Effects of State of Charge on the Physical Characteristics of V(IV)/V(V) Electrolytes and Membrane for the All Vanadium Flow Battery



Abstract
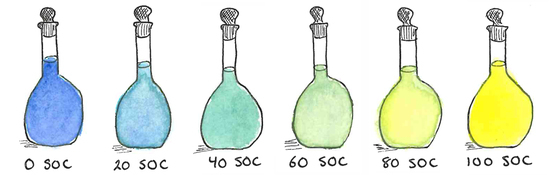
1. Introduction
2. Results and Discussion
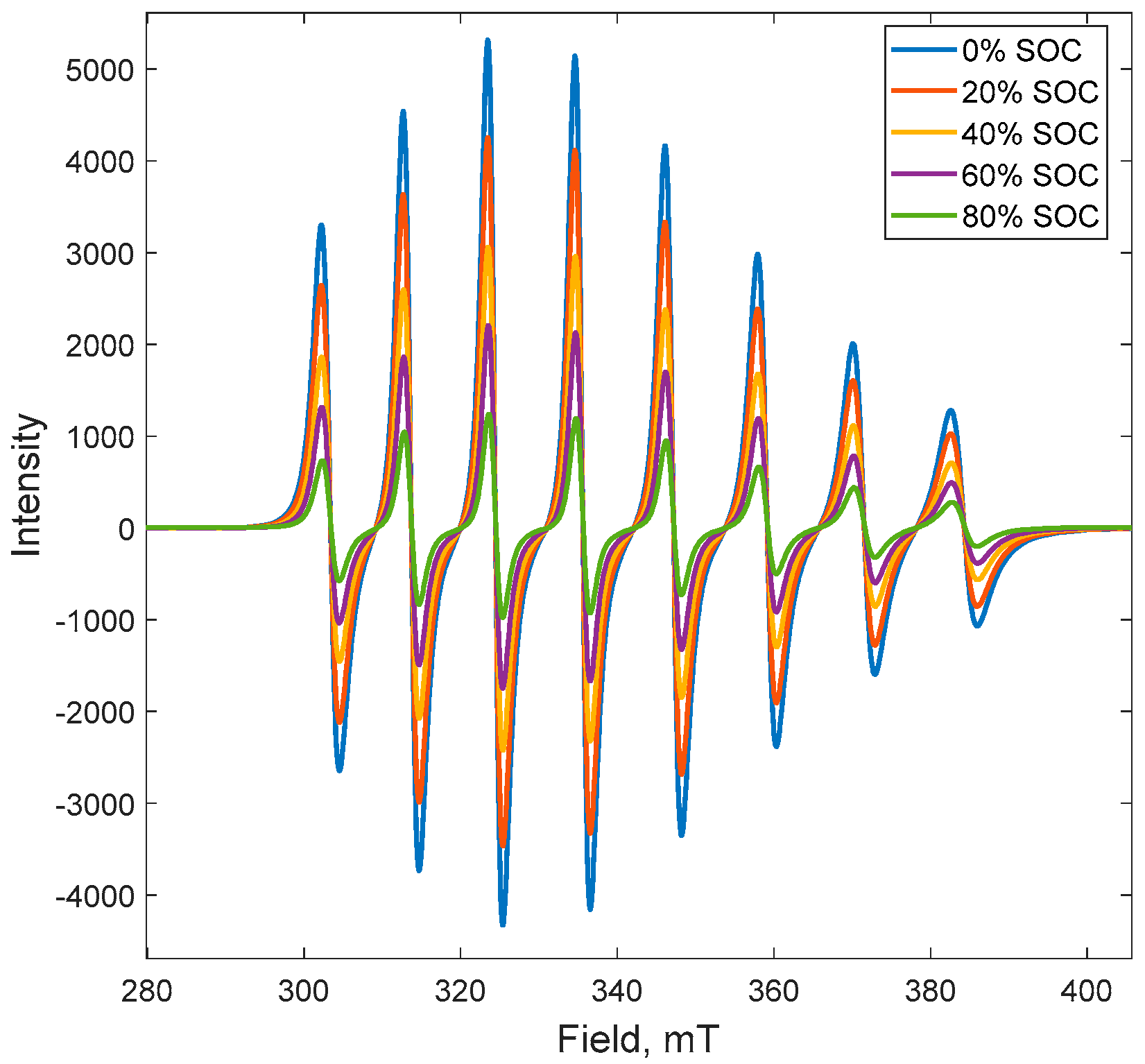
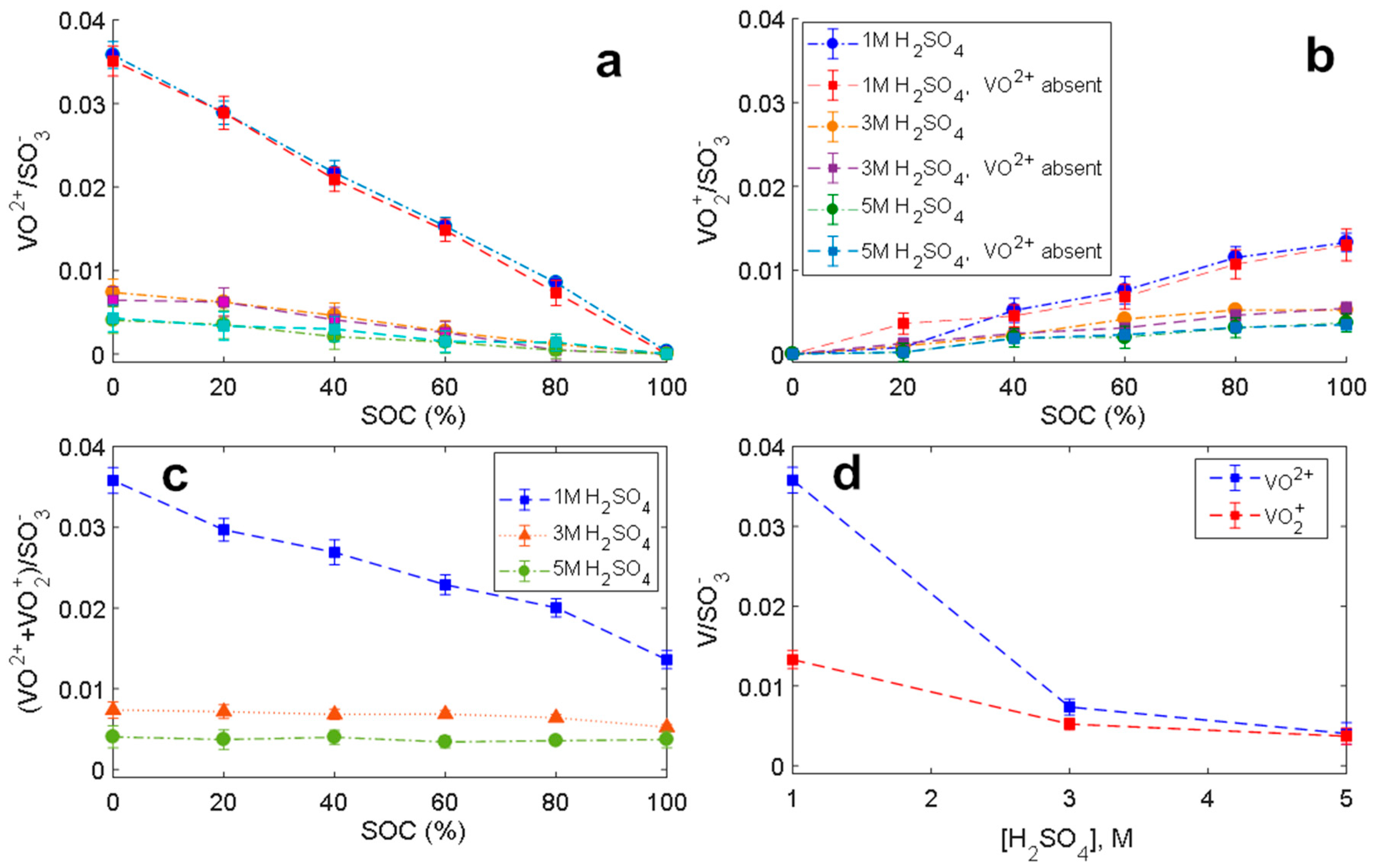
2.1. Electron Paramagnetic Resonance
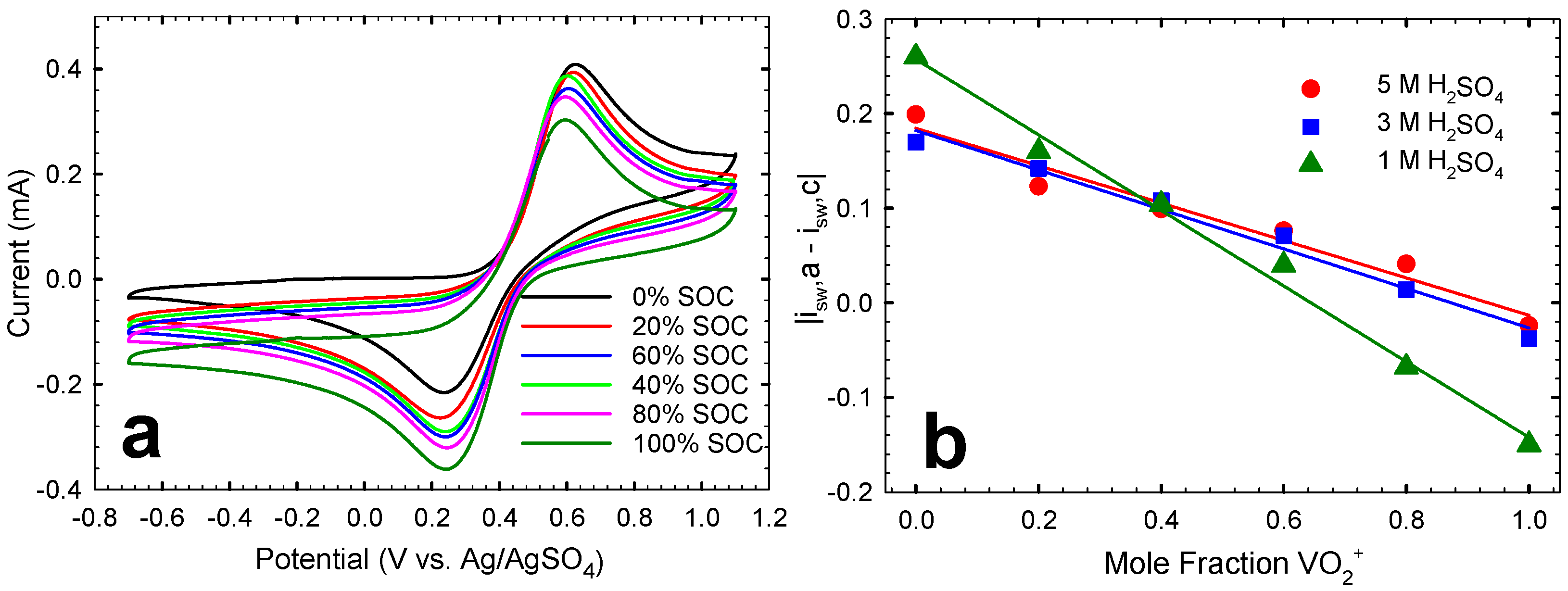
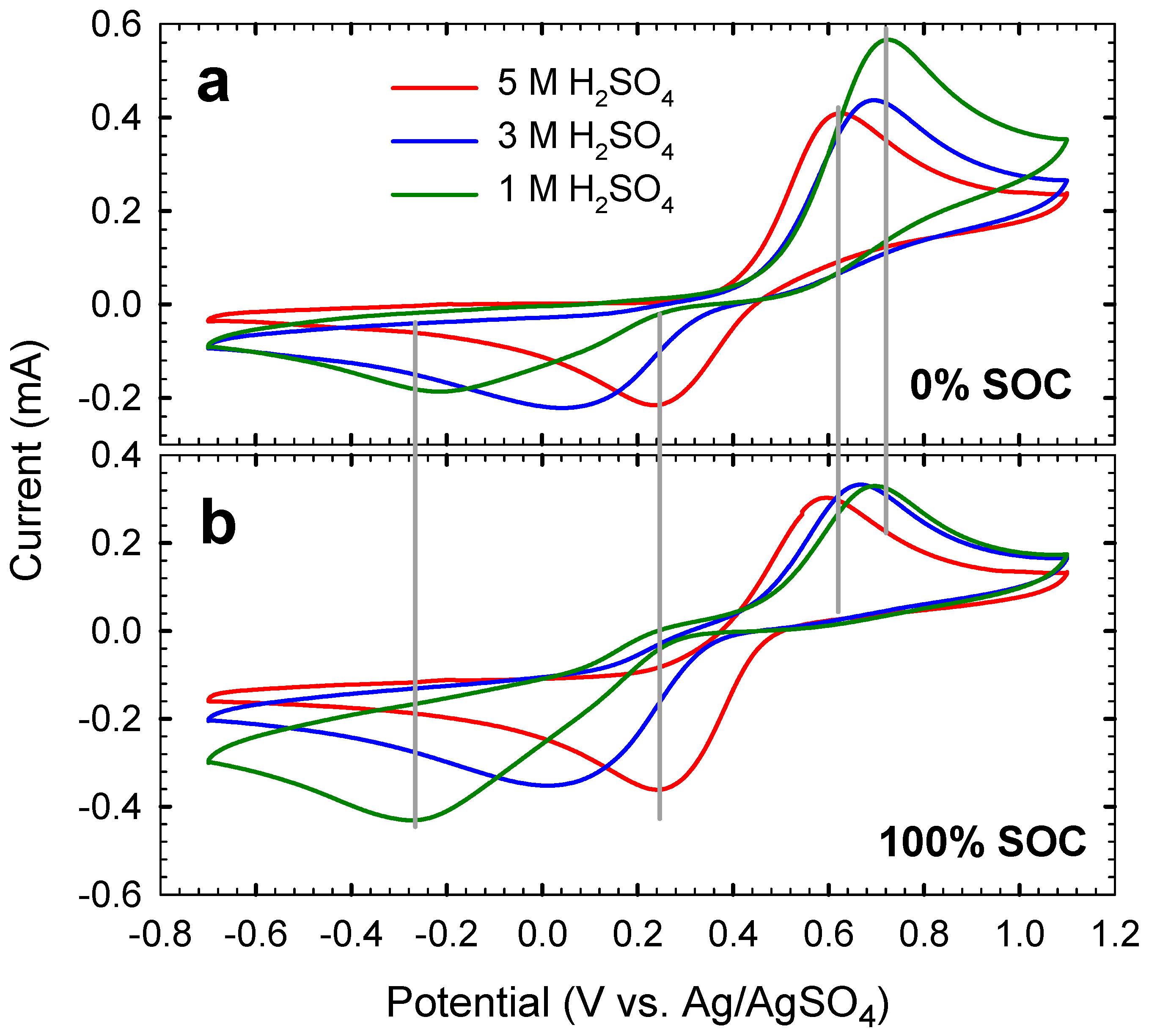
2.2. Cyclic Voltammetry
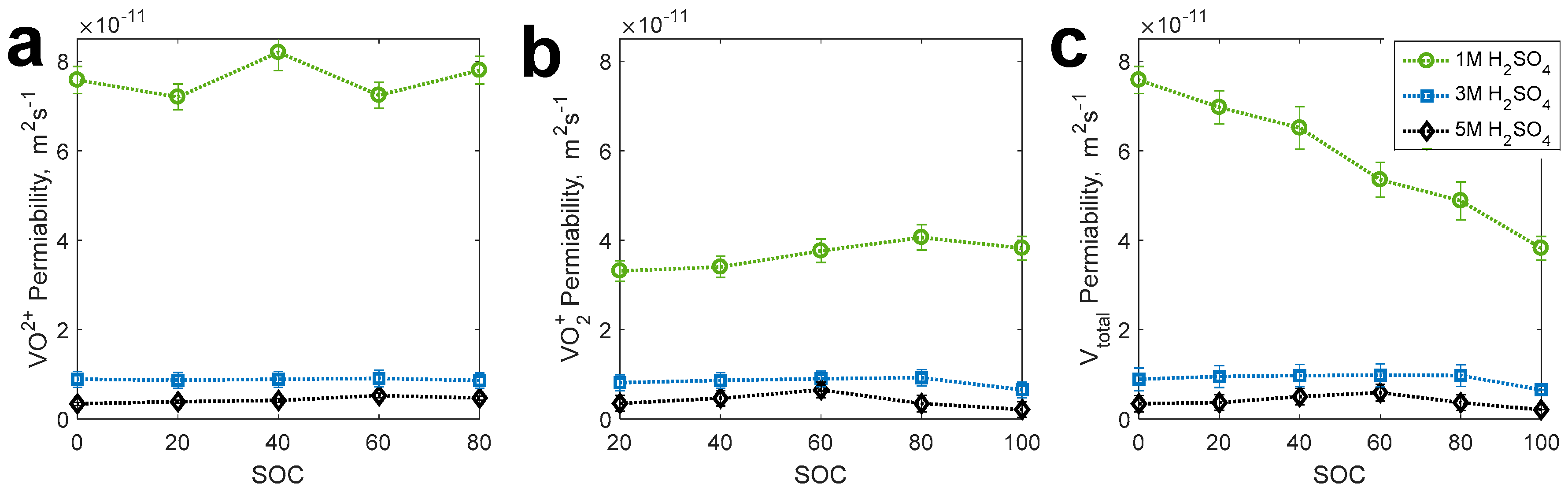
2.3. Uptake of Vanadium in Nafion Membrane
3. Materials and Methods
3.1. Preparation of Electrolyte Solutions
3.2. Electron Paramagnetic Resonance (EPR) Measurements
3.3. Membrane Pre-treatment
3.4. Membrane Uptake Experiments
3.5. Crossover/Diffusion Cell Experiments
3.6. Cyclic Voltammetry (CV) Experiments
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

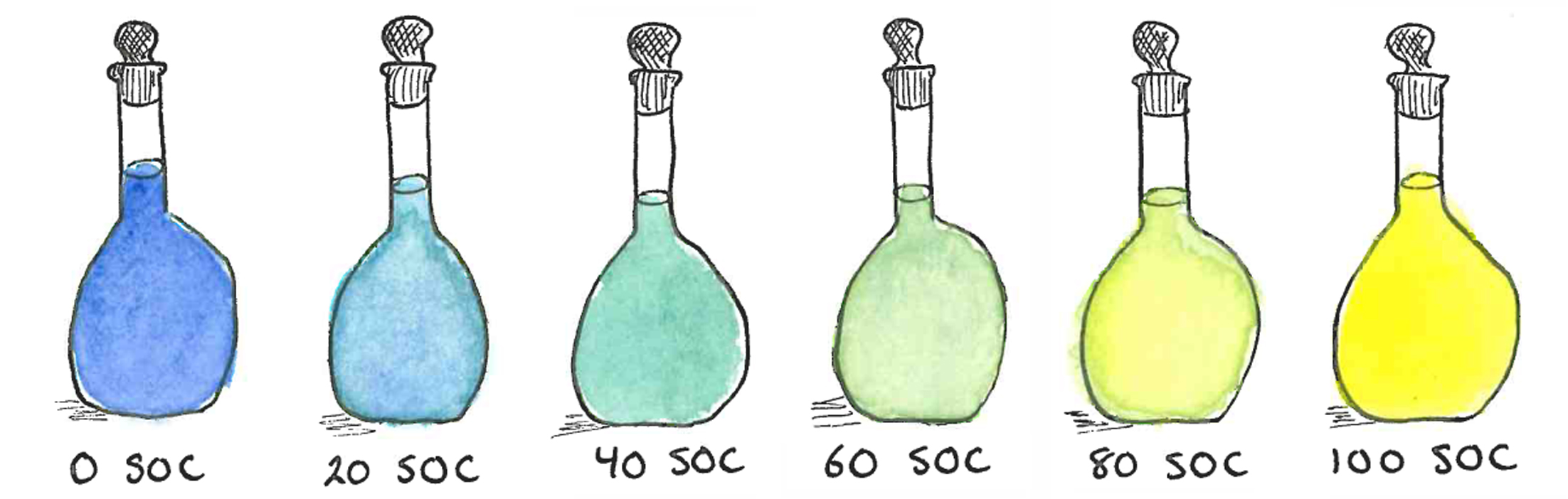{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| [H2SO4] (M) | SOC (%) | VO2+ DT, Solution, EPR, (m2s−1) ×1010 | VO2+ DT, Solution, CV, (m2s−1) ×1010 |
|---|---|---|---|
| 1 | 0 | 6.03 | 3.20 |
| 1 | 20 | 6.02 | 2.97 |
| 1 | 40 | 5.91 | 2.88 |
| 1 | 60 | 5.92 | 2.95 |
| 1 | 80 | 6.06 | 3.15 |
| 1 | 100 | - | - |
| 3 | 0 | 4.19 | 2.18 |
| 3 | 20 | 4.20 | 2.01 |
| 3 | 40 | 4.07 | 1.88 |
| 3 | 60 | 4.22 | 1.81 |
| 3 | 80 | 4.18 | 2.60 |
| 3 | 100 | - | - |
| 5 | 0 | 3.44 | 1.52 |
| 5 | 20 | 3.34 | 1.96 |
| 5 | 40 | 4.05 | 1.72 |
| 5 | 60 | 3.49 | 1.52 |
| 5 | 80 | 3.48 | 1.46 |
| 5 | 100 | - | - |
| SOC (%) | VO2+ Partition Coefficient | VO2+ Partition Coefficient | Mass of Absorbed Solvent per Gram of Membrane | VO2+ DT, Membrane (m2s−1) × 1011 | VO2+ DT, Membrane (m2s−1) × 1011 |
|---|---|---|---|---|---|
| 1 M H2SO4 | |||||
| 0 | 3.18 | - | 0.103 | 2.38 | - |
| 20 | 3.44 | 0.377 | 0.106 | 2.09 | 3.20 |
| 40 | 3.75 | 1.35 | 0.0951 | 2.18 | 2.51 |
| 60 | 3.88 | 1.28 | 0.0926 | 1.86 | 2.94 |
| 80 | 3.87 | 1.30 | 0.101 | 2.02 | 3.12 |
| 100 | - | 1.24 | 0.103 | - | 3.09 |
| 3 M H2SO4 | |||||
| 0 | 0.836 | - | 0.0946 | 1.06 | - |
| 20 | 1.01 | 0.613 | 0.0826 | 0.865 | 1.32 |
| 40 | 1.11 | 0.802 | 0.0742 | 0.802 | 1.07 |
| 60 | 0.790 | 0.817 | 0.0911 | 1.14 | 1.11 |
| 80 | 0.740 | 0.840 | 0.0837 | 1.15 | 1.10 |
| 100 | - | 0.64 | 0.0878 | - | 0.996 |
| 5 M H2SO4 | |||||
| 0 | 0.615 | - | 0.0764 | 0.551 | - |
| 20 | 0.780 | 0.164 | 0.0660 | 0.497 | 2.12 |
| 40 | 0.433 | 0.604 | 0.0924 | 0.958 | 0.768 |
| 60 | 0.574 | 0.530 | 0.0725 | 0.913 | 1.53 |
| 80 | 0.360 | 0.643 | 0.0707 | 1.31 | 0.536 |
| 100 | - | 0.552 | 0.0782 | - | 0.375 |
| Term/Symbol | Definition |
|---|---|
| A | Electrode area, cm2 |
| Co | Bulk Concentration, mol m−3 |
| DR | Rotational diffusion, s−1 |
| DT | Translational diffusion, m2 s−1 |
| Anodic peak potential, V | |
| Cathodic peak potential, V | |
| Esw,a | Anodic switching potential, V |
| Esw,c | Cathodic switching potential, V |
| F | Faraday’s constant, C mol−1 |
| i | Current, Amperes |
| ip,a | Anodic peak current, Amperes |
| ip,c | Cathodic peak current, Amperes |
| K | Partitioning coefficient, a.u. |
| kB | Boltzmann Constant, kg m2 s−2 K−1 |
| k0 | Rate constant, cm s−1 |
| n | Moles |
| ɳ | Viscosity, cP |
| P | Permeability, m2s−1 |
| r | Hydrodynamic radius, m |
| R | Gas constant, Jmol−1K−1 |
| T | Temperature, K |
| α | Electron transfer constant |
| ΔEp | Difference in peak potentials, V |
| ν | Scan rate, Vs−1 |
References
- Skyllas-Kazacas, M.; Rychcik, M.; Robins, R.G.; Fane, A.G.; Green, M.A. New All-Vanadium Redox Flow Cell. J. Electrochem. Soc. 1986, 133, 1057–1058. [Google Scholar] [CrossRef]
- Skyllas-Kazacos, M.; Cao, L.; Kazacos, M.; Kausar, N.; Mousa, A. Vanadium Electrolyte Studies for the Vanadium Redox Battery—A Review. ChemSusChem 2016, 9, 1521–1543. [Google Scholar] [CrossRef]
- Shi, Y.; Eze, C.; Xiong, B.; He, W.; Zhang, H.; Lim, T.M.; Ukil, A.; Zhao, J. Recent development of membrane for vanadium redox flow battery applications: A review. Appl. Energy 2019, 238, 202–224. [Google Scholar] [CrossRef]
- Lawton, J.S.; Budil, D.E. Investigation of water and methanol sorption in monovalent- and multivalent-ion-exchanged nafion membranes using electron spin resonance. J. Phys. Chem. B 2009, 113, 10679–10685. [Google Scholar] [CrossRef] [PubMed]
- Darling, R.M.; Saraidaridis, J.D.; Shovlin, C.; Fortin, M. Transference Numbers of Vanadium Cations in Nafion. J. Electrochem. Soc. 2020, 167, 020529. [Google Scholar] [CrossRef]
- Kushner, D.I.; Crothers, A.R.; Kusoglu, A.; Weber, A.Z. Transport phenomena in flow battery ion-conducting membranes. Curr. Opin. Electrochem. 2020, 21, 132–139. [Google Scholar] [CrossRef]
- Sun, C.; Chen, J.; Zhang, H.; Han, X.; Luo, Q. Investigations on transfer of water and vanadium ions across Nafion membrane in an operating vanadium redox flow battery. J. Power Sources 2010, 195, 890–897. [Google Scholar] [CrossRef]
- Vardner, J.T.; Edziah, J.-J.S.; West, A.C. Measurement of VO 2+ Transference Number in Nafion with Varying Concentrations of Sulfuric Acid. J. Electrochem. Soc. 2019, 166, A848. [Google Scholar] [CrossRef]
- Gurieff, N.; Keogh, D.F.; Baldry, M.; Timchenko, V.; Green, D.; Koskinen, I.; Menictas, C. Mass transport optimization for redox flow battery design. Appl. Sci. 2020, 10, 2801. [Google Scholar] [CrossRef]
- Roe, S.; Menictas, C.; Skyllas-Kazacos, M. A High Energy Density Vanadium Redox Flow Battery with 3 M Vanadium Electrolyte. J. Electrochem. Soc. 2016, 163, A5023–A5028. [Google Scholar] [CrossRef]
- Oriji, G.; Katayama, Y.; Miura, T. Investigation on V(IV)/V(V) species in a vanadium redox flow battery. Electrochim. Acta 2004, 49, 3091–3095. [Google Scholar] [CrossRef]
- Rahman, F.; Skyllas-Kazacos, M. Vanadium redox battery: Positive half-cell electrolyte studies. J. Power Sources 2009, 189, 1212–1219. [Google Scholar] [CrossRef]
- Choi, C.; Kim, S.; Kim, R.; Choi, Y.; Kim, S.; Jung, H.Y.; Yang, J.H.; Kim, H.T. A review of vanadium electrolytes for vanadium redox flow batteries. Renew. Sustain. Energy Rev. 2017, 69, 263–274. [Google Scholar] [CrossRef]
- Skyllas-Kazacos, M.; Kazacos, M. State of charge monitoring methods for vanadium redox flow battery control. J. Power Sources 2011, 196, 8822–8827. [Google Scholar] [CrossRef]
- Wu, X.; Liu, S.; Wang, N.; Peng, S.; He, Z. Influence of organic additives on electrochemical properties of the positive electrolyte for all vanadium redox flow battery. Electrochim. Acta 2012, 78, 475–482. [Google Scholar] [CrossRef]
- Skyllas-Kazacos, M.; Peng, C.; Cheng, M. Evaluation of Precipitation Inhibitors for Supersaturated Vanadyl Electrolytes for the Vanadium Redox Battery. Electrochem. Solid-State Lett. 1999, 2, 121. [Google Scholar] [CrossRef]
- Li, L.; Kim, S.; Wang, W.; Vijayakumar, M.; Nie, Z.; Chen, B.; Zhang, J.; Xia, G.; Hu, J.; Graff, G.; et al. A Stable Vanadium Redox-Flow Battery with High Energy Density for Large-Scale Energy Storage. Adv. Energy Mater. 2011, 1, 394–400. [Google Scholar] [CrossRef]
- Lawton, J.S.; Tiano, S.M.; Donnelly, D.J.; Flanagan, S.P.; Arruda, T.M. The Effect of Sulfuric Acid Concentration on Physical and Electrochemical Properties of Vanadyl Solutions. Batteries 2018, 4, 40. [Google Scholar] [CrossRef]
- Cao, L.; Skyllas-Kazacos, M.; Wang, D.-W. Effects of Surface Pretreatment of Glassy Carbon on the Electrochemical Behavior of V(IV)/V(V) Redox Reaction. J. Electrochem. Soc. 2016, 163, A1164. [Google Scholar]
- Lawton, J.S.; Jones, A.; Zawodzinski, T. Concentration dependence of VO2+ crossover of nafion for vanadium redox flow batteries. J. Electrochem. Soc. 2013, 160, A697–A702. [Google Scholar] [CrossRef]
- Lawton, J.S.; Jones, A.M.; Tang, Z.; Lindsey, M.; Zawodzinski, T. Ion effects on vanadium transport in Nafion membranes for vanadium redox flow batteries. J. Electrochem. Soc. 2017, 164, A2987. [Google Scholar]
- Tang, Z.; Svoboda, R.; Lawton, J.S.; Aaron, D.S.; Papandrew, A.B.; Zawodzinski, T.A. Composition and Conductivity of Membranes Equilibrated with Solutions of Sulfuric Acid and Vanadyl Sulfate. J. Electrochem. Soc. 2013, 160, F1040. [Google Scholar]
- Miles, D.T. Run-D.M.C.: A mnemonic aid for explaining mass transfer in electrochemical systems. J. Chem. Educ. 2013, 90, 1649–1653. [Google Scholar] [CrossRef]
- Oh, K.; Won, S.; Ju, H. A comparative study of species migration and diffusion mechanisms in all-vanadium redox flow batteries. Electrochim. Acta 2015, 181, 238–247. [Google Scholar]
- Barton, J.L.; Brushett, F.R. A one-dimensional stack model for redox flow battery analysis and operation. Batteries 2019, 5, 25. [Google Scholar]
- Lawton, J.S.; Budil, D.E. Spin Probe ESR Study of Cation Effects on Methanol and DMMP Solvation in Sulfonated Poly(styrene−isobutylene−styrene) Triblock Copolymers at High Ion-Exchange Capacities. Macromolecules 2010, 43, 652–661. [Google Scholar] [CrossRef]
- Lawton, J.S.; Smotkin, E.S.; Budil, D.E. Electron spin resonance investigation of microscopic viscosity, ordering, and polarity in nafion membranes containing methanol-water mixtures. J. Phys. Chem. B 2008, 112, 8549–8557. [Google Scholar] [PubMed]
- Lawton, J.S.; Budil, D.E. Electron spin resonance investigaion of the effects of methanol on microscopic viscosity, ordering, and polarity in different phases of ionomer membranes with sulfonated polyarylene backbones. J. Memb. Sci. 2010, 357, 47–53. [Google Scholar] [CrossRef]
- Coronado, F.; Valenzuela Soto, A.; Encinas Romero, M.A.; Hernández-Negrete, O.; Tiburcio Munive, G. Microstructural and chemical characterization of stabilized spent vanadium pentoxide catalyst in copper smelting slag. Mater. Res. Express 2020, 7, 016521. [Google Scholar]
- Mangini, L.F.K.; Valt, R.B.G.; Ponte, M.J.J.d.S.; Ponte, H.d.A. Vanadium removal from spent catalyst used in the manufacture of sulfuric acid by electrical potential application. Sep. Purif. Technol. 2020, 246, 116854. [Google Scholar]
- Rehder, D. The potentiality of vanadium in medicinal applications. Inorg. Chim. Acta 2020, 504, 119445. [Google Scholar] [CrossRef]
- Frank, P.; Carlson, R.M.K.; Carlson, E.J.; Hedman, B.; Hodgson, K.O. Biological sulfur in the blood cells of Ascidia ceratodes: XAS spectroscopy and a cellular-enzymatic hypothesis for vanadium reduction in the ascidians. J. Inorg. Biochem. 2020, 205, 110991. [Google Scholar]
- Frank, P.; Hodgson, K.O.; Carlson, R.M.K. Vanadyl Ion EPR as a Noninvasive Probe of pH in Intact Vanadocytes from Ascidia ceratodes. Inorg. Chem. 1986, 25, 470–478. [Google Scholar] [CrossRef]
- Servedio, L.T.; Lawton, J.S.; Zawodzinski, T.A. An electrochemical study of cobalt-salen (N,N′-bis(salicylidene)ethylenediaminocobalt(II) in the oxidation of syringyl alcohol in acetonitrile. J. Appl. Electrochem. 2020, 1–12. [Google Scholar] [CrossRef]
- Maurya, A. Homogeneous catalytic oxidation of alkenes employing mononuclear vanadium complex with hydrogen peroxide. J. Iran. Chem. Soc. 2020, 1–9. [Google Scholar] [CrossRef]
- Zwolak, I. Protective Effects of Dietary Antioxidants against Vanadium-Induced Toxicity: A Review. Oxid. Med. Cell. Longev. 2020, 2020, 1490316. [Google Scholar]
- Srinivasa Rao, L.; Thirmal, C.; Raghavendra Rao, P. Dielectric dispersion, linear and nonlinear optical properties of Li 2 O–WO 3 –B 2 O 3: V 2 O 5 glasses. J. Adv. Dielectr. 2020, 10, 2050006. [Google Scholar]
- Vijayakumar, M.; Li, L.; Graff, G.; Liu, J.; Zhang, H.; Yang, Z.; Zhi, J. Towards understanding the poor thermal stability of V 5 + electrolyte solution in Vanadium Redox Flow Batteries. J. Power Sources 2011, 196, 3669–3672. [Google Scholar] [CrossRef]
- Barklie, R.C.; Girard, O.; Braddell, O. EPR of vanadyl (2+) in a Nafion membrane. J. Phys. Chem. 1988, 92, 1371–1377. [Google Scholar] [CrossRef]
- Reuben, J.; Fiat, D. Proton and Deuteron Relaxation in Aqueous Solutions of Vanadyl( IV). Effects of Electron Spin Relaxation and Chemical Exchange. J. Am. Chem. Soc. 1969, 1611, 4652–4656. [Google Scholar]
- Chasteen, D.; Hanna, M. Resonance Line Widths of vanadyl(IV) hydroxycarboxylates. J. Phys. Chem. 1972, 76, 3951–3959. [Google Scholar] [CrossRef]
- Scholz, F.; Hermes, M. The determination of the redox state of a dissolved depolariser by cyclic voltammetry in the case of electrochemically reversible systems. Electrochem. Commun. 1999, 1, 345–348. [Google Scholar]
- Lawton, J.S.; Aaron, D.S.; Tang, Z.; Zawodzinski, T. Qualitative Behavior of Vanadium Ions In Nafion Membranes Using Electron Spin Resonance. J. Membrane Sci. 2013, 428, 38–45. [Google Scholar] [CrossRef]
- Gattrell, M.; Park, J.; MacDougall, B.; Apte, J.; McCarthy, S.; Wu, C.W. Study of the Mechanism of the Vanadium 4+/5+ Redox Reaction in Acidic Solutions. J. Electrochem. Soc. 2004, 151, A123–A130. [Google Scholar]
- Batchelor-Mcauley, C.; Kätelhön, E.; Barnes, E.O.; Compton, R.G.; Laborda, E.; Molina, A. Recent Advances in Voltammetry. ChemistryOpen 2015, 4, 224–260. [Google Scholar]
- Brownson, D.A.C.; Banks, C.E. The Handbook of Graphene Electrochemistry, 1st ed.; Springer: London, UK, 2014. [Google Scholar]
- Nicholson, R.S.; Shain, I. Theory of Stationary Electrode Polarography: Single Scan and Cyclic Methods Applied to Reversible, Irreversible, and Kinetic Systems. Anal. Chem. 1964, 36, 706–723. [Google Scholar]
- Aristov, N.; Habekost, A. Cyclic Voltammetry—A Versatile Electrochemical Method Investigating Electron Transfer Processes. World J. Chem. Educ. 2015, 3, 115–119. [Google Scholar]
- Arruda, T.M.; Shyam, B.; Lawton, J.S.; Ramaswamy, N.; Budil, D.E.; Ramaker, D.E.; Mukerjee, S. Fundamental Aspects of Spontaneous Cathodic Deposition of Ru onto Pt/C Electrocatalysts and Membranes under Direct Methanol Fuel Cell Operating Conditions: An in Situ X-ray Absorption Spectroscopy and Electron Spin Resonance Study. J. Phys. Chem. C 2010, 114, 1028–1040. [Google Scholar]
- Stoll, S.; Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef]
- Stoll, S.; Schweiger, A. EasySpin: Simulating cw ESR spectra. Biol. Magn. Reson. 2007, 27, 299–321. [Google Scholar]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chace, W.S.; Tiano, S.M.; Arruda, T.M.; Lawton, J.S. Effects of State of Charge on the Physical Characteristics of V(IV)/V(V) Electrolytes and Membrane for the All Vanadium Flow Battery. Batteries 2020, 6, 49. https://doi.org/10.3390/batteries6040049
Chace WS, Tiano SM, Arruda TM, Lawton JS. Effects of State of Charge on the Physical Characteristics of V(IV)/V(V) Electrolytes and Membrane for the All Vanadium Flow Battery. Batteries. 2020; 6(4):49. https://doi.org/10.3390/batteries6040049
Chicago/Turabian StyleChace, Wyndom S., Sophia M. Tiano, Thomas M. Arruda, and Jamie S. Lawton. 2020. "Effects of State of Charge on the Physical Characteristics of V(IV)/V(V) Electrolytes and Membrane for the All Vanadium Flow Battery" Batteries 6, no. 4: 49. https://doi.org/10.3390/batteries6040049
APA StyleChace, W. S., Tiano, S. M., Arruda, T. M., & Lawton, J. S. (2020). Effects of State of Charge on the Physical Characteristics of V(IV)/V(V) Electrolytes and Membrane for the All Vanadium Flow Battery. Batteries, 6(4), 49. https://doi.org/10.3390/batteries6040049