Development of Strategies to Decrease False Positive Results in Newborn Screening

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Introduction of New Disease-Specific Biomarkers: The Case of Tyrosinemia Type I
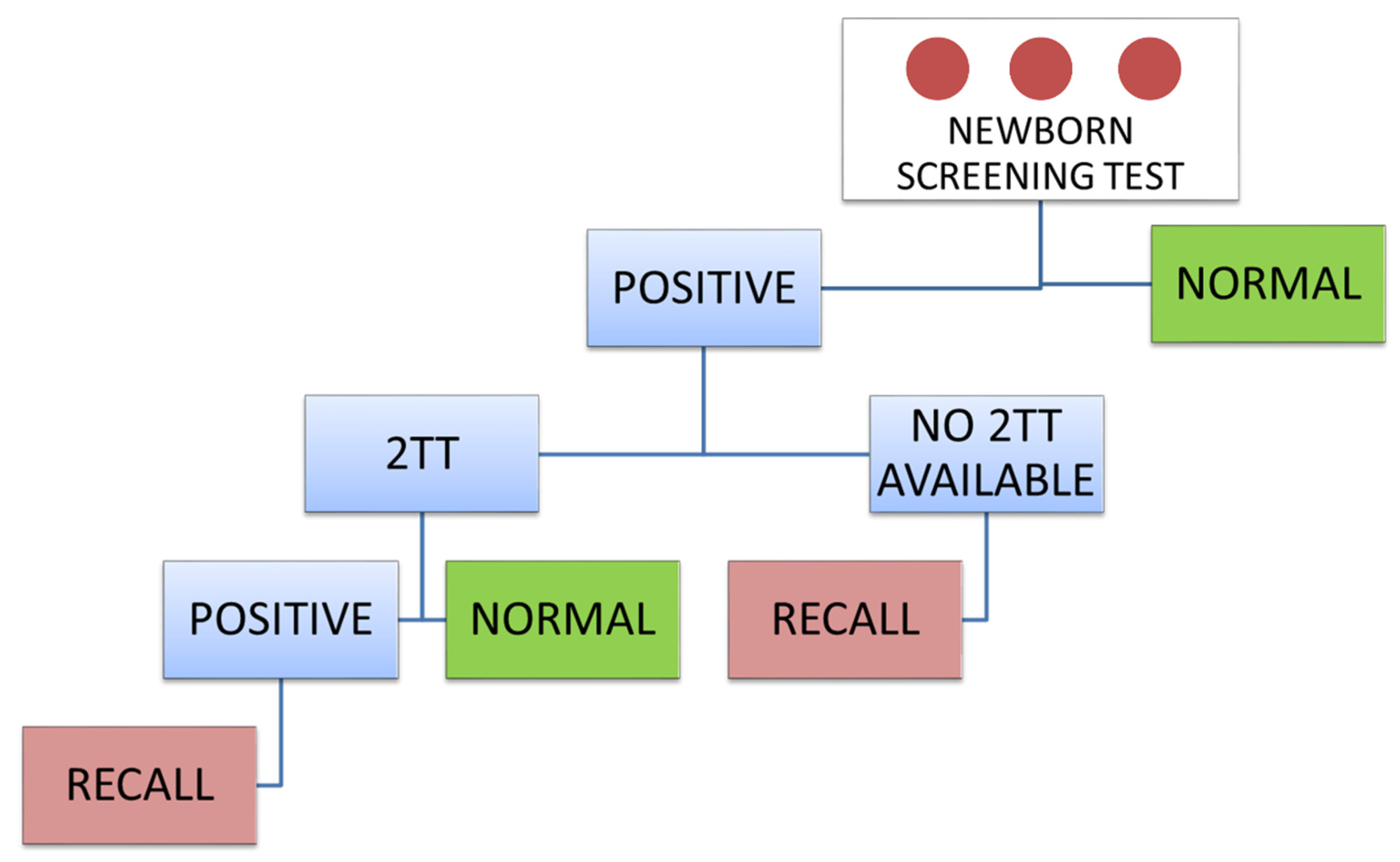
3. Second-Tier Test Strategy
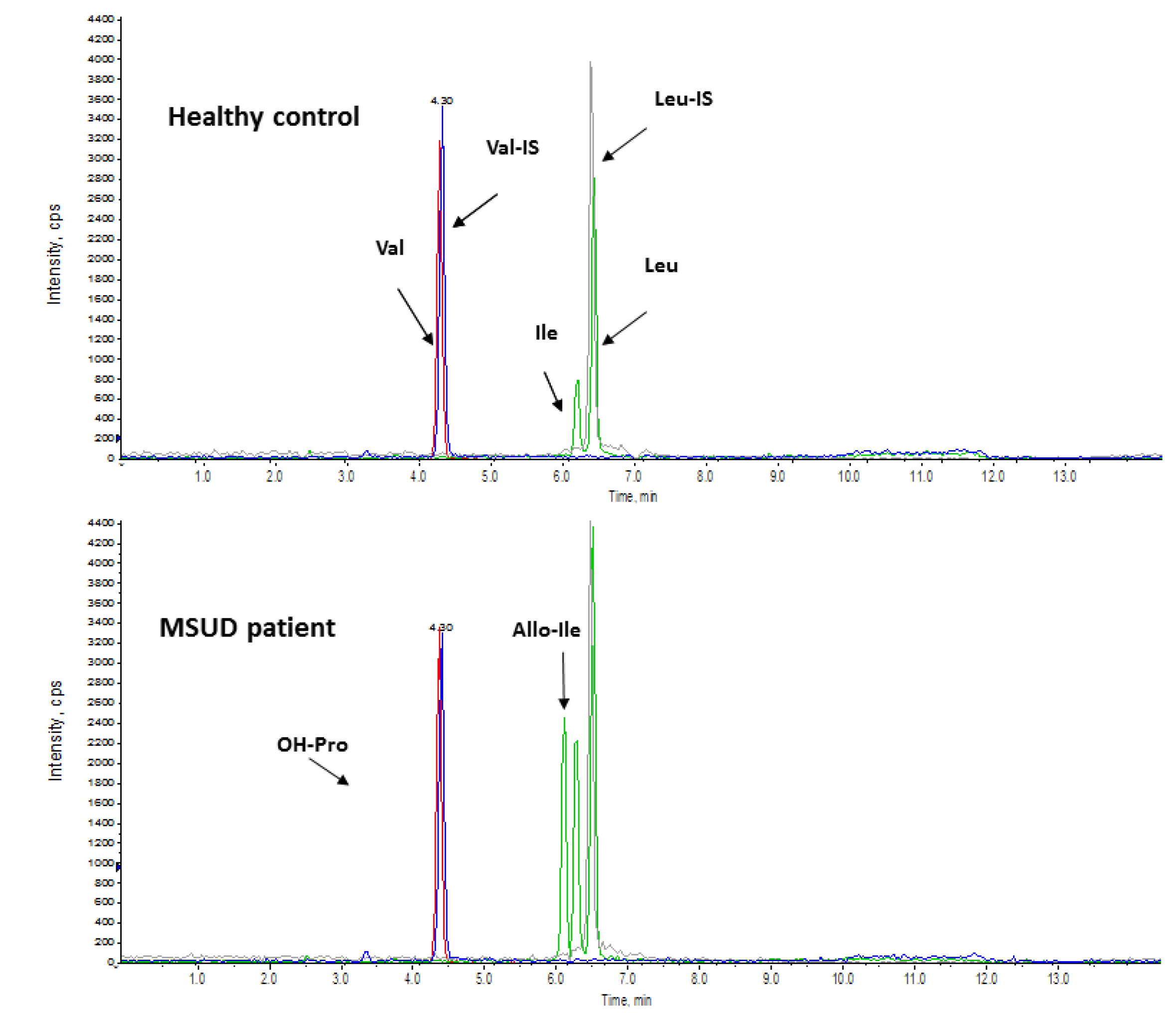
3.1. Maple Syrup Urine Disease (MSUD)
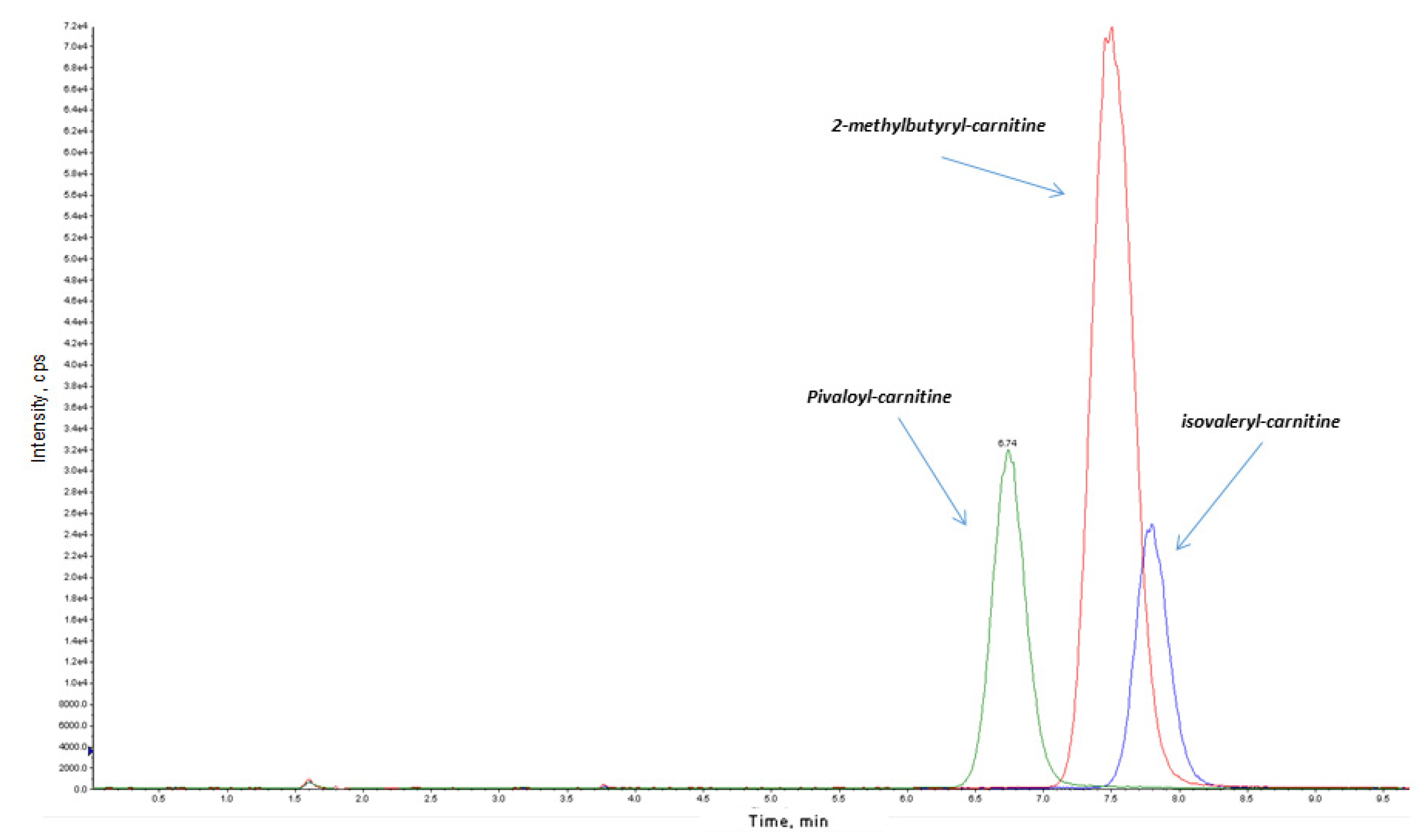
3.2. Isovaleric Acidaemia
3.3. Propionate Metabolism
3.4. Homocystinurias and Methylation Disorders
3.5. Congenital Adrenal Hyperplasia (CAH)
3.6. Adenosine Deaminase Deficiency (Ada-SCID) and Purine Nucleoside Phosphorylase Deficiency (PNP-SCID)
4. Lysosomal Storage Disorders (LSD)
4.1. Pompe Disease
4.2. Fabry Disease
4.3. Mucopolysaccharidosis Type I (MPS I)
4.4. Gaucher Disease
4.5. Krabbe Disease
5. Post-Analytical Tools
6. Molecular Testing as a 2-TT
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Saudubray, J.M.; Baumgartner, M.; Walter, J. Inborn Metabolic Diseases: Diagnosis and Treatment, 6th ed.; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Hoffmann, G.F.; Zschocke, J.; Nyhan, W.L. Inherited Metabolic Diseases A Clinical Approach, 2th ed.; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Wilcken, B.; Wiley, V. Newborn screening. Pathology 2008, 40, 104–115. [Google Scholar] [CrossRef]
- Rajabi, F. Updates in Newborn Screening. Pediatr. Ann. 2018, 47, e187–e190. [Google Scholar] [CrossRef]
- Mak, C.M.; Lee, H.C.; Chan, A.Y.; Lam, C.W. Inborn errors of metabolism and expanded newborn screening: Review and update. Crit. Rev. Clin. Lab. Sci. 2013, 50, 142–162. [Google Scholar] [CrossRef]
- Sweetman, L.; Millington, D.S.; Therrell, B.L.; Hannon, W.H.; Popovich, B.; Watson, M.S.; Mann, M.Y.; Lloyd-Puryear, M.A.; van Dyck, P.C. Naming and counting disorders (conditions) included in newborn screening panels. Pediatrics 2006, 117, S308–S314. [Google Scholar] [CrossRef]
- Therrell, B.L.; Padilla, C.D.; Loeber, J.G.; Kneisser, I.; Saadallah, A.; Borrajo, G.J.; Adams, J. Current status of newborn screening worldwide: 2015. Semin. Perinatol. 2015, 39, 171–187. [Google Scholar] [CrossRef]
- Fabie, N.A.V.; Pappas, K.B.; Feldman, G.L. The Current State of Newborn Screening in the United States. Pediatr. Clin. N. Am. 2019, 66, 369–386. [Google Scholar] [CrossRef]
- Tarini, B.A.; Christakis, D.A.; Welch, H.G. State newborn screening in the tandem mass spectrometry era: More tests, more false-positive results. Pediatrics 2006, 118, 448–456. [Google Scholar] [CrossRef]
- Gurian, E.A.; Kinnamon, D.D.; Henry, J.J.; Waisbren, S.E. Expanded newborn screening for biochemical disorders: The effect of a false positive result. Pediatrics 2006, 117, 1915–1921. [Google Scholar] [CrossRef]
- Tu, W.J.; He, J.; Chen, H.; Shi, X.D.; Li, Y. Psychological effects of false-positive results in expanded newborn screening in China. PLoS ONE 2012, 7, e36235. [Google Scholar] [CrossRef]
- Waisbren, S.E.; Albers, S.; Amato, S.; Ampola, M.; Brewster, T.G.; Demmer, L.; Eaton, R.B.; Greenstein, R.; Korson, M.; Larson, C.; et al. Effect of expanded newborn screening for biochemical genetic disorders on child outcomes and parental stress. JAMA 2003, 290, 2564–2572. [Google Scholar] [CrossRef]
- Frazier, D.M.; Millington, D.S.; McCandless, S.E.; Koeberl, D.D.; Weavil, S.D.; Chaing, S.H.; Muenzer, J. The Tandem Mass Spectrometry Newborn Screening Experience in North Carolina: 1997–2005. J. Inherit. Metab. Dis. 2006, 29, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Wilcken, B.; Wiley, V.; Hammond, J.; Carpenter, K. Screening Newborns for Inborn Errors of Metabolism by Tandem Mass Spectrometry. N. Engl. J. Med. 2003, 348, 2304–2312. [Google Scholar] [CrossRef]
- La Marca, G.; Malvagia, S.; Pasquini, E.; Cavicchi, C.; Morrone, A.; Ciani, F.; Funghini, S.; Villanelli, F.; Zammarchi, E.; Guerrini, R. Newborn Screening for Tyrosinemia Type I: Further Evidence that Succinylacetone Determination on Blood Spot Is Essential. JIMD Rep. 2011, 1, 107–109. [Google Scholar] [PubMed]
- La Marca, G.; Malvagia, S.; Pasquini, E.; Innocenti, M.; Fernandez, M.R.; Donati, M.A.; Zammarchi, E. The inclusion of succinylacetone as marker for tyrosinemia type I in expanded newborn screening programs. Rapid Commun. Mass Spectrom. 2008, 22, 812–818. [Google Scholar] [CrossRef] [PubMed]
- De Jesús, V.R.; Adam, B.W.; Mandel, D.; Cuthbert, C.D.; Matern, D. Succinylacetone as primary marker to detect tyrosinemia type I in newborns and its measurement by newborn screening programs. Mol. Genet. Metab. 2014, 113, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Chinsky, J.M.; Singh, R.; Ficicioglu, C.; van Karnebeek, C.D.M.; Grompe, M.; Mitchell, G.; Waisbren, S.E.; Gucsavas-Calikoglu, M.; Wasserstein, M.P.; Coakley, K.; et al. Diagnosis and treatment of tyrosinemia type I: A US and Canadian consensus group review and recommendations. Genet. Med. 2017, 19. [Google Scholar] [CrossRef]
- Stinton, C.; Geppert, J.; Freeman, K.; Clarke, A.; Johnson, S.; Fraser, H.; Sutcliffe, P.; Taylor-Phillips, S. Newborn screening for Tyrosinemia type 1 using succinylacetone—A systematic review of test accuracy. Orphanet J. Rare Dis. 2017, 12, 48. [Google Scholar] [CrossRef]
- La Marca, G.; Malvagia, S.; Casetta, B.; Pasquini, E.; Donati, M.A.; Zammarchi, E. Progress in expanded newborn screening for metabolic conditions by LC-MS/MS in Tuscany: Update on methods to reduce false tests. J. Inherit. Metab. Dis. 2008, 31, S395–S404. [Google Scholar] [CrossRef]
- Gavrilov, D.K.; Piazza, A.L.; Pino, G.; Turgeon, C.; Matern, D.; Oglesbee, D.; Raymond, K.; Tortorelli, S.; Rinaldo, P. The Combined Impact of CLIR Post-Analytical Tools and Second Tier Testing on the Performance of Newborn Screening for Disorders of Propionate, Methionine, and Cobalamin Metabolism. Int. J. Neonatal Screen. 2020, 6, 33. [Google Scholar] [CrossRef]
- National Newborn Screening and Global Resource Center. Available online: https://genes-r-us.uthscsa.edu/resources/consumer/statemap.htm (accessed on 21 October 2020).
- Chuang, D.T.; Shih, V.E. Disorders of branched-chain amino acid and keto acid metabolism. In The Metabolic and Molecular Basis of Inherited Disease, 7th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 1995; pp. 1239–1277. [Google Scholar]
- Oglesbee, D.; Sanders, K.A.; Lacey, J.M.; Magera, M.J.; Casetta, B.; Strauss, K.A.; Tortorelli, S.; Rinaldo, P.; Matern, D. Second-tier test for quantification of alloisoleucine and branched-chain amino acids in dried blood spots to improve newborn screening for maple syrup urine disease (MSUD). Clin. Chem. 2008, 54, 542–549. [Google Scholar] [CrossRef]
- Puckett, R.L.; Lorey, F.; Rinaldo, P.; Lipson, M.H.; Matern, D.; Sowa, M.E.; Levine, S.; Chang, R.; Wang, R.Y.; Abdenur, J.E. Maple syrup urine disease: Further evidence that newborn screening may fail to identify variant forms. Mol. Genet. Metab. 2010, 100, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Boemer, F.; Schoos, R.; de Halleux, V.; Kalenga, M.; Debray, F.G. Surprising causes of C5-carnitine false positive results in newborn screening. Mol. Genet. Metab. 2014, 111, 52–54. [Google Scholar] [CrossRef]
- Poggiali, S.; Ombrone, D.; Forni, G.; Malvagia, S.; Funghini, S.; Mura, M.; Pasquini, E.; Santoro, L.; Bellavia, V.; Granata, O.M.; et al. Reducing the False-Positive Rate for Isovalerylcarnitine in Expanded Newborn Screening: The Application of a Second-Tier Test. J. Inborn Errors Metab. Screen. 2016, 4, 1–7. [Google Scholar] [CrossRef]
- Carling, R.S.; Burden, D.; Hutton, I.; Randle, R.; John, K.; Bonham, J.R. Introduction of a Simple Second Tier Screening Test for C5 Isobars in Dried Blood Spots: Reducing the False Positive Rate for Isovaleric Acidaemia in Expanded Newborn Screening. JIMD Rep. 2018, 38, 75–80. [Google Scholar]
- Ombrone, D.; Giocaliere, E.; Forni, G.; Malvagia, S.; la Marca, G. Expanded newborn screening by mass spectrometry: New tests, future perspectives. Mass Spectrom. Rev. 2016, 35, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, M.R.; Hörster, F.; Dionisi-Vici, C.; Haliloglu, G.; Karall, D.; Chapman, K.A.; Huemer, M.; Hochuli, M.; Assoun, M.; Ballhausen, D.; et al. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J. Rare Dis. 2014, 9, 130. [Google Scholar] [CrossRef]
- Estrella, J.; Wilcken, B.; Carpenter, K.; Bhattacharya, K.; Tchan, M.; Wiley, V. Expanded newborn screening in New South Wales: Missed cases. J. Inherit. Metab. Dis. 2014, 37, 881–887. [Google Scholar] [CrossRef]
- la Marca, G.; Malvagia, S.; Pasquini, E.; Innocenti, M.; Donati, M.A.; Zammarchi, E. Rapid 2nd-tier test for measurement of 3-OH-propionic and methylmalonic acids on dried blood spots: Reducing the false-positive rate for propionylcarnitine during expanded newborn screening by liquid chromatography-tandem mass spectrometry. Clin. Chem. 2007, 53, 1364–1369. [Google Scholar] [CrossRef]
- Turgeon, C.T.; Magera, M.J.; Cuthbert, C.D.; Loken, P.R.; Gavrilov, D.K.; Tortorelli, S.; Raymond, K.M.; Oglesbee, D.; Rinaldo, P.; Matern, D. Determination of total homocysteine, methylmalonic acid, and 2-methylcitric acid in dried blood spots by tandem mass spectrometry. Clin. Chem. 2010, 56, 1686–1695. [Google Scholar] [CrossRef]
- Malvagia, S.; Haynes, C.A.; Grisotto, L.; Ombrone, D.; Funghini, S.; Moretti, E.; McGreevy, K.S.; Biggeri, A.; Guerrini, R.; Yahyaoui, R.; et al. Heptadecanoylcarnitine (C17) a novel candidate biomarker for newborn screening of propionic and methylmalonic acidemias. Clin. Chim. Acta 2015, 450, 342–348. [Google Scholar] [CrossRef]
- Huemer, M.; Kožich, V.; Rinaldo, P.; Baumgartner, M.R.; Merinero, B.; Pasquini, E.; Ribes, A.; Blom, H.J. Newborn screening for homocystinurias and methylation disorders: Systematic review and proposed guidelines. J. Inherit. Metab. Dis. 2015, 38, 1007–1019. [Google Scholar] [CrossRef] [PubMed]
- Ryckman, K.K.; Cook, D.E.; Berberich, S.L.; Shchelochkov, O.A.; Berends, S.K.; Busch, T.; Dagle, J.M.; Murray, J.C. Replication of clinical associations with 17-hydroxyprogesterone in preterm newborns. J. Pediatr. Endocrinol. Metab. 2012, 25, 301–305. [Google Scholar] [CrossRef]
- Vats, P.; Dabas, A.; Jain, V.; Seth, A.; Yadav, S.; Kabra, M.; Gupta, N.; Singh, P.; Sharma, R.; Kumar, R.; et al. Newborn Screening and Diagnosis of Infants with Congenital Adrenal Hyperplasia. Indian Pediatr. 2020, 57, 49–55. [Google Scholar] [CrossRef]
- Fingerhut, R. False positive rate in newborn screening for congenital adrenal hyperplasia (CAH)-ether extraction reveals two distinct reasons for elevated 17alpha-hydroxyprogesterone (17-OHP) values. Steroids 2009, 74, 662–665. [Google Scholar] [CrossRef]
- Schwarz, E.; Liu, A.; Randall, H.; Haslip, C.; Keune, F.; Murray, M.; Longo, N.; Pasquali, M. Use of steroid profiling by UPLC-MS/MS as a second tier test in newborn screening for congenital adrenal hyperplasia: The Utah experience. Pediatr. Res. 2009, 66, 230–235. [Google Scholar] [CrossRef]
- Seo, J.Y.; Park, H.D.; Kim, J.W.; Oh, H.J.; Yang, J.S.; Chang, Y.S.; Park, W.S.; Lee, S.Y. Steroid profiling for congenital adrenal hyperplasia by tandem mass spectrometry as a second-tier test reduces follow-up burdens in a tertiary care hospital: A retrospective and prospective evaluation. J. Perinat. Med. 2014, 42, 121–127. [Google Scholar] [CrossRef]
- Puck, J.M. The case for newborn screening for severe combined immunodeficiency and related disorders. Ann. N. Y. Acad. Sci. 2011, 1246, 108–117. [Google Scholar] [CrossRef]
- Van Der Spek, J.; Groenwold, R.H.; Van Der Burg, M.; van Montfrans, J.M. TREC Based Newborn Screening for Severe Combined Immunodeficiency Disease: A Systematic Review. J. Clin. Immunol. 2015, 35, 416–430. [Google Scholar] [CrossRef] [PubMed]
- La Marca, G.; Giocaliere, E.; Malvagia, S.; Funghini, S.; Ombrone, D.; Della Bona, M.L.; Vanessa, C.; Lippi, F.; Romano, F.; Guerrini, R.; et al. The inclusion of ADA-SCID in expanded newborn screening by tandem mass spectrometry. J. Pharm. Biomed. Anal. 2014, 88, 201–206. [Google Scholar] [CrossRef] [PubMed]
- La Marca, G.; Giocoliere, E.; Malvagia, S.; Villanelli, F.; Funghini, S.; Ombrone, D.; Della Bona, M.; Forni, G.; Canessa, C.; Ricci, S.; et al. Development and validation of a 2nd tier test for identification of purine nucleoside phosphorylase deficiency patients during expanded newborn screening by liquid chromatography-tandem mass spectrometry. Clin. Chem. Lab. Med. 2016, 54, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Hattori, K.; Endo, F. Newborn screening for lysosomal storage disorders. Am. J. Med. Genet. C Semin. Med. Genet. 2011, 157C, 63–71. [Google Scholar] [CrossRef]
- Hopkins, P.V.; Campbell, C.; Klug, T.; Rogers, S.; Raburn-Miller, J.; Kiesling, J. Lysosomal storage disorder screening implementation: Findings from the first six months of full population pilot testing in Missouri. J. Pediatr. 2015, 166, 172–177. [Google Scholar] [CrossRef]
- Camargo Neto, E.; Schulte, J.; Pereira, J.; Bravo, H.; Sampaio-Filho, C.; Giugliani, R. Neonatal screening for four lysosomal storage diseases with a digital microfluidics platform: Initial results in Brazil. Genet. Mol. Biol. 2018, 41, 414–416. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.D.; Bainbridge, M.N.; Parad, R.B.; Bhattacharjee, A. Second Tier Molecular Genetic Testing in Newborn Screening for Pompe Disease: Landscape and Challenges. Int. J. Neonatal Screen. 2020, 6, 32. [Google Scholar] [CrossRef]
- Gelb, M.H. Newborn Screening for Lysosomal Storage Diseases: Methodologies, Screen Positive Rates, Normalization of Datasets, Second-Tier Tests, and Post-Analysis Tools. Int. J. Neonatal Screen. 2018, 4, 23. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.C.; Hwu, W.L.; Lee, N.C.; Hsu, L.W.; Chien, Y.H. Algorithm for Pompe disease newborn screening: Results from the Taiwan screening program. Mol. Genet. Metab. 2012, 106, 281–286. [Google Scholar] [CrossRef]
- Tortorelli, S.; Eckerman, J.S.; Orsini, J.J.; Stevens, C.; Hart, J.; Hall, P.L.; Alexander, J.J.; Gavrilov, D.; Oglesbee, D.; Raymond, K.; et al. Moonlighting newborn screening markers: The incidental discovery of a second-tier test for Pompe disease. Genet. Med. 2018, 20, 840–846. [Google Scholar] [CrossRef]
- Carducci, C.; Santagata, S.; Leuzzi, V.; Carducci, C.; Artiola, C.; Giovanniello, T.; Battini, R.; Antonozzi, I. Quantitative determination of guanidinoacetate and creatine in dried blood spot by flow injection analysis-electrospray tandem mass spectrometry. Clin. Chim. Acta 2006, 364, 180–187. [Google Scholar] [CrossRef]
- Van der Tol, L.; Smid, B.E.; Poorthuis, B.J.; Biegstraaten, M.; Deprez, R.H.; Linthorst, G.E.; Hollak, C.E. A systematic review on screening for Fabry disease: Prevalence of individuals with genetic variants of unknown significance. J. Med. Genet. 2014, 51, 1–9. [Google Scholar] [CrossRef]
- Aerts, J.M.; Groener, J.E.; Kuiper, S.; Donker-Koopman, W.E.; Strijland, A.; Ottenhoff, R.; van Roomen, C.; Mirzaian, M.; Wijburg, F.A.; Linthorst, G.E.; et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc. Natl. Acad. Sci. USA 2008, 105, 2812–2817. [Google Scholar] [CrossRef] [PubMed]
- Rombach, S.M.; Dekker, N.; Bouwman, M.G.; Linthorst, G.E.; Zwinderman, A.H.; Wijburg, F.A.; Kuiper, S.; Vd Bergh Weerman, M.A.; Groener, J.E.M.; Poorthuis, B.J.; et al. Plasma globotriaosylsphingosine: Diagnostic value and relation to clinical manifestations of Fabry disease. Biochim. Biophys. Acta 2010, 1802, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Gold, H.; Mirzaian, M.; Dekker, N.; Joao Ferraz, M.; Lugtenburg, J.; Codee, J.D.C.; van der Marel, G.A.; Overkleeft, H.S.; Linthorst, G.E.; Groener, J.E.; et al. Quantification of Globotriaosylsphingosine in Plasma and Urine of Fabry Patients by Stable Isotope Ultraperformance Liquid Chromatography Tandem Mass Spectrometry. Clin. Chem. 2013, 59, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Boutin, M.; Lavoie, P.; Abaoui, M.; Auray-Blais, C. Tandem Mass Spectrometry Quantitation of LysoGb3 and Six Related Analogs in Plasma for Fabry Disease Patients. Curr. Protoc. Hum. Genet. 2016, 90. [Google Scholar]
- Dajnoki, A.; Fekete, G.; Keutzer, J.; Orsini, J.J.; De Jesus, V.R.; Chien, Y.H.; Hwu, W.L.; Lukacs, Z.; Mühl, A.; Zhang, X.K.; et al. Newborn screening for Fabry disease by measuring GLA activity using tandem mass spectrometry. Clin. Chim. Acta. 2010, 411, 1428–1431. [Google Scholar] [CrossRef]
- Johnson, B.; Mascher, H.; Mascher, D.; Legnini, E.; Hung, C.Y.; Dajnoki, A.; Chien, Y.H.; Maródi, L.; Hwu, W.L.; Bodamer, O.A. Analysis of lyso-globotriaosylsphingosine in dried blood spots. Ann. Lab. Med. 2013, 33, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.J.; Ceuterick, C. Prenatal pathology in mucopolysaccharidoses: A comparison with postnatal cases. Clin. Neuropathol. 1983, 2, 122–127. [Google Scholar]
- Gabrielli, O.; Clarke, L.A.; Ficcadenti, A.; Santoro, L.; Zampini, L.; Volpi, N.; Coppa, G.V. 12 year follow up of enzyme-replacement therapy in two siblings with attenuated mucopolysaccharidosis I: The important role of early treatment. BMC Med. Genet. 2016, 17, 19. [Google Scholar] [CrossRef]
- de Ruijter, J.; de Ru, M.H.; Wagemans, T.; Ijlst, L.; Lund, A.M.; Orchard, P.J.; Schaefer, G.B.; Wijburg, F.A.; van Vlies, N. Heparan sulfate and dermatan sulfate derived disaccharides are sensitive markers for newborn screening for mucopolysaccharidoses types I, II and III. Mol. Genet. Metab. 2012, 107, 705–710. [Google Scholar] [CrossRef]
- Kubaski, F.; Mason, R.W.; Nakatomi, A.; Shintaku, H.; Xie, L.; van Vlies, N.N.; Church, H.; Giugliani, R.; Kobayashi, H.; Yamaguchi, S.; et al. Newborn screening for mucopolysaccharidoses: A pilot study of measurement of glycosaminoglycans by tandem mass spectrometry. J. Inherit. Metab. Dis. 2017, 40, 151–158. [Google Scholar] [CrossRef]
- Kubaski, F.; Suzuki, Y.; Orii, K.; Giugliani, R.; Church, H.J.; Mason, R.W.; Dũng, V.C.; Ngoc, C.T.; Yamaguchi, S.; Kobayashi, H.; et al. Glycosaminoglycan Levels in Dried Blood Spots of Patients with Mucopolysaccharidoses and Mucolipidoses. Mol. Genet. Metab. 2017, 120, 247–254. [Google Scholar] [CrossRef]
- Tomatsu, S.; Montaño, A.M.; Oguma, T.; Dung, V.C.; Oikawa, H.; de Carvalho, T.G.; Gutiérrez, M.L.; Yamaguchi, S.; Suzuki, Y.; Fukushi, M.; et al. Dermatan sulfate and heparan sulfate as a biomarker for mucopolysaccharidosis I. J. Inherit. Metab. Dis. 2010, 33, 141–150. [Google Scholar] [CrossRef]
- Burlina, A.B.; Polo, G.; Rubert, L.; Gueraldi, D.; Cazzorla, C.; Duro, G.; Salviati, L.; Burlina, A.P. Implementation of Second-Tier Tests in Newborn Screening for Lysosomal Disorders in North Eastern Italy. Int. J. Neonatal Screen. 2019, 5, 24. [Google Scholar] [CrossRef]
- Stapleton, M.; Kubaski, F.; Mason, R.W.; Shintaku, H.; Kobayashi, H.; Yamaguchi, S.; Taketani, T.; Suzuki, Y.; Orii, K.; Orii, T.; et al. Newborn Screening for Mucopolysaccharidoses: Measurement of Glycosaminoglycans by LC-MS/MS. Mol. Genet. Metab. Rep. 2020, 22, 100563. [Google Scholar] [CrossRef]
- Polo, G.; Gueraldi, D.; Giuliani, A.; Rubert, L.; Cazzorla, C.; Salviati, L.; Marzollo, A.; Biffi, A.; Burlina, A.P.; Burlina, A.B. The combined use of enzyme activity and metabolite assays as a strategy for newborn screening of mucopolysaccharidosis type I. Clin. Chem. Lab. Med. 2020. [Google Scholar] [CrossRef]
- Dekker, N.; van Dussen, L.; Hollak, C.E.; Overkleeft, H.; Scheij, S.; Ghauharali, K.; van Breemen, M.J.; Ferraz, M.J.; Groener, J.E.; Maas, M.; et al. Elevated plasma glucosylsphingosine in Gaucher disease: Relation to phenotype, storage cell markers, and therapeutic response. Blood 2011, 118, e118–e127. [Google Scholar] [CrossRef]
- Rolfs, A.; Giese, A.K.; Grittner, U.; Mascher, D.; Elstein, D.; Zimran, A.; Böttcher, T.; Lukas, J.; Hübner, R.; Gölnitz, U.; et al. Glucosylsphingosine is a highly sensitive and specific biomarker for primary diagnostic and follow-up monitoring in Gaucher disease in a non-Jewish, Caucasian cohort of Gaucher disease patients. PLoS ONE 2013, 8, e79732. [Google Scholar] [CrossRef]
- Fuller, M.; Szer, J.; Stark, S.; Fletcher, J.M. Rapid, Single-Phase Extraction of Glucosylsphingosine From Plasma: A Universal Screening and Monitoring Tool. Clin. Chim. Acta 2015, 450, 6–10. [Google Scholar] [CrossRef]
- Duffner, P.K.; Caggana, M.; Orsini, J.J.; Wenger, D.A.; Patterson, M.C.; Crosley, C.J.; Kurtzberg, J.; Arnold, G.L.; Escolar, M.L.; Adams, D.J.; et al. Newborn screening for Krabbe disease: The New York State model. Pediatr. Neurol. 2009, 40, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Orsini, J.J.; Saavedra-Matiz, C.A.; Gelb, M.H.; Caggana, M. Newborn screening for Krabbe’s disease. J. Neurosci. Res. 2016, 94, 1063–1075. [Google Scholar] [CrossRef] [PubMed]
- Chuang, W.L.; Pacheco, J.; Zhang, X.K.; Martin, M.M.; Biski, C.K.; Keutzer, J.M.; Wenger, D.A.; Caggana, M.; Orsini, J.J., Jr. Determination of psychosine concentration in dried blood spots from newborns that were identified via newborn screening to be at risk for Krabbe disease. Clin. Chim. Acta 2013, 419, 73–76. [Google Scholar] [CrossRef]
- Escolar, M.L.; Kiely, B.T.; Shawgo, E.; Hong, X.; Gelb, M.H.; Orsini, J.J.; Matern, D.; Poe, M.D. Psychosine, a marker of Krabbe phenotype and treatment effect. Mol. Genet. Metab. 2017, 121, 271–278. [Google Scholar] [CrossRef]
- Turgeon, C.T.; Orsini, J.J.; Sanders, K.A.; Magera, M.J.; Langan, T.J.; Escolar, M.L.; Duffner, P.; Oglesbee, D.; Gavrilov, D.; Tortorelli, S.; et al. Measurement of psychosine in dried blood spots—a possible improvement to newborn screening programs for Krabbe disease. J. Inherit. Metab. Dis. 2015, 38, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Eastman, J.W.; Sherwin, J.E.; Wong, R.; Liao, C.L.; Currier, R.J.; Lorey, F.; Cunningham, G. Use of the phenylalanine:tyrosine ratio to test newborns for phenylketonuria in a large public health screening programme. J. Med. Screen. 2000, 7, 131–135. [Google Scholar] [CrossRef]
- Marquardt, G.; Currier, R.; McHugh, D.M.; Gavrilov, D.; Magera, M.J.; Matern, D.; Oglesbee, D.; Raymond, K.; Rinaldo, P.; Smith, E.H.; et al. Enhanced interpretation of newborn screening results without analyte cutoff values. Genet. Med. 2012, 14, 648–655. [Google Scholar] [CrossRef]
- Hall, P.L.; Marquardt, G.; McHugh, D.M.S.; Currier, R.J.; Tang, H.; Stoway, S.D.; Rinaldo, P. Postanalytical tools improve performance of newborn screening by tandem mass spectrometry. Genet. Med. 2014, 16, 889–895. [Google Scholar] [CrossRef]
- Mørkrid, L.; Rowe, A.D.; Elgstoen, K.B.; Olesen, J.H.; Ruijter, G.; Hall, P.L.; Tortorelli, S.; Schulze, A.; Kyriakopoulou, L.; Wamelink, M.M.; et al. Continuous age- and sex-adjusted reference intervals of urinary markers for cerebral creatine deficiency syndromes: A novel approach to the definition of reference intervals. Clin. Chem. 2015, 61, 760–768. [Google Scholar] [CrossRef]
- Saavedra-Matiz, C.A.; Isabelle, J.T.; Biski, C.K.; Duva, S.J.; Sweeney, M.L.; Parker, A.L.; Young, A.J.; Diantonio, L.L.; Krein, L.M.; Nichols, M.J.; et al. Cost-effective and scalable DNA extraction method from dried blood spots. Clin. Chem. 2013, 59, 1045–1051. [Google Scholar] [CrossRef]
- Jinks, D.C.; Minter, M.; Tarver, D.A.; Vanderford, M.; Hejtmancik, J.F.; McCabe, E.R.B. Molecular genetic diagnosis of sickle cell disease using dried blood specimens on blotters used for newborn screening. Hum. Genet. 1989, 81, 363–366. [Google Scholar] [CrossRef]
- Gregg, R.G.; Wilfond, B.S.; Farrell, P.M.; Laxova, A.; Hassemer, D.; Mischler, E.H. Application of DNA analysis in a population-screening program for neonatal diagnosis of cystic fibrosis (CF): Comparison of screening protocols. Am. J. Hum. Genet. 1993, 52, 616–626. [Google Scholar]
- Southern, K.W.; Littlewood, J.M. Newborn screening programmes for cystic fibrosis. Paediatr. Respir. Rev. 2003, 4, 299–305. [Google Scholar] [CrossRef]
- Sartippour, M.R.; Doroudian, R.; Frampton, G.; Lorey, F.; Helmer, G.; Ho, T.; Bhandal, A. Identification of galactose-1-phosphate uridyl transferase gene common mutations in dried blood spots. Clin. Chim. Acta 2014, 436, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Strand, J.; Gul, K.A.; Erichsen, H.C.; Lundman, E.; Berge, M.C.; Trømborg, A.K.; Sørgjerd, L.K.; Ytre-Arne, M.; Hogner, S.; Halsne, R.; et al. Second-Tier Next Generation Sequencing Integrated in Nationwide Newborn Screening Provides Rapid Molecular Diagnostics of Severe Combined Immunodeficiency. Front. Immunol. 2020, 11, 1417. [Google Scholar] [CrossRef]
- Tangeraas, T.; Sæves, I.; Klingenberg, C.; Jørgensen, J.; Kristensen, E.; Gunnarsdottir, G.; Hansen, E.V.; Strand, J.; Lundman, E.; Ferdinandusse, S.; et al. Performance of Expanded Newborn Screening in Norway Supported by Post-Analytical Bioinformatics Tools and Rapid Second-Tier DNA Analyses. Int. J. Neonatal Screen. 2020, 6, 51. [Google Scholar] [CrossRef] [PubMed]
- Barendsen, R.W.; Dijkstra, I.M.E.; Visser, W.F.; Alders, M.; Bliek, J.; Boelen, A.; Bouva, M.J.; van der Crabben, S.N.; Elsinghorst, E.; van Gorp, A.G.M.; et al. Adrenoleukodystrophy Newborn Screening in the Netherlands (SCAN Study): The X-Factor. Front. Cell Dev. Biol. 2020, 8, 499. [Google Scholar] [CrossRef]
- Almannai, M.; Marom, R.; Sutton, V.R. Newborn screening: A review of history, recent advancements, and future perspectives in the era of next generation sequencing. Curr. Opin. Pediatr. 2016, 28, 694–699. [Google Scholar] [CrossRef]
- Baker, M.W.; Atkins, A.E.; Cordovado, S.K.; Hendrix, M.; Earley, M.C.; Farrell, P.M. Improving newborn screening for cystic fibrosis using next-generation sequencing technology: A technical feasibility study. Genet. Med. 2016, 18, 231–238. [Google Scholar] [CrossRef]
- Narravula, A.; Garber, K.B.; Askree, S.H.; Hegde, M.; Hall, P.L. Variants of uncertain significance in newborn screening disorders: Implications for large-scale genomic sequencing. Genet. Med. 2017, 19, 77–82. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malvagia, S.; Forni, G.; Ombrone, D.; la Marca, G. Development of Strategies to Decrease False Positive Results in Newborn Screening. Int. J. Neonatal Screen. 2020, 6, 84. https://doi.org/10.3390/ijns6040084
Malvagia S, Forni G, Ombrone D, la Marca G. Development of Strategies to Decrease False Positive Results in Newborn Screening. International Journal of Neonatal Screening. 2020; 6(4):84. https://doi.org/10.3390/ijns6040084
Chicago/Turabian StyleMalvagia, Sabrina, Giulia Forni, Daniela Ombrone, and Giancarlo la Marca. 2020. "Development of Strategies to Decrease False Positive Results in Newborn Screening" International Journal of Neonatal Screening 6, no. 4: 84. https://doi.org/10.3390/ijns6040084
APA StyleMalvagia, S., Forni, G., Ombrone, D., & la Marca, G. (2020). Development of Strategies to Decrease False Positive Results in Newborn Screening. International Journal of Neonatal Screening, 6(4), 84. https://doi.org/10.3390/ijns6040084