
Hepcidin Deficiency Disrupts Iron Homeostasis and Induces Ferroptosis in Zebrafish Liver

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Zebrafish Husbandry
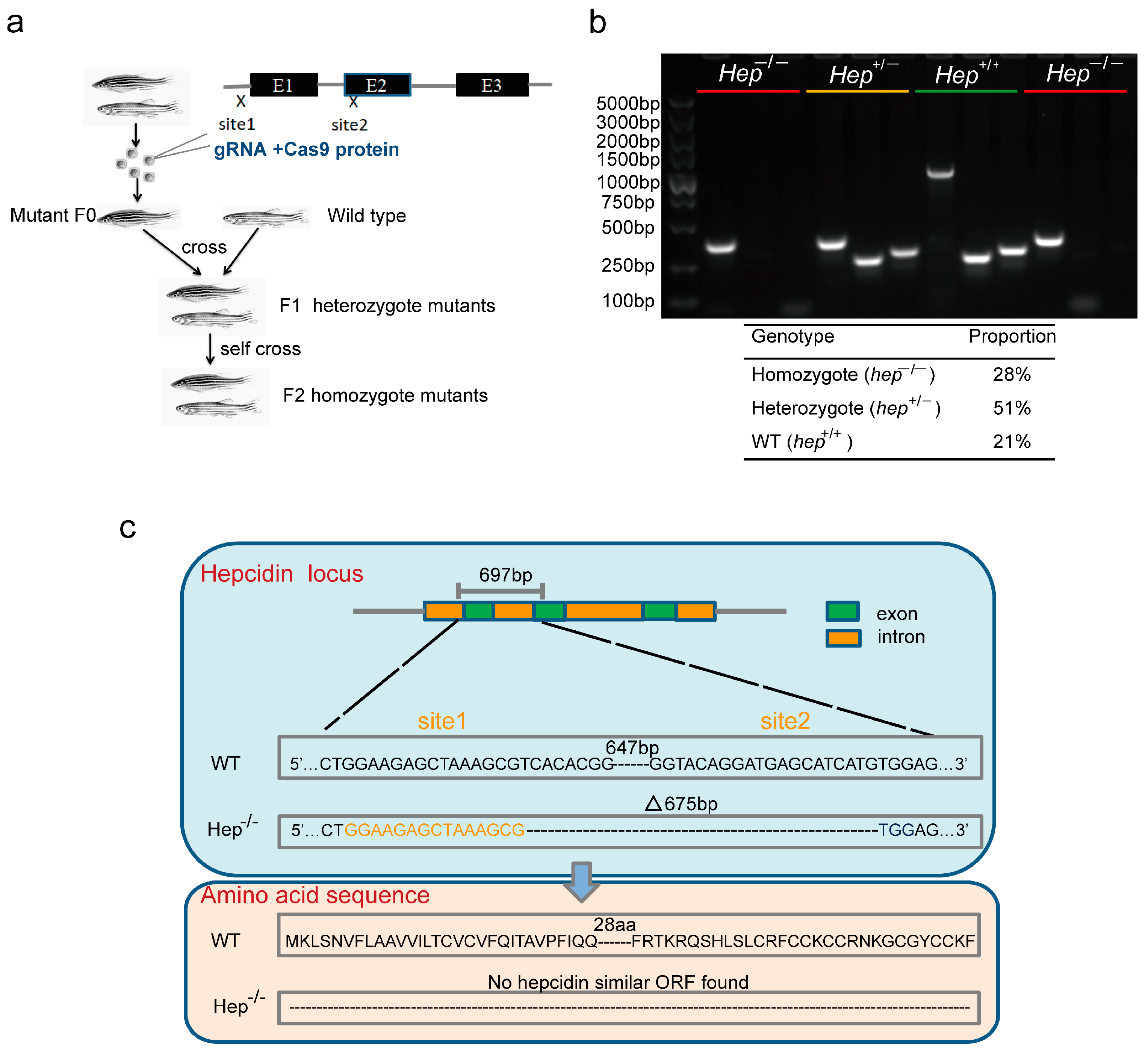
2.2. Construction of Zebrafish Mutant by CRISPR/cas9 Genome Editing
2.3. Genotyping
2.4. RNA Isolation and Sequencing
2.5. Liver Bulk RNA Sequencing Data Processing
2.6. Enrichment Analysis
2.7. Visualization of Differential Gene Expression
2.8. Statistical Analysis
2.9. Western Blot Assay
2.10. Iron Measurement by ICP-MS
2.11. Tissue Section Preparation and Perls’ Prussian Blue Staining
3. Results
3.1. Zebrafish Lacking Hepcidin Exhibit Iron Overload
3.2. RNA-Seq Reveals Ferroptosis and Key Regulatory Genes in hep−/− Zebrafish Livers
3.3. Validation of Key Differentially Expressed Genes via Western Blotting
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DEGs | Differentially expressed genes |
| WT | Wild-type |
| hep−/− | Homozygote of hepcidin knockout zebrafish |
| hep+/− | Heterozygote of hepcidin knockout zebrafish |
| FPN | Ferroportin |
| SLC40A1 | Solute carrier family 40 member 1 |
| GPX4 | Glutathione peroxidase 4 |
| Fth1 | Ferritin heavy chain 1 |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| TFR | Transferrin receptors |
| DMT1 | Divalent metal transporter 1 |
| ORF | Open reading frame |
| ICP-MS | Inductively coupled plasma mass spectrometry |
| PCA | Principal component analysis |
Appendix A
References
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to tango: Regulation of Mammalian iron metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Ru, Q.; Li, Y.S.; Chen, L.; Wu, Y.X.; Min, J.X.; Wang, F.D. Iron homeostasis and ferroptosis in human diseases: Mechanisms and therapeutic prospects. Signal Transduct Target Ther. 2024, 9, 271–336. [Google Scholar] [PubMed]
- Li, L.; Holscher, C.; Chen, B.B.; Zhang, Z.F.; Liu, Y.Z. Hepcidin treatment modulates the expression of divalent metal transporter-1, ceruloplasmin, and ferroportin-1 in the rat cerebral cortex and hippocampus. Biol. Trace Elem. Res. 2011, 143, 1581–1593. [Google Scholar] [CrossRef]
- Park, C.H.; Valore, E.V.; Waring, A.J.; Ganz, T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J. Biol. Chem. 2001, 276, 7806–7810. [Google Scholar] [CrossRef]
- Krause, A.; Neitz, S.; Mägert, H.J.; Schulz, A.; Forssmann, W.G.; Knappe, P.S.; Adermann, K. LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett. 2000, 480, 147–150. [Google Scholar] [CrossRef]
- Fleming, R.E.; Sly, W.S. Hepcidin: A putative iron-regulatory hormone relevant to hereditary hemochromatosis and the anemia of chronic disease. Proc. Natl. Acad. Sci. USA 2001, 98, 8160. [Google Scholar] [CrossRef]
- Weinstein, D.A.; Roy, C.N.; Fleming, M.D.; Loda, M.F.; Wolfsdorf, J.I.; Andrews, N.C. Inappropriate expression of hepcidin is associated with iron refractory anemia: Implications for the anemia of chronic disease. Blood 2002, 100, 3776–3781. [Google Scholar] [CrossRef]
- Nicolas, G.; Bennoun, M.; Devaux, I.; Vaulont, S. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc. Natl. Acad. Sci. USA 2001, 98, 8780–8785. [Google Scholar] [CrossRef]
- Lin, W.; Liu, S.S.; Hu, L.L.; Zhang, S.C. Characterization and bioactivity of hepcidin-2 in zebrafish: Dependence of antibacterial activity upon disulfide bridges. Peptides 2014, 57, 36–42. [Google Scholar] [CrossRef]
- Lauth, X.; Babon, J.J.; Stannard, J.A.; Singh, S.; Nizet, V.; Carlberg, J.M.; Ostland, V.E.; Pennington, M.W.; Norton, R.S.; Westerman, M.E. Bass hepcidin synthesis, solution structure, antimicrobial activities and synergism, and in vivo hepatic response to bacterial infections. J. Biol. Chem. 2005, 280, 9272–9282. [Google Scholar] [CrossRef]
- Domenico, I.D.; Ward, D.M.; Patti, M.C.B.; Jeong, S.Y.; David, S.; Musci, G.; Kaplan, J. Ferroxidase activity is required for the stability of cell surface ferroportin in cells expressing GPI-ceruloplasmin. Embo J. 2007, 26, 2823–2831. [Google Scholar] [CrossRef] [PubMed]
- Fleming, M.D. The regulation of hepcidin and its effects on systemic and cellular iron metabolism. Hematol. Am. Soc. Hematol. Educ. Program 2008, 2008, 151–158. [Google Scholar] [CrossRef]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef]
- Viatte, L.; Lesbordes-Brion, J.C.; Lou, D.Q.; Bennoun, M.; Nicolas, G.; Kahn, A.; Canonne-Hergaux, F.; Vaulont, S. Deregulation of proteins involved in iron metabolism in hepcidin-deficient mice. Blood 2005, 105, 4861–4864. [Google Scholar] [CrossRef]
- Hu, R.; Li, G.F.; Hu, P.; Niu, H.B.; Li, W.H.; Jiang, S.W.; Guan, G.J.; Xu, Q.H.; Liu, M.L.; Chen, L.B. bmp10 maintains cardiac function by regulating iron homeostasis. J. Genet. Genom. 2024, 51, 1459–1473. [Google Scholar] [CrossRef]
- Skjørringe, T.; Burkhart, A.; Johnsen, K.B.; Moos, T. Divalent metal transporter 1 (DMT1) in the brain: Implications for a role in iron transport at the blood-brain barrier, and neuronal and glial pathology. Front. Mol. Neurosci. 2015, 8, 1–13. [Google Scholar]
- Lim, D.; Kim, K.S.; Jeong, J.H.; Marques, O.; Kim, H.J.; Song, M.; Le, T.H.; Kim, J.; Choi, H.S.; Min, J.J.; et al. The hepcidin-ferroportin axis controls the iron content of Salmonella-containing vacuoles in macrophages. Nat. Commun. 2018, 9, 2091–2104. [Google Scholar] [CrossRef]
- Brunet, S.; Thibault, L.; Delvin, E.; Yotov, W.; Bendayan, M.; Levy, E. Dietary iron overload and induced lipid peroxidation are associated with impaired plasma lipid transport and hepatic sterol metabolism in rats. Hepatology 1999, 29, 1809–1817. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Stockwell, B.R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 2022, 185, 2401–2421. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Lei, G.; Mao, C.; Yan, Y.L.; Zhuang, L.; Gan, B.Y. Ferroptosis, radiotherapy, and combination therapeutic strategies. Protein Cell 2021, 12, 836–857. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef]
- Yang, W.Y.; Peng, M.J.; Wang, Y.Q.; Zhang, X.W.; Li, W.; Zhai, X.; Wu, Z.C.; Hu, P.; Chen, L.B. Deletion of hepcidin disrupts iron homeostasis and hematopoiesis in zebrafish embryogenesis. Development 2025, 152, dev204307. [Google Scholar] [CrossRef]
- Wilkinson, R.N.; Elworthy, S.; Ingham, P.W.; van Eeden, F.J.M. A method for high-throughout PCR-based genotyping of larval zebrafish tail biopsies. Bio Tech. 2013, 55, 314–316. [Google Scholar]
- Xin, Y.J.; Gao, H.; Wang, J.; Qiang, Y.Z.; Imam, M.U.; Li, Y.; Wang, J.Y.; Zhang, R.; Zhang, H.; Yingying, Y.; et al. Manganese transporter Slc39a14 deficiency revealed its key role in maintaining manganese homeostasis in mice. Cell Discov. 2017, 3, 17025. [Google Scholar] [CrossRef]
- Fei, Y.Y.; Wang, Q.; Lu, J.G.; Ouyang, L.Y.; Hu, Q.Q.; Chen, L.B. New insights into the antimicrobial mechanism of LEAP2 mutant zebrafish under Aeromonas hydrophila infection using transcriptome analysis. Fish Shellfish. Immunol. 2023, 143, 109225. [Google Scholar] [CrossRef]
- Gupta, A.; Balakrishnan, B.; Karki, S.; Slayton, M.; Jash, S.; Banerjee, S.; Grahn, T.H.M.; Jambunathan, S.; Disney, S.; Hussein, H.; et al. Human CIDEC transgene improves lipid metabolism and protects against high-fat diet-induced glucose intolerance in mice. J. Biol. Chem. 2022, 298, 102347. [Google Scholar] [CrossRef]
- Gao, G.; Li, J.; Zhang, Y.T.; Chang, Y.Z. Cellular Iron Metabolism and Regulation. Adv. Exp. Med. Biol. 2019, 1173, 21–32. [Google Scholar]
- Jiang, Y.; Yan, L.L.; Wang, X.; Zhu, G.X.; Xu, Y.J. Hepcidin inhibition on the effect of osteogenesis in zebrafish. Biochem. Biophys. Res. Commun. 2016, 476, 1–6. [Google Scholar] [CrossRef]
- Das, B.K.; Wang, L.; Fujiwara, T.; Zhou, J.; Burns, N.A.; Krager, K.J.; Lan, R.; Mackintosh, S.G.; Edmondson, R.; Jennings, M.L.; et al. Transferrin receptor 1-mediated iron uptake regulates bone mass in mice via osteoclast mitochondria and cytoskeleton. Elife 2022, 11, 73539–73570. [Google Scholar] [CrossRef] [PubMed]
- Kubo, Y. A new world of heme function. Pflügers Arch. Eur. J. Physiol. 2020, 472, 547–548. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence (5′-3′) | Location in GRCz11 | Primer Binding Location | Product Size (bp) |
|---|---|---|---|---|
| Hep-F1 | GATTAAAGGGACTACGCCGAATG | chr16:50,289,518-50,289,540 | 5′ UTR | 986 |
| Hep-R1 | TAGGCCAAAATCATATCCCTGT | chr16:50,290,678-50,290,699 | Intron2 | |
| Hep-F2 | CTTCCAGATCACAGCCGTTCCCTTCATACAG | chr16:50,290,064-50,290,094 | Exon1 | 170 |
| Hep-R2 | GTGAATGTGTATTGTCTATATGTCCCCATAG | chr16:50,290,203-50,290,233 | Intron1 | |
| Hep-F3 | AAATATCAGAGCCGAGCAGAAG | chr16:50,289,922-50,289,943 | Exon1 | 306 |
| Hep-R3 | GTGTATTGTCTATATGTCCCCATAGG | chr16:50,290,202-50,290,227 | Intron1 | |
| sgRNA1 | GATCACTAATACGACTCACTAT GGAAGAGCTAAAGCGTCACAAGTTTTAGAGCTAGAAATAGC | chr16:50,289,873-50,289,892 | 5’ UTR | 123 |
| sgRNA-scaffold | AAAAGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAAC | |||
| sgRNA2 | GATCACTAATACGACTCACTAT GGTACAGGATGAGCATCATGAGTTTTAGAGCTAGAAATAGC | chr16:50,290,543-50,290,562 | Exon2 | 123 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, M.; Peng, M.; Ma, J.; Hu, R.; Xu, Q.; Hu, P.; Chen, L. Hepcidin Deficiency Disrupts Iron Homeostasis and Induces Ferroptosis in Zebrafish Liver. Fishes 2025, 10, 243. https://doi.org/10.3390/fishes10050243
Liu M, Peng M, Ma J, Hu R, Xu Q, Hu P, Chen L. Hepcidin Deficiency Disrupts Iron Homeostasis and Induces Ferroptosis in Zebrafish Liver. Fishes. 2025; 10(5):243. https://doi.org/10.3390/fishes10050243
Chicago/Turabian StyleLiu, Mingli, Mingjian Peng, Jingwen Ma, Ruiqin Hu, Qianghua Xu, Peng Hu, and Liangbiao Chen. 2025. "Hepcidin Deficiency Disrupts Iron Homeostasis and Induces Ferroptosis in Zebrafish Liver" Fishes 10, no. 5: 243. https://doi.org/10.3390/fishes10050243
APA StyleLiu, M., Peng, M., Ma, J., Hu, R., Xu, Q., Hu, P., & Chen, L. (2025). Hepcidin Deficiency Disrupts Iron Homeostasis and Induces Ferroptosis in Zebrafish Liver. Fishes, 10(5), 243. https://doi.org/10.3390/fishes10050243