layered oxide cathode. ACS Energy Lett. 2019, 4, 2836–2842. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Tuccillo, M.; Palumbo, O.; Pavone, M.; Muñoz-García, A.; Paolone, A.; Brutti, S. Analysis of the Phase Stability of LiMO2 Layered Oxides (M = Co, Mn, Ni). Crystals 2000, 10, 526. [Google Scholar] [CrossRef]
- Wang, L.; Maxisch, T.; Ceder, G. Analysis of the Phase Stability of LiMO2 Layered Oxides (M = Co, Mn, Ni). Phys. Rev. B 2006, 73, 195107. [Google Scholar] [CrossRef]
- Jain, A.; Hautier, G.; Ping Ong, S.; Moore, C.; Fischer, C.; Persson, K.; Ceder, G. Formation enthalpies by mixing GGA and GGA + U calculations. Phys. Rev. B 2011, 84, 045115. [Google Scholar] [CrossRef]
- Sun, J.; Ruzsinszky, A.; Perdew, J.P. Strongly Constrained and Appropriately Normed Semilocal Density Functional. Phys. Rev. Lett. 2015, 115, 036402. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Remsing, R.C.; Zhang, Y.; Sun, Z.; Ruzsinszky, A.; Peng, H.; Yang, Z.; Paul, A.; Waghmare, U.; Wu, X.; et al. Accurate first-principles structures and energies of diversely bonded systems from an efficient density functional. Nat. Chem. 2016, 8, 831–836. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Gajdoš, M.; Hummer, K.; Kresse, G.; Furthmüller, J.; Bechstedt, F. Linear optical properties in the projector-augmented wave methodology. Phys. Rev. B 2006, 73, 045112. [Google Scholar] [CrossRef]
- Liu, Y.; Lian, J.; Sun, Z.; Zhao, M.; Shi, Y.; Song, H. The first-principles study for the novel optical properties of LiTi2O4, Li4Ti5O12, Li2Ti2O4 and Li7Ti5O12. Chem. Phys. Lett. 2017, 677, 114–119. [Google Scholar] [CrossRef]
- Altarelli, M.; Dexter, D.L.; Nussenzveig, H.M.; Smith, D.Y. Superconvergence and Sum Rules for the Optical Constants. Phys. Rev. B 1972, 6, 4502–4509. [Google Scholar] [CrossRef]
- Laubach, S.; Laubach, S.; Schmidt, P.C.; Ensling, D.; Schmid, S.; Jaegermann, W.; Thißen, A.; Nikolowski, K.; Ehrenberg, H. Changes in the crystal and electronic structure of LiCoO2 and LiNiO2 upon Li intercalation and de-intercalation. Phys. Chem. Chem. Phys. 2009, 11, 3278–3289. [Google Scholar] [CrossRef]
- Hoang, K.; Johannes, M.D. Defect chemistry in layered transition-metal oxides from screened hybrid density functional calculations. J. Mater. Chem. A 2014, 2, 5224–5235. [Google Scholar] [CrossRef]
- Hoang, K.; Johannes, M.D. Defect physics in complex energy materials. J. Phys. Condens. Matter 2018, 30, 293001. [Google Scholar] [CrossRef]
- Chakraborty, A.; Dixit, M.; Aurbach, D.; Major, D.T. Predicting accurate cathode properties of layered oxide materials using the SCAN meta-GGA density functional. NPJ Comput. Mater. 2018, 4, 1–9. [Google Scholar] [CrossRef]
- Jain, A.; Ong, S.P.; Hautier, G.; Chen, W.; Richards, W.D.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G.; et al. Commentary: The Materials Project: A materials genome approach to accelerating materials innovation. APL Mater. 2013, 1, 011002. [Google Scholar] [CrossRef]
- Välikangas, J.; Laine, P.; Hietaniemi, M.; Hu, T.; Tynjälä, P.; Lassi, U. Precipitation and Calcination of High-Capacity LiNiO2 Cathode Material for Lithium-Ion Batteries. Appl. Sci. 2020, 10, 8988. [Google Scholar] [CrossRef]
- Sicolo, S.; Mock, M.; Bianchini, M.; Albe, K. Furthermore, Yet It Moves: LiNiO2, a Dynamic Jahn–Teller System. Chem. Mater. 2020, 32, 10096–10103. [Google Scholar] [CrossRef]
- Setyawan, W.; Curtarolo, S. High-throughput electronic band structure calculations: Challenges and tools. Comput. Mater. Sci. 2010, 49, 299–312. [Google Scholar] [CrossRef]
- Fox, M. Optical Properties of Solids, 2nd ed.; Oxford Master Series in Condensed Matter Physics; Oxford University Press: Oxford, UK, 2010. [Google Scholar]
- Craco, L.; Leoni, S. Electrodynamics and quantum capacity of LixFePO4 battery material. Appl. Phys. Lett. 2011, 99, 192103. [Google Scholar] [CrossRef]
- Radha, S.K.; Lambrecht, W.R.L.; Cunningham, B.; Grüning, M.; Pashov, D.; van Schilfgaarde, M. Optical response and band structure of LiCoO2 including electron-hole interaction effects. Phys. Rev. B 2021, 104, 115120. [Google Scholar] [CrossRef]
- Scafetta, M.D.; Cordi, A.M.; Rondinelli, J.M.; May, S.J. Band structure and optical transitions in LaFeO3: Theory and experiment. J. Phys. Condens. Matter 2014, 26, 505502. [Google Scholar] [CrossRef]
- Tan, G.; DeNoyer, L.; French, R.; Guittet, M.; Gautier-Soyer, M. Kramers–Kronig transform for the surface energy loss function. J. Electron Spectrosc. Relat. Phenom. 2005, 142, 97–103. [Google Scholar] [CrossRef]
- Jia, Y.; Ye, Y.; Liu, J.; Zheng, S.; Lin, W.; Wang, Z.; Li, S.; Pan, F.; Zheng, J. Breaking the energy density limit of LiNiO2: Li2NiO3 or Li2NiO2? Sci. China Mater. 2022, 65, 913–919. [Google Scholar] [CrossRef]
- Prange, M.P.; Rehr, J.J.; Rivas, G.; Kas, J.J.; Lawson, J.W. Real space calculation of optical constants from optical to x-ray frequencies. Phys. Rev. B 2009, 80, 155110. [Google Scholar] [CrossRef]
- Hafiz, H.; Suzuki, K.; Barbiellini, B.; Tsuji, N.; Yabuuchi, N.; Yamamoto, K.; Orikasa, Y.; Uchimoto, Y.; Sakurai, Y.; Sakurai, H.; et al. Tomographic reconstruction of oxygen orbitals in lithium-rich battery materials. Nature 2021, 564, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Chabaud, S.; Bellin, C.; Mauri, F.; Loupias, G.; Rabii, S.; Croguennec, L.; Pouillerie, C.; Delmas, C.; Buslaps, T. Electronic density distorsion of NiO2 due to intercalation by Li. J. Phys. Chem. Solids 2004, 65, 241–243. [Google Scholar] [CrossRef]
- Suzuki, K.; Otsuka, Y.; Hoshi, K.; Sakurai, H.; Tsuji, N.; Yamamoto, K.; Yabuuchi, N.; Hafiz, H.; Orikasa, Y.; Uchimoto, Y.; et al. Magnetic Compton Scattering Study of Li-Rich Battery Materials. Condens. Matter 2022, 7, 4. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lattice Parameters | GGA | GGA+U | SCAN | SCAN+U | Exp. |
|---|---|---|---|---|---|
| a (Å) | 4.839 | 4.799 | 4.779 | 4.749 | 4.99 |
| b (Å) | 2.799 | 2.789 | 2.778 | 2.765 | 2.83 |
| c (Å) | 5.109 | 5.129 | 5.093 | 5.054 | 5.07 |
| () | 112.701 | 112.429 | 112.064 | 112.195 | 109.7 |
| V (Å/atom) | 7.980 | 7.932 | 7.833 | 7.681 | 8.426 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kothalawala, V.N.; Sasikala Devi, A.A.; Nokelainen, J.; Alatalo, M.; Barbiellini, B.; Hu, T.; Lassi, U.; Suzuki, K.; Sakurai, H.; Bansil, A. First Principles Calculations of the Optical Response of LiNiO2. Condens. Matter 2022, 7, 54. https://doi.org/10.3390/condmat7040054
Kothalawala VN, Sasikala Devi AA, Nokelainen J, Alatalo M, Barbiellini B, Hu T, Lassi U, Suzuki K, Sakurai H, Bansil A. First Principles Calculations of the Optical Response of LiNiO2. Condensed Matter. 2022; 7(4):54. https://doi.org/10.3390/condmat7040054
Chicago/Turabian StyleKothalawala, Veenavee Nipunika, Assa Aravindh Sasikala Devi, Johannes Nokelainen, Matti Alatalo, Bernardo Barbiellini, Tao Hu, Ulla Lassi, Kosuke Suzuki, Hiroshi Sakurai, and Arun Bansil. 2022. "First Principles Calculations of the Optical Response of LiNiO2" Condensed Matter 7, no. 4: 54. https://doi.org/10.3390/condmat7040054
APA StyleKothalawala, V. N., Sasikala Devi, A. A., Nokelainen, J., Alatalo, M., Barbiellini, B., Hu, T., Lassi, U., Suzuki, K., Sakurai, H., & Bansil, A. (2022). First Principles Calculations of the Optical Response of LiNiO2. Condensed Matter, 7(4), 54. https://doi.org/10.3390/condmat7040054