Quantitative Microdialysis: Experimental Protocol and Software for Small Molecule Protein Affinity Determination and for Exclusion of Compounds with Poor Physicochemical Properties



Abstract
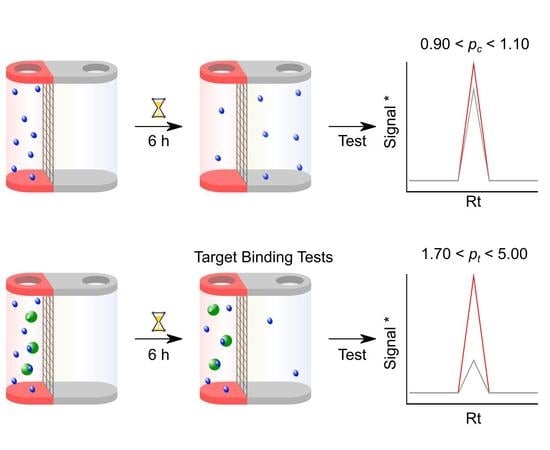
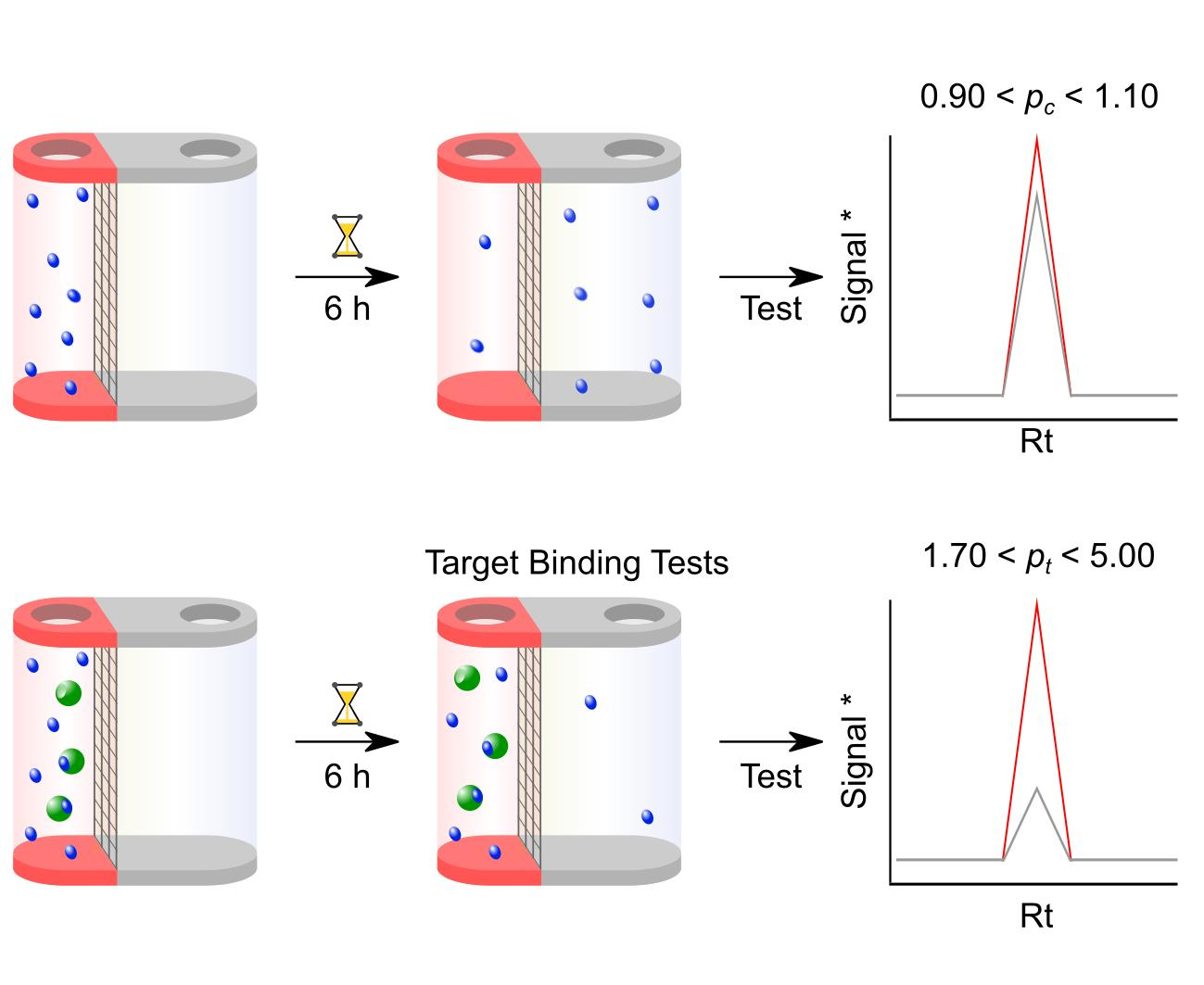
:
1. Introduction
2. Experimental and Computational Design
2.1. Experimental Design
2.2. Materials
- Self-sticking plate seal (Sigma-Aldrich, Cat no.: Z369667-100EA);
- Rapid equilibrium dialysis (RED) device reusable base plate (Thermo Fisher Scientific, Cat. no.: 89811);
- RED device inserts, 8.0 kDa (Thermo Fisher Scientific, Cat. no.: 89809);
- RED device insert removal tool (Thermo Fisher Scientific, Cat. no.: 89812), or a pair of non-pointy tweezers;
- Assay buffer. A buffer that is compatible with the target protein;
- 20% ethanol. Made from ethanol (Fisher Scientific, product code 10437341, purity ≥ 99.8%) and Milli Q water;
- Pluronic F127, low UV absorbance, (Thermo Fisher Scientific, Cat. no.: P6867);
- Dimethyl sulfoxide (DMSO) anhydrous, (Sigma Aldrich, Cat. no: 276855, purity ≥ 99.9%;
- Formic acid (FA) for LCMS LiChropur, (Sigma Aldrich, Cat no.: 5.33002, purity 98–100%);
- HPLC grade H2O and Acetonitrile. H2O: Milli Q water with a resistivity ≥ 18 MΩ cm. Acetonitrile HPLC Plus (Sigma Aldrich, Cat. no.: 34998, purity ≥ 99.9%);
- Trifluoroacetic acid (TFA) for HPLC, (Sigma Aldrich, Cat. no.: 302031, purity ≥ 99.0%);
- 1.5 mL Eppendorf safe-lock tubes (Eppendorf, Cat. no.: 0030120086) or 1 mL glass vials (VWR, Cat no.: 548-0385, and lids, Cat. no.: 548-0389);
- Chromacol glass insert vials, (VWR, Cat. no.:548-0260);
- Insert vial snap caps (VWR, Cat. no.:548-0265);
- Standard laboratory orbital shaker, such as Ika Schüttler MTS 4 S 2 Microtiter Shaker.
2.3. Equipment
- Thermo FisherTM LTQ Orbitrap XLTM.
- Waters Acquity UPLCTM system.
2.4. Computational Design-Simulating System Behaviour
3. Procedure
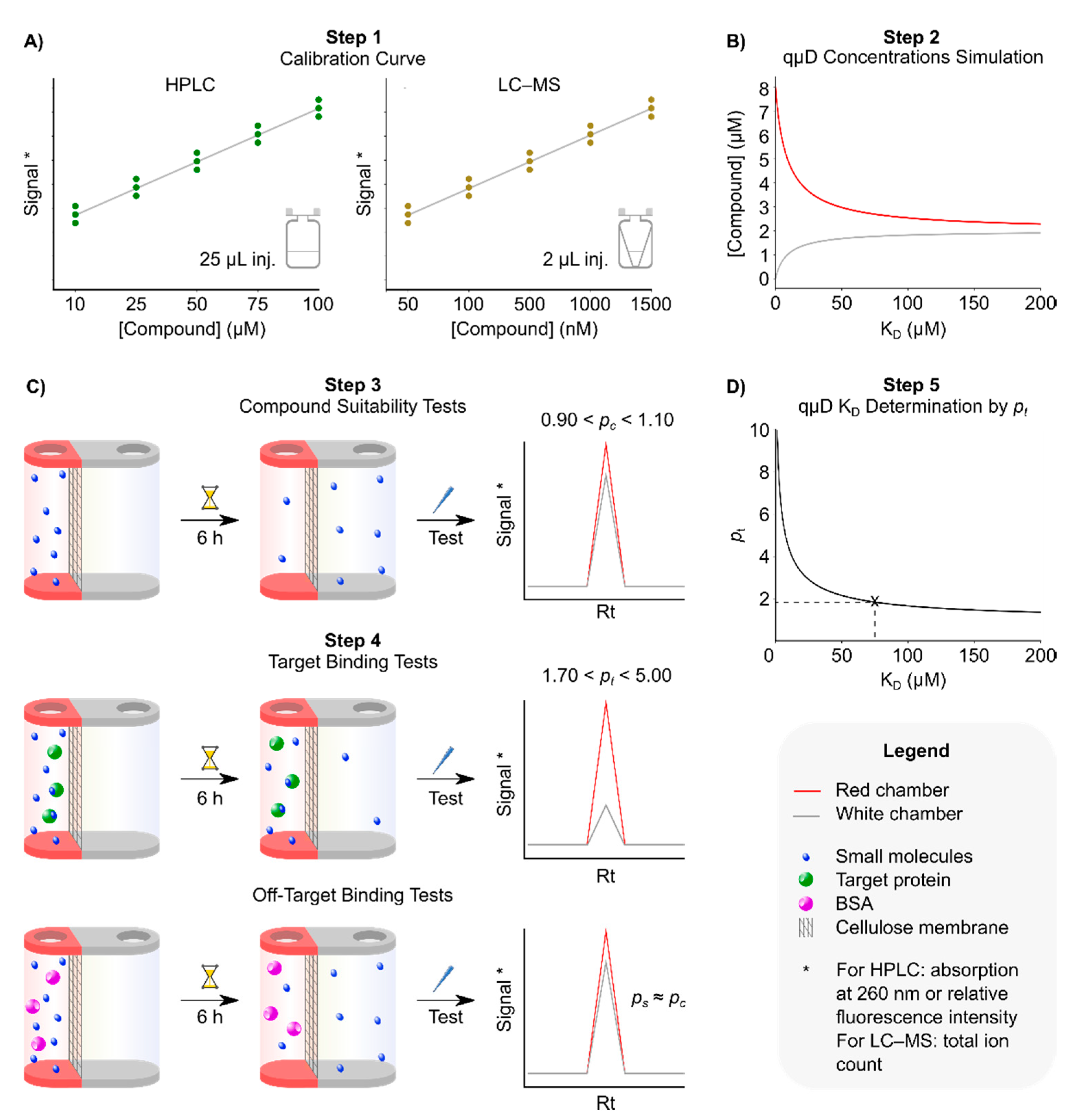
3.1. Step 1: Obtaining Calibration Curves
- Run a RP-HPLC analysis of the small molecules under investigation at 200 µM concentration, using the method above to determine the purity and retention time (Rt) of the compound. Additionally, identifying the absorption wavelength that gives the strongest absorption and most stable baseline. We found that this is usually the optical density (OD) at 210 nm or at 260 nm. Alternatively, for LC–MS, we ran an analysis of the compound at 5 µM using the method above with an injection volume of 2 µL. We determined the Rt and the mass of the protonated molecular ion of the compound [M+H]+ in the ion chromatogram, with M being the monoisotopic mass of the compound.
- Create a series of dilutions of the small molecules to be tested in the assay buffer with 5% DMSO and 0.1% Pluronic f127 in HPLC insert vials, giving a total volume per concentration of 100 µL. The concentrations needed to depend on each individual compound. However, we found that a versatile initial choice of concentrations, applicable to most compounds, is 10, 25, 50, 75 and 100 μM. If LC–MS is to be used instead, a series of dilutions of 50, 100, 500, 1000 and 1500 nM are recommended, in an assay buffer with 5% DMSO in HPLC insert vials, at a total volume per concentration of 50 µL. Pluronic f127 is omitted in this instance as detergents ionise very well so can cause problems in mass spectrometry.
- Run RP-HPLC or LC–MS analyses of the dilution series of compounds, in triplicate, using the method described above. Injection volume 25 µL (RP-HPLC) or 2 µL (LC–MS) for each concentration.
- Analyse the data. For RP-HPLC, determine the area under the elution peak at the predetermined Rt in the particular chromatogram chosen in 3.1.1, and plot peak area versus each of the applied small molecule concentrations in triplicate. The graph should show a straight line if the analysis was performed with compound concentrations in the linear range of the detector. Based on our own analyses, we concluded that the linear range of peak area versus concentration of > 90% of small molecules tested covered a range between 25 and 200 µM. Fit the data to a linear y = kx + d equation using a suitable software (Excel, Origin, GraFit, etc.). These preanalysis runs will serve as reference information for the expected peak area for each test compound concentration. Alternatively for LC–MS, we obtained the ion chromatogram of the compound and determine the area under the [M+H]+ mass ion peak at its Rt, which was identified in 3.1.1. This ion peak area represents the total ion count; we plotted this against each of the small molecule concentrations in triplicate and followed the same procedure as for HPLC above to obtain a calibration curve. Based on our analyses, the linear range of our LC–MS covered a range between 100 and 1000 nM.
3.2. Step 2: Identification of Appropriate Target Protein Concentration
- Open the Python program entitled “02_plot_KDvsConcentrations.py” in a text editor and assign simulation concentrations to the following variables:
- a.
- l0 = 50 → Compound (ligand) concentration achieved at equilibrium in both chambers in the absence of the protein. A starting concentration of 200 µM in the red chamber would equilibrate to 50 µM in both chambers in a system with standard volumes of 100 and 300 µL for the red and white chambers respectively.
- b.
- t0 = 80 → Protein (target) concentration in the red chamber.
- c.
- redvol = 100 → Volume of the red chamber.
- d.
- whitevol = 300 → Volume of the white chamber.
- e.
- KD_beginning = 0 → Lower boundary of the KD range for the X-axis.
- f.
- KD_end = 500 → Upper boundary of the KD range for the X-axis.
- g.
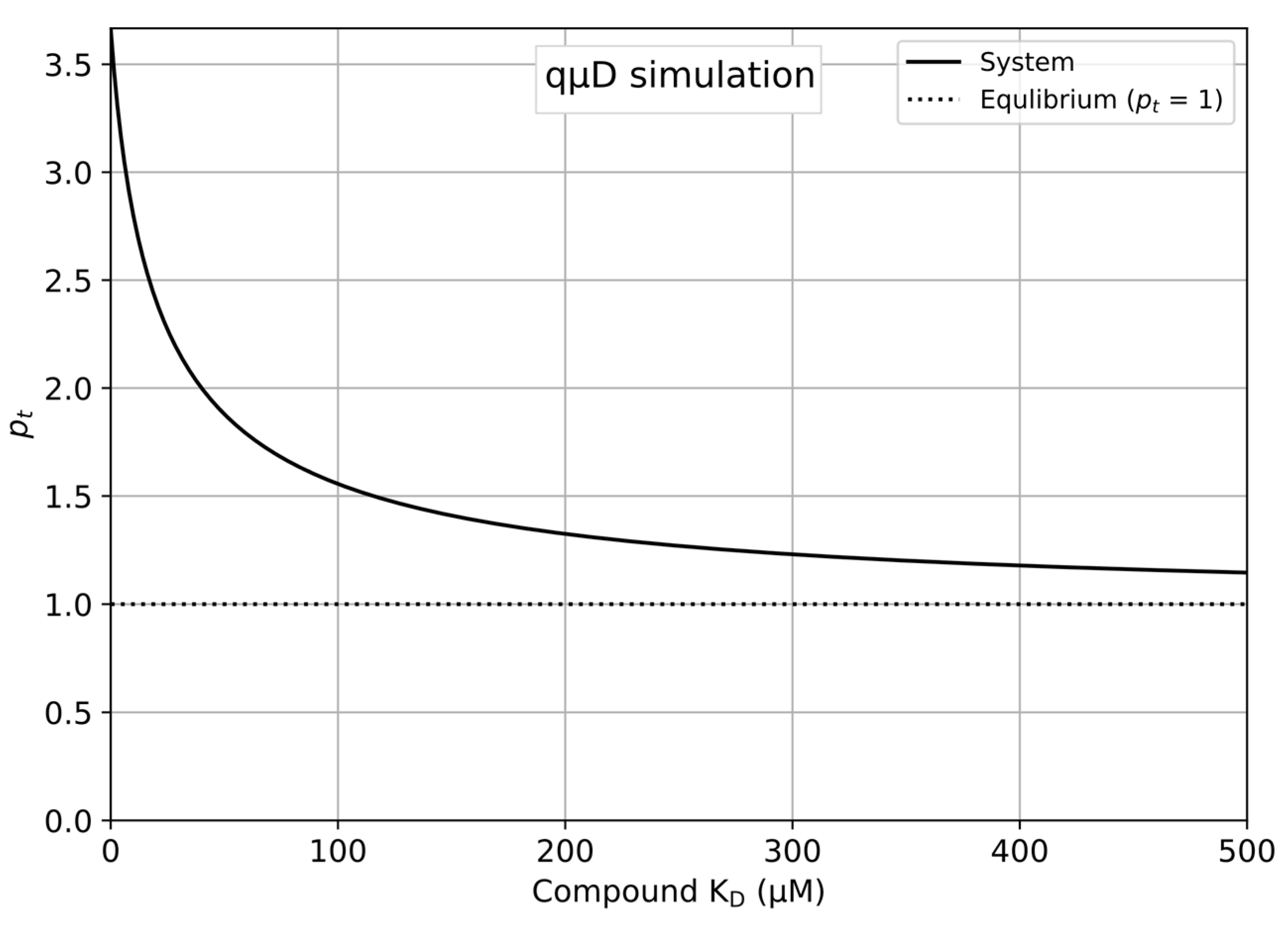
- pc = 1.0 → pc = 1 indicates that the compound equilibrates perfectly in the control qµD run.
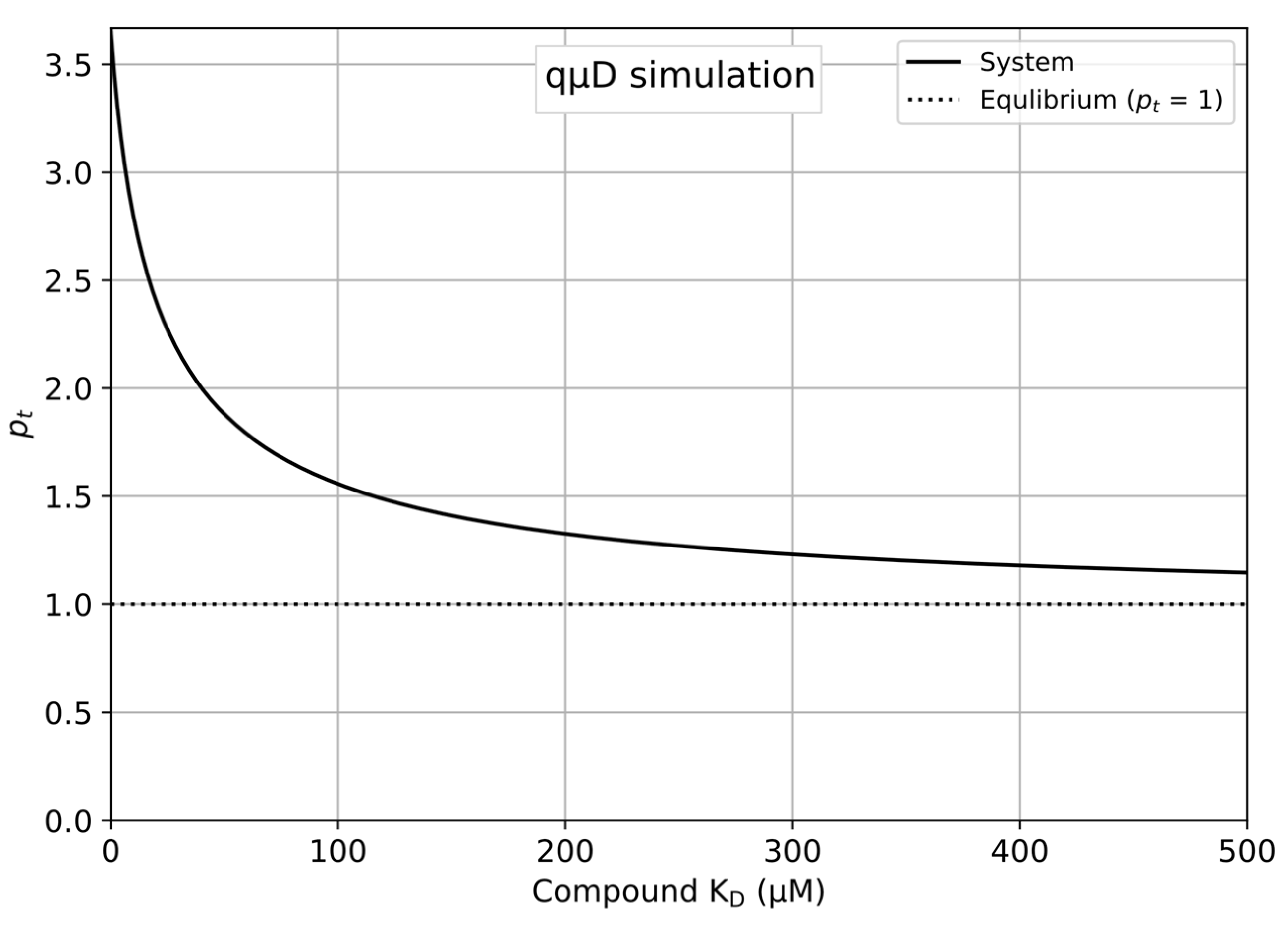
- Save and run the newly edited Python program to produce a plot as shown in Figure 1.
- To perfom single point accurate measurements and read out the data, edit the Python program “01_simulate_qud_concentrations.py” and set l0, t0, red and whitevol as defined in the Python part of Section 3.2, additionally, set kdtl to be the precise value of KD at which to read out the red and white volume concentrations.
- Open the Excel workbook file entitled “quDSimulation_v1.xlsx” in Microsoft Excel, LibreOffice calc or a similar compatible spreadsheet program. Fields that should be edited by users to simulate a system are coloured red, and readout cells are green.
- Open the sheet entitled ‘Simulation’ and edit the following fields as shown below to simulate the standard qµD system defined above. The plot produced automatically updates upon eachvariable manipulation.
- a.
- l0 = 50 → Compound (ligand) concentration achieved at equilibrium in both chambers in the absence of the protein. A starting concentration of 200 µM in the red chamber would equilibrate to 50 µM across chambers in a system with standard volumes of 100 and 300 µL for red and white chambers respectively.
- b.
- t0 = 80 → Protein (target) concentration in the red chamber.
- c.
- redvol = 100 → Volume of the red chamber.
- d.
- whitevol = 300 → Volume of the white chamber.
- e.
- KD_end = 500 → Upper boundary of the KD range for the X-axis.
- f.
- pc = 1.0 → pc = 1 indicates that the compound equilibrates perfectly in the control qµD run.
- The updated plot is displayed automatically on the sheet, producing the plot shown in Figure 1.
- To perfom accurate single-point measurements and read out the data, select the ‘KD to conc+pValue’ sheet and fill in the same variables as listed in the Excel part of Section 3.2, adding a new value for ‘KD’ indicating the KD at which to read out the red and white chamber ligand concentrations.
3.3. Step 3: Compound-only Dialisability Test
- Define the compound concentration to use based on the peak area as a function of the concentration analysis performed in 3.1.4; it should lie in the middle of the linear range determined in 3.1.4. Keep in mind that after full equilibration only 25% of the starting concentration will be detectable (see qµD process outline above). For example, if the centre of the linear range found is 50 µM for the RP-HPLC analysis, then one needs to use a starting compound concentration of 200 µM. This is a direct consequence of the volumes present in the RED device.
- Prepare at least 900 µL of the assay buffer. When RP-HPLC is used for analysis, this includes 5% DMSO and 0.1% pluronic f127. For LC–MS, Pluronic f127 is omitted.
- To produce the standard qµD system modelled in Section 3.2, step 2, prepare a 350 µL solution of test compounds at 200 µM in the assay buffer with 5% DMSO and 0.1% pluronic f127. Apply sonication and vortexing to support compound solubilisation, and visually inspect the solution for precipitation. For LC–MS, prepare a 350 µL solution of test compounds at 40 µM in the assay buffer with 5% DMSO and use the same methods as above to make sure that the compound is fully in solution.
- Clean the base plate thoroughly with 20% ethanol and allow it to dry.
- Place 3 RED devices into the base plate for each compound to be tested.
- Add 100 µL of compound solution into the red chamber of each RED device and 300 µL of assay buffer with 5% DMSO and 0.1% pluronic f127 in the white chamber, respectively. For LC–MS: add 100 µL of compound solution into the red chamber of each RED device and 300 µL of assay buffer with 5% DMSO.
- Seal the base plate with a self-adhesive plate seal to inhibit evaporation.
- Place the base plate onto an orbital shaker at 250 rpm for 6 h for equilibration. Up to 6 h is the suggested time by the manufacturers but this needs to be investigated for the compound under study; some compounds may require an overnight equilibration.
- After > 6 h equilibration time, harvest 50 µL from each chamber of each RED tube and run RP-HPLC using the identical gradient and 25 µL injection volume. If fully equilibrated, the compound concentration in either chamber should be 50 µM. For LC–MS, harvest 10 µL from each chamber of the RED tube, and dilute 20 × with 0.1% formic acid solution. Run the diluted solution using the LC–MS method above, injection volume 2 µL. If fully equilibrated, the compound concentration in either chamber should be 10 µM and after 20 × dilution it should be 500 nM.
- Check that the Rt in the chromatographic elution profile of each test sample has not changed from the condition-setting experiments. Determine the peak area for the compounds in the solutions taken from the red and the white chamber for each HPLC run. Determine pc from the ratio of these two peak areas. A well-equilibrated compound should result in a pc of 0.90–1.10. Any compound showing larger than 10% deviation from 1.00 should be considered for exclusion from further analysis. For LC–MS, check that the Rt in the ion chromatogram of each sample has not changed from the condition setting experiments. Determine the mass ion peak area for the compounds in the solutions taken from the red and white chamber for each LC–MS run. Perform a data analysis as above using this total ion count.
- Additionally, if a compound shows full equilibration with 0.90 < pc < 1.10, check for compound losses, which could occur for a variety of reasons described in the introduction section. If the area under the chromatographic peaks (or mass ion peak in LC–MS) is within 10% of the one found during the initial calibration experiment using a 50 µM (500 nM for LC–MS) concentration, it can be assumed that compound losses are negligible.
3.4. Step 4: Microdialysis Target Protein–Small Molecule Binding assay
- Place 6 RED devices (3 RED devices containing compounds only, and 3 containing compounds + target protein) into the cleaned base plate.
- Prepare 2 mL of assay buffer with 5% DMSO and 0.1% pluronic f127 when HPLC is used for analysis. When LC–MS is used for analysis, prepare 2 mL of the assay buffer containing 5% DMSO.
- Prepare 350 µL of 200 µM small molecule solution in the assay buffer with 5% DMSO and 0.1% pluronic f127, and 350 µL of 200 µM small molecule plus 80 µM protein in the same buffer. For LC–MS, prepare 350 µL of 40 µM small molecule solution in the assay buffer with 5% DMSO and 350 µL of 40 µM small molecule plus 40 µM protein in the same buffer.
- Add 3 × 100 µL of 200 µM (or 40 µM) compound solution to 3 red chambers of the RED tubes followed by adding 3 × 300 µL dialysis buffer in the corresponding white chambers.
- Add 3 × 100 µL of 200 µM + 80 µM protein (or 40 µM compound + 40 µM protein) solution into 3 red chambers of the RED tubes and add 3 × 300 µL of dialysis buffer in the corresponding white chambers.
- Seal plate with the self-adhesive plate seal and place on shaker at 250 rpm for 6 h.
- After 6 h or overnight incubation, remove the plate from the shaker and harvest 50 µL each from the red and white chamber of the compound-only samples, as well as 50 µL from the white chambers of the compound-protein binding samples; and add them to insert vials for the HPLC analysis. For the LC–MS analysis, take out 10 µL from each red and white chamber of the compound alone samples as well as from the compound and protein samples.
- Run these samples on RP-HPLC under the same conditions as above, with an injection volume of 25 µL, or for LC–MS, dilute the 10 µL aliquot 20 × with 0.1% FA solution and run the LC–MS analysis, injection volume 2 µL.
- From the resulting chromatograms, obtain peak areas at known Rts for the samples for RP-HPLC. For LC–MS, obtain mass ion peak areas of the compounds at known Rts.
- 10.
- Determine pc = compound peak area from red chamber/white chamber or determine pc = mass ion peak area from red chamber/white chamber.
- 11.
- This should resemble the data achieved from the compound equilibration test experiments and should therefore be close to 1.0.
- 12.
- For determination of the total amount of compounds in the qµD. Convert the compound peak area or mass ion peak area obtained in Section 3.3. 11 into compound concentration by using the calibration curve in Section 3.1. 4. The amount of compound in the chambers can be obtained by multiplication of the converted compound concentration in the red chamber by 100 µL plus the converted compound concentration of the white chamber multiplied by 300 µL. This should yield close to the amount of introduced compound at the start of the dialysis (200 µM × 100 µL = 20 × 10−9 mol for HPLC and 40 µM × 100 µL = 4 × 10−9 mol for LC–MS) indicating no losses due to adhesion, precipitation or aggregation effects. Note that for LC–MS the solutions were diluted 20 × before analysis. Therefore, one needs to multiply by a factor of 20 to obtain the real concentrations in the RED tube.
- 13.
- From the HPLC chromatograms of the white chambers, determine the peak area and convert to compound concentration by using the calibration curve above.
- 14.
- Determine the amount of compound in the white chamber by multiplication of the determined compound concentration with the volume of the white chamber, 300 µL.
- 15.
- From Section 3.3. 13 the total amount of compound is known. Therefore, the amount of compound in the red chamber is the difference between the total amount of compound and the amount of compound in the white chamber.
- 16.
- Calculated the compound concentration in the red chamber by dividing the amount of compound in the red chamber by 100 µL.
- 17.
- Determine pt by dividing the compound concentration of the red chamber by that of the white chamber. For LC–MS, determine pt by dividing the compound mass ion peak area of the red chamber by that of the white chamber obtained in Section 3.3. 9.
3.5. Step 5: Use the Software to Determine the Dissociation Constant (KD) of the Compound to the Target Protein by Using the Red/White Chamber Concentrations or pt Values
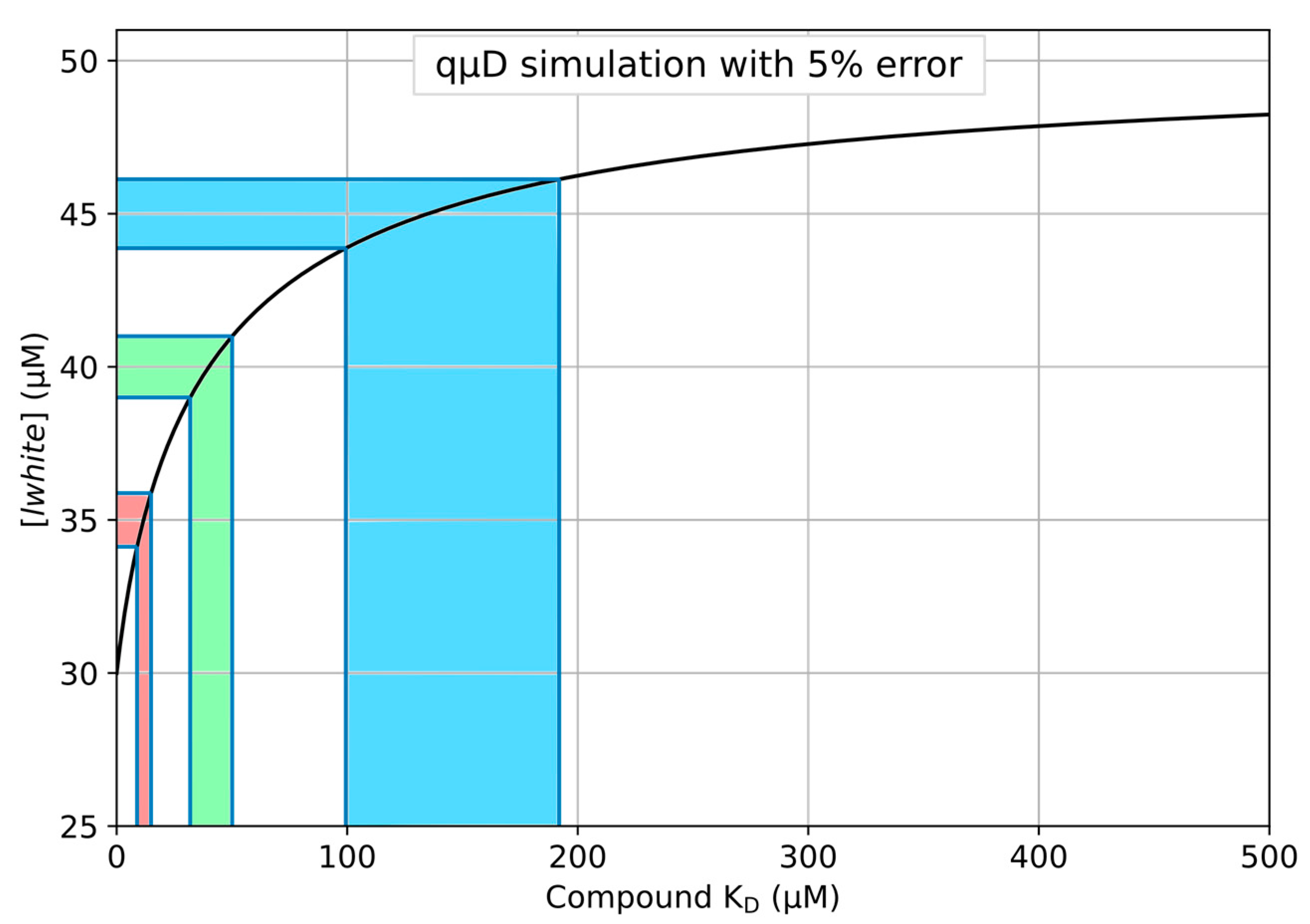
- To determine a KD based on only a single point measurement of the white chamber concentration, open the Python file entitled ‘03_deriveKD_from_lwhite.py’ in a text or code editor and assign the following variables, taking care to input the correct value for lwhite as the measured concentration of ligand in the white chamber and run the program to read out the KD:
- l0 = 50, Compound (ligand) concentration achieved at equilibrium in both chambers in the absence of the protein. A starting concentration of 100 µM in the red chamber would equilibrate to 25 µM across chambers in a system with standard volumes of 100 and 300 µL for red and white chambers respectively.
- t0 = 80, Protein (target) concentration present in the red chamber.
- redvol = 100, Volume of the red chamber.
- whitevol = 300, Volume of the white chamber.
- lwhite, The ligand concentration as measured in the white chamber.
- pc, pc = 1 indicates that the compound equilibrates perfectly in the control qµD run.
- To determine a KD based on only a single point measurement of the red chamber concentration, open the Python file entitled ‘03_deriveKD_from_lred.py’ in a text or code editor and assign the following variables, taking care to input the correct value for lred as the measured concentration of ligand in the red chamber and run the program to read out the KD:
- l0 = 50, Compound (ligand) concentration achieved at equilibrium in both chambers in the absence of the protein. A starting concentration of 100 µM in the red chamber would equilibrate to 25 µM across chambers in a system with standard volumes of 100 and 300 µL for red and white chambers respectively.
- t0 = 80, Protein (target) concentration present in the red chamber.
- redvol = 100, Volume of the red chamber.
- whitevol = 300, Volume of the white chamber.
- lred, The ligand concentration as measured in the red chamber.
- pc, pc = 1 indicates that the compound equilibrates perfectly in the control qµD run.
- To determine a KD based on a pt derived from single point measurements open the Python file entitled ‘03_deriveKD_from_pt.py’ in a text or code editor and assign the following variables, taking care to input the correct value for pt as the ratio of compound concentration the the red versus white chambers and run the program to read out the KD:
- l0 = 50, Compound (ligand) concentration achieved after equilibrium in both chambers in the absence of the protein. A starting concentration of 100 µM in the red chamber would equilibrate to 25 µM across chambers in a system with standard volumes of 100 and 300 µL for red and white chambers respectively.
- t0 = 80, Protein (target) concentration present in the red chamber.
- redvol = 100, Volume of the red chamber.
- whitevol = 300, Volume of the white chamber.
- pt, The pt value, which is the concentration of ligand in the red chamber divided by the concentration of ligand in the white chamber.
- pc, pc = 1 indicates that the compound equilibrates perfectly in the control qµD run.
- To determine a KD based on multiple replicate measurements of compound concentration in the white chamber, open ‘04_deriveKD_from_multiple_lwhite.py’ in a text or code editor. Comments in the file provide instructions in line with the following:
- Assign the experimental variables as descibed in Section 3.5. 1 above, setting l0, t0, redvol and whitevol.
- If 3 measurements of compound concentration lwhite have been made, calculate the mean and standard deviation of these variables using numpy (val1, val2 and val3 bellow are measurement replicates):
- lwhite_mean = np.mean([val1, val2, val3]);
- lwhite_std = np.std([val1, val2, val3]).
- Assign lwhite to be a newly constructed Python uncertainties package ufloat object.
- lwhite = ufloat(lwhite_mean, lwhite_std).
- If multiple pc values have been derived, the pc variable may be assigned to a ufloat object constructed as:
- pc_mean = np.mean([val1, val2, val3]);
- pc_std = np.std([val1, val2, val3]);
- pc = ufloat(pc_mean, pc_std).
- Alternatively, a single pc value may be assigned as in 3.5.
- To determine a KD based on multiple replicate measurements of compound concentration in the red chamber, open ‘04_deriveKD_from_multiple_lred.py’ in a text or code editor. Comments in the file provide instructions in line with the following:
- Assign the experimental variables as descibed in Section 3.5. 1 above, setting l0, t0, redvol and whitevol.
- If 3 measurements of compound concentration lred have been made, calculate the mean and standard deviation of these variables using numpy (val1, val2 and val3 bellow are measurement replicates):
- lred_mean = np.mean([val1, val2, val3]);
- lred_std = np.std([val1, val2, val3]).
- Assign lwhite to be a newly constructed Python uncertainties package ufloat object.
- lred = ufloat(lred_mean, lred_std).
- If multiple pc values have been derived, the pc variable may be assigned to a ufloat object constructed as:
- pc_mean = np.mean([val1, val2, val3]);
- pc_std = np.std([val1, val2, val3]);
- pc = ufloat(pc_mean, pc_std).
- Alternatively, a single pc value may be assigned as in Section 3.5. 1.
- To determine a KD based on multiple replicate measurements of pt, open ‘04_deriveKD_from_multiple_pt.py’ in a text or code editor. Comments in the file provide instructions in line with the following:
- Assign the experimental variables as descibed in Section 3.5. 1 above, setting l0, t0, redvol, and whitevol.
- If 3 measurements of pt have been made, calculate the mean and standard deviation of these variables using numpy (val1, val2 and val3 bellow are measurement replicates):
- pt_mean = np.mean([val1, val2, val3]);
- pt_std = np.std([val1, val2, val3]).
- Assign lwhite to be a newly constructed Python uncertainties package ufloat object.
- pt = ufloat(pt_mean, pt_std).
- If multiple pc values have been derived, the pc variable may be assigned to a ufloat object constructed as:
- pc_mean = np.mean([val1, val2, val3]);
- pc_std = np.std([val1, val2, val3]);
- pc = ufloat(pc_mean, pc_std).
- Alternatively, a single pc value may be assigned as in Section 3.5. 1.
- To determine a KD based on only the white chamber concentration, open the Excel file entitled ‘quDSimulation_v1.xlsx’ in Microsoft Excel, LibreOffice calc, or similar compatible spreadsheet program. Go to the tab named ‘lwhite to KD’ and assign the following variables, taking care to input the correct value for lwhite as the measured concentration of ligand in the white chamber. The readout of KD will update with every change of input variable:
- l0 = 50 → Compound (ligand) concentration achieved at equilibrium in both chambers in the absence of the protein. A starting concentration of 100 µM in the red chamber would equilibrate to 25 µM across chambers in a system with standard volumes of 100 and 300 µL for red and white chambers respectively.
- t0 = 80 → Protein (target) concentration present in the red chamber.
- redvol = 100 → Volume of the red chamber.
- whitevol = 300 → Volume of the white chamber.
- lwhite → The ligand concentration as measured in the white chamber.
- pc → pc = 1 indicates that the compound equilibrates perfectly in the control qµD run.
- To determine a KD based on only the red chamber concentration, open the Excel file entitled ‘quDSimulation_v1.xlsx’ in Microsoft Excel, LibreOffice calc, or similar compatible spreadsheet program. Go to the tab named ‘lred to KD’ and assign the following variables, taking care to input the correct value for LRED as the measured concentration of ligand in the red chamber. The readout of KD will update with every change of input variable:
- l0 = 50 → Compound (ligand) concentration achieved at equilibrium in both chambers in the absence of the protein. A starting concentration of 100 µM in the red chamber would equilibrate to 25 µM across chambers in a system with standard volumes of 100 and 300 µL for red and white chambers respectively.
- t0 = 80 → Protein (target) concentration present in the red chamber.
- redvol = 100 → Volume of the red chamber.
- whitevol = 300 → Volume of the white chamber.
- lred → The ligand concentration as measured in the red chamber.
- pc → pc = 1 indicates that the compound equilibrates perfectly in the control qµD run.
- To determine a KD based on pt, open the Excel file entitled ‘quDSimulation_v1.xlsx’ in Microsoft Excel, LibreOffice calc or a similar compatible spreadsheet program. Go to the tab named ‘pt to KD’ and assign the following variables, taking care to input the correct value for PT_VALUE as the measured and calculated pt value. The readout of KD will update with every change of input variable:
- l0 = 50 → Compound (ligand) concentration achieved at equilibrium in both chambers in the absence of the protein. A starting concentration of 100 µM in the red chamber would equilibrate to 25 µM across chambers in a system with standard volumes of 100 and 300 µL for red and white chambers respectively.
- t0 = 80 → Protein (target) concentration present in the red chamber.
- redvol = 100 → Volume of the red chamber.
- whitevol = 300 → Volume of the white chamber.
- pt → The pt value, which is concentration of the ligand in the red chamber divided by the concentration of the ligand in the white chamber.
- pc → pc = 1 indicates that the compound equilibrates perfectly in the control qµD run.
4. Expected Results
4.1. Simulation of the qµD Experiment as a Function of the Concentrations and KDs
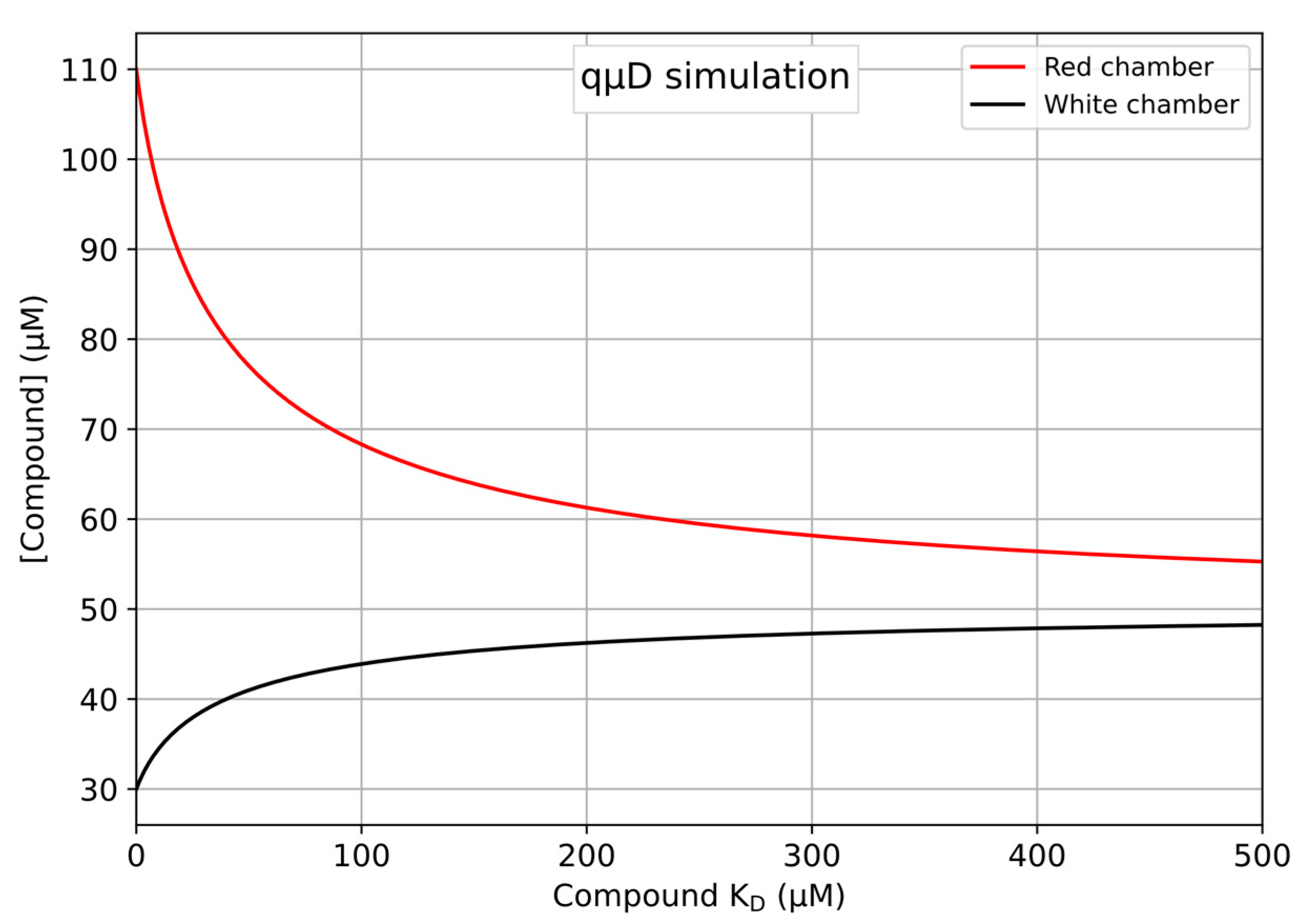
4.2. Exemplaric Considerations on the Accuracy of KD Determinations
4.3. Experimental Results
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nimmo, I.A.; Atkins, G.L.; Strange, R.C.; Percy-Robb, I.W. An evaluation of ways of using equilibrium dialysis to quantify the binding of ligand to macromolecules. Biochem. J. 1977, 165, 107–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, A.; He, D.; Nie, L.H.; Yao, S.Z. Determination of the binding parameters of drug to protein by equilibrium dialysis/piezoelectric quartz crystal sensor. Anal. Biochem. 2000, 282, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Kariv, I.; Cao, H.; Oldenburg, K.R. Development of a high throughput equilibrium dialysis method. J. Pharm. Sci. 2001, 90, 580–587. [Google Scholar] [CrossRef]
- Arkin, M.R.; Wells, J.A. Small-molecule inhibitors of protein-protein interactions: Progressing towards the dream. Nat. Rev. Drug Discov. 2004, 3, 301–317. [Google Scholar] [CrossRef] [PubMed]
- Ward, L.D. Measurement of ligand binding to proteins by fluorescence spectroscopy. Methods Enzymol. 1985, 117, 400–414. [Google Scholar] [PubMed]
- Meyer, B.; Peters, T. NMR spectroscopy techniques for screening and identifying ligand binding to protein receptors. Angew. Chem. Int. Ed. 2003, 42, 864–890. [Google Scholar] [CrossRef] [PubMed]
- Höfner, G.; Wanner, K.T. Competitive binding assays made easy with a native marker and mass spectrometric quantification. Angew. Chem. Int. Ed. 2003, 42, 5235–5237. [Google Scholar] [CrossRef] [PubMed]
- Annis, D.A.; Nickbarg, E.; Yang, X.; Ziebell, M.R.; Whitehurst, C.E. Affinity selection-mass spectrometry screening techniques for small molecule drug discovery. Curr. Opin. Chem. Biol. 2007, 11, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Loken, H.F.; Havel, R.J.; Gordan, G.S.; Whittington, S.L. Ultracentrifugal analysis of protein-bound and free calcium in human serum. J. Biol. Chem. 1960, 235, 3654–3658. [Google Scholar] [PubMed]
- Zehender, H.; Le Goff, F.; Lehmann, N.; Filipuzzi, I.; Mayr, L.M. SpeedScreen: The “missing link” between genomics and lead discovery. J. Biomol. Screen. 2004, 9, 498–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Auer Lab. Affinity Selection. Available online: https://sites.google.com/view/the-auer-lab-uoe/screening/affinity-selection (accessed on 28 June 2020).
- Weidemann, T.; Seifert, J.-M.; Hintersteiner, M.; Auer, M. Analysis of protein-small molecule interactions by microscale equilibrium dialysis and its application as a secondary confirmation method for on-bead screening. J. Comb. Chem. 2010, 12, 647–654. [Google Scholar] [CrossRef]
- Waters, N.J.; Jones, R.; Williams, G.; Sohal, B. Validation of a rapid equilibrium dialysis approach for the measurement of plasma protein binding. J. Pharm. Sci. 2008, 97, 4586–4595. [Google Scholar] [CrossRef]
- Van Liempd, S.; Morrison, D.; Sysmans, L.; Nelis, P.; Mortishire-Smith, R. Development and validation of a higher-throughput equilibrium dialysis assay for plasma protein binding. J. Assoc. Lab. Autom. 2011, 16, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Zetterberg, C.; Gao, H. Automation of plasma protein binding assay using rapid equilibrium dialysis device and Tecan workstation. J. Pharm. Biomed. Anal. 2017, 140, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Hoefliger, N.; De Smet, R.; Ringoir, S.; Vogeleere, P. Extraction of protein bound ligands from azotemic sera: Comparison of 12 deproteinization methods. Kidney Int. 1992, 41, 1707–1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathematica; Version 12.1; Wolfram Research, Inc.: Champaign, IL, USA, 2020.
- Python Software Foundation. Python Language Reference, Version 3.8. Available online: http://www.python.org (accessed on 4 June 2020).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| lred | lwhite | pt | ||||
|---|---|---|---|---|---|---|
| KD | Concentration (µM) | ∂KD/∂lred (Fold Change from 1 µM) | Concentration (µM) | ∂lKD/∂lwhite (Fold Change from 1 µM) | Value | ∂KD/∂pt (Fold Change from 1 µM) |
| 1 | 108.1032 | −0.5553 (1) | 30.6323 | 1.6659 (1) | 3.5291 | −7.8158 (1) |
| 100 | 68.304 | −8.6209 (16) | 43.8987 | 25.8628 (16) | 1.5559 | −249.2 (32) |
| 200 | 61.2679 | −23.2953 (42) | 46.244 | 69.8859 (42) | 1.3249 | −747.259 (96) |
| 300 | 58.1682 | −44.6309 (80) | 47.2773 | 133.8926 (80) | 1.2304 | −1496.34 (192) |
| 400 | 56.4121 | −72.6318 (131) | 47.8626 | 217.8954 (131) | 1.1786 | −2495.81 (319) |
| 500 | 55.2795 | −107.299 (193) | 48.2402 | 321.8969 (193) | 1.1459 | −3745.46 (479) |
| Compound # | [l0] (µM) | [t0] (µM) | pc | pt | ps (BSA) | Comment |
|---|---|---|---|---|---|---|
| 1 | 25 | 80 | 1.41 ± 0.02 | NA | NA | Non-equilibrating compound. |
| 2 | 25 | 80 | 1.02 ± 0.01 | 1.15 ± 0.03 | 1.29 ± 0.05 | Equilibrating compound, but it binds BSA stronger than to the target protein; 603 ± 154 µM vs. 278 ± 57 µM–assuming 1:1 binding. |
| 3 | 25 | 80 | 1.02 ± 0.01 | 1.21 ± 0.01 | 1.11 ± 0.01 | Equilibrating compound, KD = 405 ± 35 μM to target protein, and 882 ± 57 µM to BSA–assuming 1:1 binding. |
| 4 | 10 | 40 | 0.99 ± 0.03 | 1.21 ± 0.04 | NA | Well-equilibrating compound, KD = 171 ± 34 μM. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shave, S.; Pham, N.T.; Śmieja, C.B.; Auer, M. Quantitative Microdialysis: Experimental Protocol and Software for Small Molecule Protein Affinity Determination and for Exclusion of Compounds with Poor Physicochemical Properties. Methods Protoc. 2020, 3, 55. https://doi.org/10.3390/mps3030055
Shave S, Pham NT, Śmieja CB, Auer M. Quantitative Microdialysis: Experimental Protocol and Software for Small Molecule Protein Affinity Determination and for Exclusion of Compounds with Poor Physicochemical Properties. Methods and Protocols. 2020; 3(3):55. https://doi.org/10.3390/mps3030055
Chicago/Turabian StyleShave, Steven, Nhan T. Pham, Connor B. Śmieja, and Manfred Auer. 2020. "Quantitative Microdialysis: Experimental Protocol and Software for Small Molecule Protein Affinity Determination and for Exclusion of Compounds with Poor Physicochemical Properties" Methods and Protocols 3, no. 3: 55. https://doi.org/10.3390/mps3030055
APA StyleShave, S., Pham, N. T., Śmieja, C. B., & Auer, M. (2020). Quantitative Microdialysis: Experimental Protocol and Software for Small Molecule Protein Affinity Determination and for Exclusion of Compounds with Poor Physicochemical Properties. Methods and Protocols, 3(3), 55. https://doi.org/10.3390/mps3030055