Translating Molecular Technologies into Routine Newborn Screening Practice



Abstract
1. Introduction
2. Molecular Marker Identification as Primary Screening Methods in NBS for Severe Combined Immunodeficiency (SCID) and Spinal Muscular Atrophy (SMA)
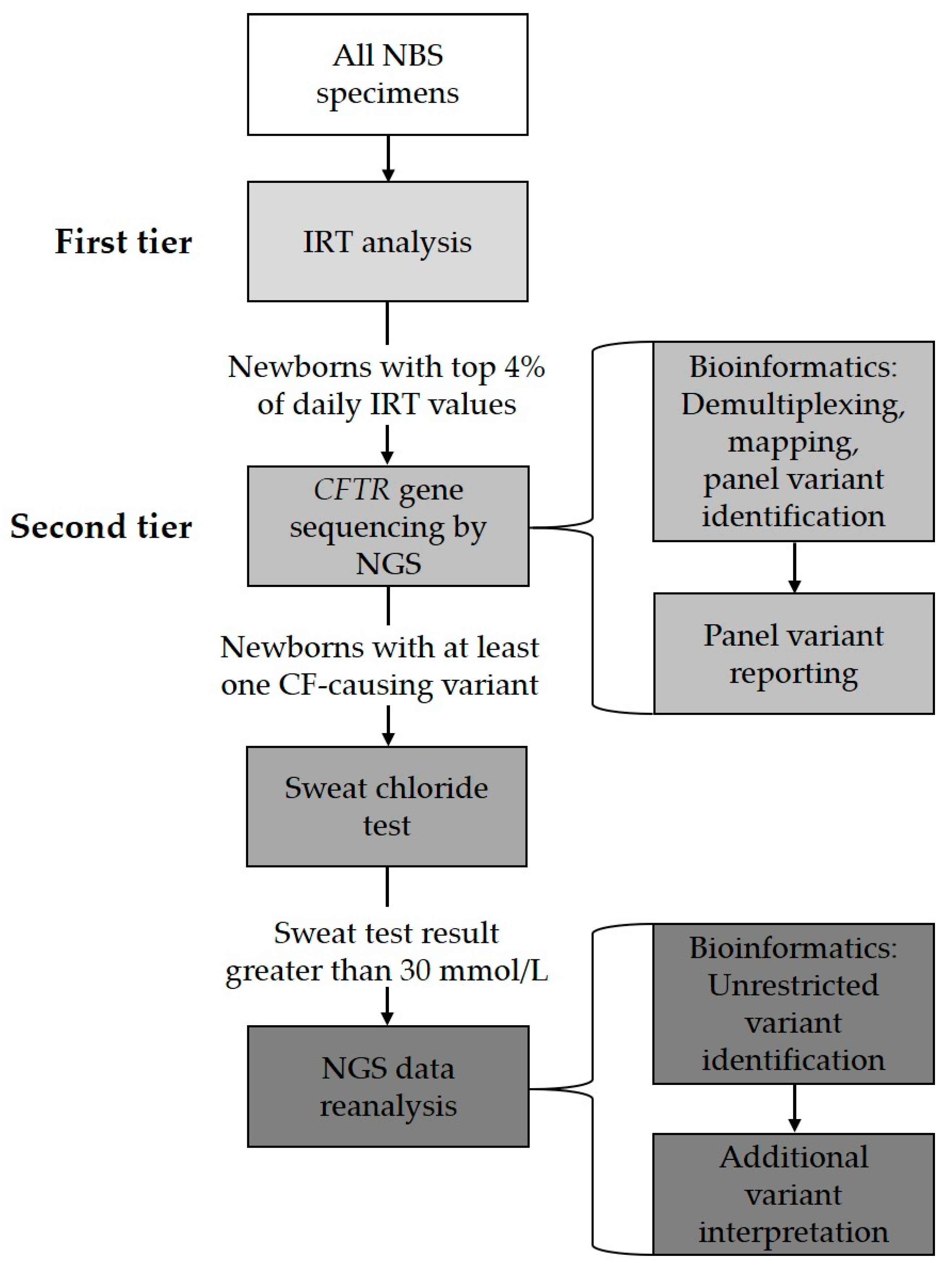
3. Targeted Gene Variant Panel as Second-Tier Testing
4. Targeted Gene Variant or Variants as “Just-In-Time” Information
5. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Centers for Disease Control and Prevention. CDC grand rounds: Newborn screening and improved outcomes. MMWR Morbid. Mortal. Wkly. Rep. 2012, 61, 390–393. [Google Scholar]
- Jinks, D.C.; Minter, M.; Tarver, D.A.; Vanderford, M.; Hejtmancik, J.F.; McCabe, E.R. Molecular genetic diagnosis of sickle cell disease using dried blood specimens on blotters used for newborn screening. Hum. Genet. 1989, 81, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Gregg, R.; Wilfond, B.; Farrell, P.; Laxova, A.; Hassemer, D.; Mischler, E. Application of DNA analysis in a population-screening program for neonatal diagnosis of cystic fibrosis (CF): Comparison of screening protocols. Am. J. Hum. Genet. 1993, 52, 616–626. [Google Scholar] [PubMed]
- Baker, M.W.; Groose, M.; Hoffman, G.; Rock, M.; Levy, H.; Farrell, P.M. Optimal DNA tier for the IRT/DNA algorithm determined by CFTR mutation results over 14 years of newborn screening. J. Cyst. Fibros. 2011, 10, 278–281. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, E.; Ryall, R.G.; Morris, C.P.; Nelson, P.V.; Carey, W.F.; Pollard, A.C.; Robertson, E.F. Neonatal screening strategy for cystic fibrosis using immunoreactive trypsinogen and direct gene analysis. Brit. Med. J. 1991, 302, 1237–1240. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Southern, K.W.; Munck, A.; Pollitt, R.; Travert, G.; Zanolla, L.; Dankert-Roelse, J.; Castellani, C.; ECFS CF Neonatal Screening Working Group. A survey of newborn screening for cystic fibrosis in Europe. J. Cyst. Fibros. 2007, 6, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Puck, J.M.; SCID Newborn Screening Group. Population-based newborn screening for severe combined immunodeficiency: Steps toward implementation. J. Allergy Clin. Immun. 2007, 120, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Lindegren, M.L.; Kobrynski, L.; Rasmussen, S.A.; Moore, C.A.; Grosse, S.D.; Vanderford, M.L.; Spira, T.J.; McDougal, J.S.; Vogt, R.F., Jr.; Hannon, W.H. Applying public health strategies to primary immunodeficiency diseases: A potential approach to genetic disorders. MMWR Recomm. Rep. 2004, 53, 1–29. [Google Scholar]
- Chan, K.; Puck, J.M. Development of population-based newborn screening for severe combined immunodeficiency. J. Allergy Clin. Immun. 2005, 115, 391–398. [Google Scholar] [CrossRef]
- Douek, D.C.; McFarland, R.D.; Keiser, P.H.; Gage, E.A.; Massey, J.M.; Haynes, B.F.; Polis, M.A.; Haase, A.T.; Feinberg, M.B.; Sullivan, J.L. Changes in thymic function with age and during the treatment of HIV infection. Nature 1998, 396, 690–695. [Google Scholar] [CrossRef]
- Baker, M.W.; Grossman, W.J.; Laessig, R.H.; Hoffman, G.L.; Brokopp, C.D.; Kurtycz, D.F.; Cogley, M.F.; Litsheim, T.J.; Katcher, M.L.; Routes, J.M. Development of a routine newborn screening protocol for severe combined immunodeficiency. J. Allergy Clin. Immun. 2009, 124, 522–527. [Google Scholar] [CrossRef]
- Verbsky, J.W.; Baker, M.W.; Grossman, W.J.; Hintermeyer, M.; Dasu, T.; Bonacci, B.; Reddy, S.; Margolis, D.; Casper, J.; Gries, M.; et al. Newborn screening for severe combined immunodeficiency; The Wisconsin experience (2008–2011). J. Clin. Immunol. 2012, 32, 83–88. [Google Scholar] [CrossRef]
- Newborn Screening for Severe Combined Immunodeficiency a Summary of the Evidence and Advisory Committee Decision. Available online: https://www.hrsa.gov/sites/default/files/hrsa/advisory-committees/heritable-disorders/rusp/previous-nominations/scid-27-june-2018.pdf (accessed on 21 September 2020).
- NewSTEPs. Newborn Screening Status for All Disorder. Available online: https://www.newsteps.org/resources/data-visualizations/newborn-screening-status-all-disorders (accessed on 21 September 2020).
- Kwan, A.; Abraham, R.S.; Currier, R.; Brower, A.; Andruszewski, K.; Abbott, J.K.; Baker, M.; Ballow, M.; Bartoshesky, L.E.; Bonagura, V.R. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA 2014, 312, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Al-Mousa, H.; Al-Dakheel, G.; Jabr, A.; Elbadaoui, F.; Abouelhoda, M.; Baig, M.; Monies, D.; Meyer, B.; Hawwari, A.; Dasouki, M. High incidence of severe combined immunodeficiency disease in Saudi Arabia detected through combined T cell receptor excision circle and next generation sequencing of newborn dried blood spots. Front. Immunol. 2018, 9, 782. [Google Scholar] [CrossRef]
- Argudo-Ramírez, A.; Martín-Nalda, A.; Marín-Soria, J.L.; López-Galera, R.M.; Pajares-García, S.; González de Aledo-Castillo, J.M.; Martínez-Gallo, M.; García-Prat, M.; Colobran, R.; Riviere, J.G. First universal newborn screening program for severe combined immunodeficiency in Europe. Two-years’ experience in Catalonia (Spain). Front. Immunol. 2019, 10, 2406. [Google Scholar] [CrossRef] [PubMed]
- Audrain, M.A.; Léger, A.J.; Hémont, C.A.; Mirallié, S.M.; Cheillan, D.; Rimbert, M.G.; Le Thuaut, A.M.; Sébille-Rivain, V.A.; Prat, A.; Pinel, E.M. Newborn screening for severe combined immunodeficiency: Analytic and clinical performance of the T cell receptor excision circle assay in France (DEPISTREC study). J. Clin. Immunol. 2018, 38, 778–786. [Google Scholar] [CrossRef]
- Barbaro, M.; Ohlsson, A.; Borte, S.; Jonsson, S.; Zetterström, R.H.; King, J.; Winiarski, J.; von Döbeln, U.; Hammarström, L. Newborn screening for severe primary immunodeficiency diseases in Sweden—A 2-year pilot TREC and KREC screening study. J. Clin. Immunol. 2017, 37, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Blom, M.; Bredius, R.G.; Weijman, G.; Dekkers, E.H.; Kemper, E.A.; den Akker-van Marle, V.; Elske, M.; Van der Ploeg, C.P.; Van der Burg, M.; Schielen, P.C. Introducing newborn screening for severe combined immunodeficiency (SCID) in the Dutch neonatal screening program. Int. J. Neonatal Screen. 2018, 4, 40. [Google Scholar] [CrossRef]
- Chien, Y.-H.; Yu, H.-H.; Lee, N.-C.; Ho, H.-C.; Kao, S.-M.; Lu, M.-Y.; Jaing, T.-H.; Lee, W.-I.; Chang, K.-W.; Shieh, C.-C. Newborn screening for severe combined immunodeficiency in Taiwan. Int. J. Neonatal Screen. 2017, 3, 16. [Google Scholar] [CrossRef]
- Kanegae, M.P.P.; Barreiros, L.A.; Sousa, J.L.; Brito, M.A.S.; de Oliveira, E.B. Newborn screening for severe combined immunodeficiencies using TRECs and KRECs: Second pilot study in Brazil. Rev. Paul. Pediatr. 2017, 35, 25–32. [Google Scholar] [CrossRef]
- Muramatsu, H.; Kojima, D.; Okuno, Y.; Kataoka, S.; Nakajima, Y.; Ito, T.; Tsuge, I.; Yoshimi, S.; Kato, T.; Kojima, S. Combination of TREC measurement and next-generation sequencing in newborn screening for severe combined immunodeficiency: A pilot program in Japan. Blood 2018, 132, 3717. [Google Scholar] [CrossRef]
- Rechavi, E.; Lev, A.; Saraf-Levy, T.; Etzioni, A.; Almashanu, S.; Somech, R. Newborn screening for severe combined immunodeficiency in Israel. Int. J. Neonatal Screen. 2017, 3, 13. [Google Scholar] [CrossRef]
- Hazenberg, M.D.; Verschuren, M.C.; Hamann, D.; Miedema, F.; Dongen, J.J. T cell receptor excision circles as markers for recent thymic emigrants: Basic aspects, technical approach, and guidelines for interpretation. J. Mol. Med. 2001, 79, 631–640. [Google Scholar] [CrossRef]
- Association of Public Health Laboratories; NewSTEPs. Expanding the Reach of SCID Testing: A Report on the Severe Combined Immunodeficiency Newborn Screening Implementation Experience. Available online: https://www.aphl.org/aboutAPHL/publications/Documents/NBS-2018Nov-SCID-Implementation-Report.pdf (accessed on 21 September 2020).
- Strand, J.; Gul, K.A.; Erichsen, H.C.; Lundman, E.; Berge, M.C.; Trømborg, A.K.; Sørgjerd, L.K.; Ytre-Arne, M.; Hogner, S.; Halsne, R. Second-tier next generation sequencing integrated in nationwide newborn screening provides rapid molecular diagnostics of severe combined immunodeficiency. Front. Immunol. 2020, 11, 1417. [Google Scholar] [CrossRef] [PubMed]
- Kolb, S.J.; Kissel, J.T. Spinal muscular atrophy. Neurol. Clin. 2015, 33, 831–846. [Google Scholar] [CrossRef] [PubMed]
- Calucho, M.; Bernal, S.; Alías, L.; March, F.; Venceslá, A.; Rodríguez-Álvarez, F.J.; Aller, E.; Fernández, R.M.; Borrego, S.; Millán, J.M. Correlation between SMA type and SMN2 copy number revisited: An analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscul. Disord. 2018, 28, 208–215. [Google Scholar] [CrossRef]
- Zerres, K.; Rudnik-Schöneborn, S. Natural history in proximal spinal muscular atrophy: Clinical analysis of 445 patients and suggestions for a modification of existing classifications. Ann. Neurol. 1995, 52, 518–523. [Google Scholar] [CrossRef]
- Newborn Screening for Spinal Muscular Atrophy a Summary of the Evidence and Advisory Committee Decision. Available online: https://www.hrsa.gov/sites/default/files/hrsa/advisory-committees/heritable-disorders/rusp/previous-nominations/sma-consumer-summary.pdf (accessed on 21 September 2020).
- Boemer, F.; Caberg, J.-H.; Dideberg, V.; Dardenne, D.; Bours, V.; Hiligsmann, M.; Dangouloff, T.; Servais, L. Newborn screening for SMA in southern Belgium. Neuromuscul. Disord. 2019, 29, 343–349. [Google Scholar] [CrossRef]
- Chien, Y.-H.; Chiang, S.-C.; Weng, W.-C.; Lee, N.-C.; Lin, C.-J.; Hsieh, W.-S.; Lee, W.-T.; Jong, Y.-J.; Ko, T.-M.; Hwu, W.-L. Presymptomatic diagnosis of spinal muscular atrophy through newborn screening. J. Pediatr. 2017, 190, 124–129. [Google Scholar] [CrossRef]
- Kariyawasam, D.S.; Russell, J.S.; Wiley, V.; Alexander, I.E.; Farrar, M.A. The implementation of newborn screening for spinal muscular atrophy: The Australian experience. Genet. Med. 2020, 22, 557–565. [Google Scholar] [CrossRef]
- Kay, D.M.; Stevens, C.F.; Parker, A.; Saavedra-Matiz, C.A.; Sack, V.; Chung, W.K.; Chiriboga, C.A.; Engelstad, K.; Laureta, E.; Farooq, O. Implementation of population-based newborn screening reveals low incidence of spinal muscular atrophy. Genet. Med. 2020, 22, 1296–1302. [Google Scholar] [CrossRef]
- Vill, K.; Kölbel, H.; Schwartz, O.; Blaschek, A.; Olgemöller, B.; Harms, E.; Burggraf, S.; Röschinger, W.; Durner, J.; Gläser, D. One year of newborn screening for SMA–Results of a German pilot project. J. Neuromuscul. Dis. 2019, 6, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Comeau, A.M.; Parad, R.B.; Dorkin, H.L.; Dovey, M.; Gerstle, R.; Haver, K.; Lapey, A.; O’Sullivan, B.P.; Waltz, D.A.; Zwerdling, R.G. Population-based newborn screening for genetic disorders when multiple mutation DNA testing is incorporated: A cystic fibrosis newborn screening model demonstrating increased sensitivity but more carrier detections. Pediatrics 2004, 113, 1573–1581. [Google Scholar] [CrossRef]
- Wilcken, B. Newborn screening for cystic fibrosis: Techniques and strategies. J. Inherit. Metab. Dis. 2007, 30, 537–543. [Google Scholar] [CrossRef]
- Baker, M.W.; Atkins, A.E.; Cordovado, S.K.; Hendrix, M.; Earley, M.C.; Farrell, P.M. Improving newborn screening for cystic fibrosis using next-generation sequencing technology: A technical feasibility study. Genet. Med. 2016, 18, 231–238. [Google Scholar] [CrossRef]
- United States Cystic Fibrosis Foundation; John Hopkins University; The Hospital for Sick Kids. CFTR2: Clinical and Functional Translation of CFTR. Available online: https://cftr2.org/ (accessed on 21 September 2020).
- Munck, A.; Mayell, S.; Winters, V.; Shawcross, A.; Derichs, N.; Parad, R.; Barben, J.; Southern, K. Cystic fibrosis screen positive, inconclusive diagnosis (CFSPID): A new designation and management recommendations for infants with an inconclusive diagnosis following newborn screening. J. Cyst. Fibros. 2015, 14, 706–713. [Google Scholar] [CrossRef] [PubMed]
- Currier, R.J.; Sciortino, S.; Liu, R.; Bishop, T.; Koupaei, R.A.; Feuchtbaum, L. Genomic sequencing in cystic fibrosis newborn screening: What works best, two-tier predefined CFTR mutation panels or second-tier CFTR panel followed by third-tier sequencing? Genet. Med. 2017, 19, 1159–1163. [Google Scholar] [CrossRef]
- Skov, M.; Bækvad-Hansen, M.; Hougaard, D.M.; Skogstrand, K.; Lund, A.M.; Pressler, T.; Olesen, H.V.; Duno, M. Cystic fibrosis newborn screening in Denmark: Experience from the first 2 years. Pediatr. Pulm. 2020, 55, 549–555. [Google Scholar] [CrossRef]
- Ye, S.; Dhillon, S.; Ke, X.; Collins, A.R.; Day, I.N. An efficient procedure for genotyping single nucleotide polymorphisms. Nucleic Acids Res. 2001, 29, e88. [Google Scholar] [CrossRef]
- Carlock, G.; Fischer, S.T.; Lynch, M.E.; Potter, N.L.; Coles, C.D.; Epstein, M.P.; Mulle, J.G.; Kable, J.A.; Barrett, C.E.; Edwards, S.M. Developmental outcomes in Duarte galactosemia. Pediatrics 2019, 143, e20182516. [Google Scholar] [CrossRef]
- Therrell, B.L., Jr.; Lloyd-Puryear, M.A.; Camp, K.M.; Mann, M.Y. Inborn errors of metabolism identified via newborn screening: Ten-year incidence data and costs of nutritional interventions for research agenda planning. Mol. Genet. Metab. 2014, 113, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Strauss, K.A.; Carson, V.J.; Soltys, K.; Young, M.E.; Bowser, L.E.; Puffenberger, E.G.; Brigatti, K.W.; Williams, K.B.; Robinson, D.L.; Hendrickson, C. Branched-chain α-ketoacid dehydrogenase deficiency (maple syrup urine disease): Treatment, biomarkers, and outcomes. Mol. Genet. Metab. 2020, 129, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Erasmus University Medical Center. Pompe Disease GAA Variant Database. Available online: http://cluster15.erasmusmc.nl/klgn/pompe/mutations.html?lang=en (accessed on 21 September 2020).
- Reuser, A.J.; van der Ploeg, A.T.; Chien, Y.H.; Llerena, J., Jr.; Abbott, M.A.; Clemens, P.R.; Kimonis, V.E.; Leslie, N.; Maruti, S.S.; Sanson, B.J. GAA variants and phenotypes among 1079 patients with Pompe disease: Data from the Pompe Registry. Hum. Mutat. 2019, 40, 2146–2164. [Google Scholar] [CrossRef] [PubMed]
- Beswick, R.; David, M.; Higashi, H.; Thomas, D.; Nourse, C.; Koh, G.; Koorts, P.; Jardine, L.A.; Clark, J.E. Integration of congenital cytomegalovirus screening within a newborn hearing screening programme. J. Paediatr Child. H. 2019, 55, 1381–1388. [Google Scholar] [CrossRef]
- Gantt, S.; Dionne, F.; Kozak, F.K.; Goshen, O.; Goldfarb, D.M.; Park, A.H.; Boppana, S.B.; Fowler, K. Cost-effectiveness of universal and targeted newborn screening for congenital cytomegalovirus infection. JAMA Pediatr. 2016, 170, 1173–1180. [Google Scholar] [CrossRef]
- Baker, M.; Griggs, R.; Byrne, B.; Connolly, A.M.; Finkel, R.; Grajkowska, L.; Haidet-Phillips, A.; Hagerty, L.; Ostrander, R.; Orlando, L. Maximizing the benefit of life-saving treatments for Pompe disease, spinal muscular atrophy, and Duchenne muscular dystrophy through newborn screening: Essential steps. JAMA Neurol. 2019, 76, 978–983. [Google Scholar] [CrossRef]
- Scully, M.A.; Farrell, P.M.; Ciafaloni, E.; Griggs, R.C.; Kwon, J.M. Cystic fibrosis newborn screening: A model for neuromuscular disease screening? Ann. Neurol. 2015, 77, 189–197. [Google Scholar] [CrossRef]


{kind=link}
| Molecular Application | Example Condition | Molecular Marker | Technology |
|---|---|---|---|
| First-tier markers | Severe combined immunodeficiency | T-cell receptor excision circles | Real-time polymerase chain reaction (PCR) |
| Spinal muscular atrophy | Homozygous SMN1 exon 7 deletion | Real-time PCR | |
| Second-tier markers | Cystic fibrosis | CFTR variants | Next generation sequencing |
| Supplemental “just-in-time” 1 information | Galactosemia | GALT c.563A>G, c.404C>T, and c.940A>G | Tetra-primer amplification refractory mutation system (Tetra-primer ARMS)–PCR |
| Maple syrup urine disease | BCKDHA c.1325 T>A | Tetra-primer ARMS–PCR | |
| Sickle cell disease | HBB c.20 A>T | Sanger sequencing | |
| Pompe disease | GAA coding region and intron-exon junctions | Sanger sequencing | |
| Spinal muscular atrophy | SMN2 copy number | Droplet digital PCR |
| Reference | Region | Screening Method | SMN2 Inclusion | Number of Newborns Screened | Reported Incidence in Sample | Study Type |
|---|---|---|---|---|---|---|
| Chien et al. 2017 33 | Taiwan | Real-time PCR SMN1 assay to detect homozygous exon 7 deletion; verified by droplet digital PCR assay | Droplet digital PCR assay to assess SMN2 copy number | 120,267 | 1 in 17,181 | Pilot |
| Boemer et al. 2019 32 | Belgium | Real-time PCR SMN1 assay to detect homozygous exon 7 deletion | No | Not applicable | Not applicable | Pilot |
| Vill et al. 2019 36 | Germany | Real-time PCR SMN1 assay to detect homozygous exon 7 deletion; verified by multiplex ligation-dependent probe amplification (MLPA) | MLPA to assess SMN2 copy number | 165,525 | 1 in 7524 | Pilot |
| Kariyawasam et al. 2020 34 | Australia | Real-time PCR SMN1 assay to detect homozygous exon 7 deletion | Droplet digital PCR assay to assess SMN2 copy number | 103,903 | 1 in 10,390 | Pilot |
| Kay et al. 2020 35 | New York | Real-time PCR SMN1 assay to detect homozygous exon 7 deletion | Real time PCR assay to assess SMN2 copy number | 225,093 | 1 in 28,137 | Routine |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Furnier, S.M.; Durkin, M.S.; Baker, M.W. Translating Molecular Technologies into Routine Newborn Screening Practice. Int. J. Neonatal Screen. 2020, 6, 80. https://doi.org/10.3390/ijns6040080
Furnier SM, Durkin MS, Baker MW. Translating Molecular Technologies into Routine Newborn Screening Practice. International Journal of Neonatal Screening. 2020; 6(4):80. https://doi.org/10.3390/ijns6040080
Chicago/Turabian StyleFurnier, Sarah M., Maureen S. Durkin, and Mei W. Baker. 2020. "Translating Molecular Technologies into Routine Newborn Screening Practice" International Journal of Neonatal Screening 6, no. 4: 80. https://doi.org/10.3390/ijns6040080
APA StyleFurnier, S. M., Durkin, M. S., & Baker, M. W. (2020). Translating Molecular Technologies into Routine Newborn Screening Practice. International Journal of Neonatal Screening, 6(4), 80. https://doi.org/10.3390/ijns6040080