Vanadium Electrolyte for All-Vanadium Redox-Flow Batteries: The Effect of the Counter Ion


 ,
, 
Abstract
1. Introduction
2. Results and Discussion
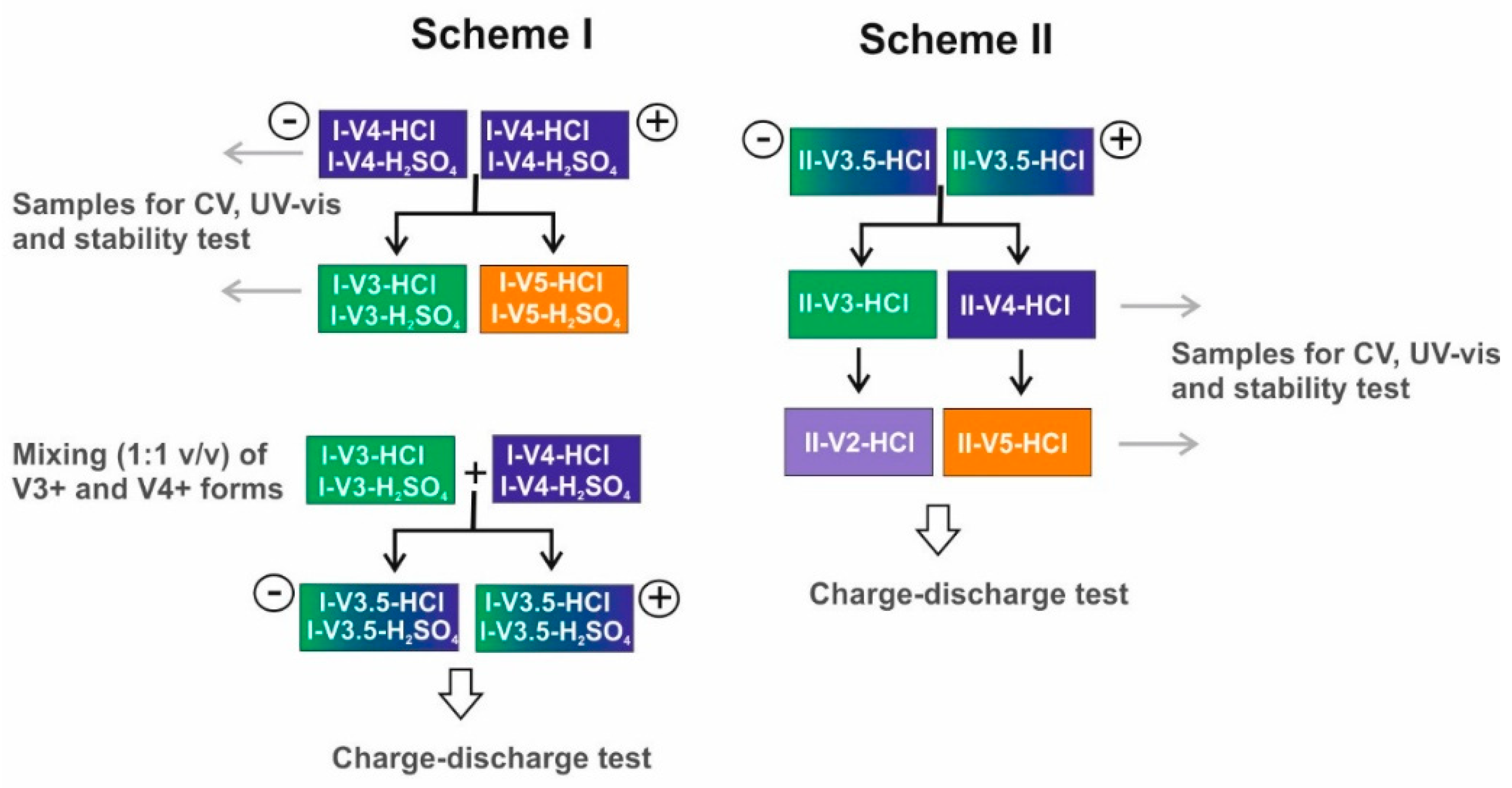
2.1. Electrolyte Preparation
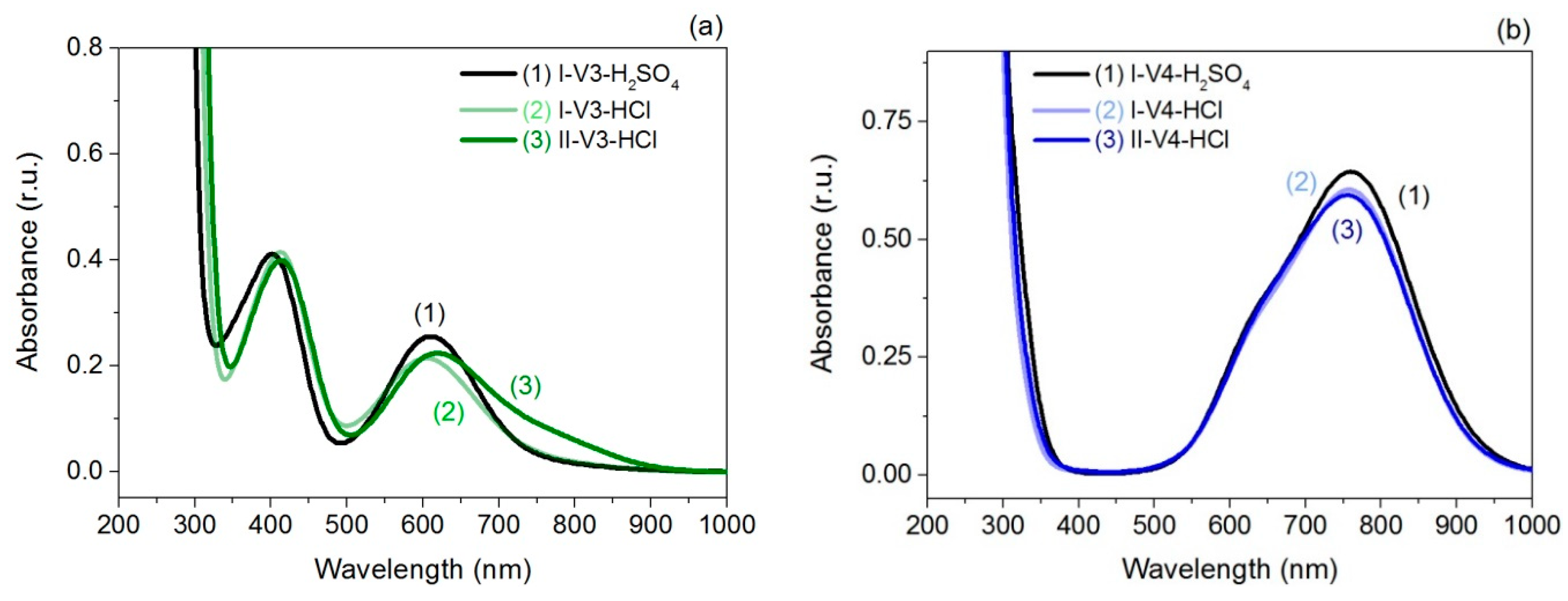
2.2. UV-Vis Spectroscopy
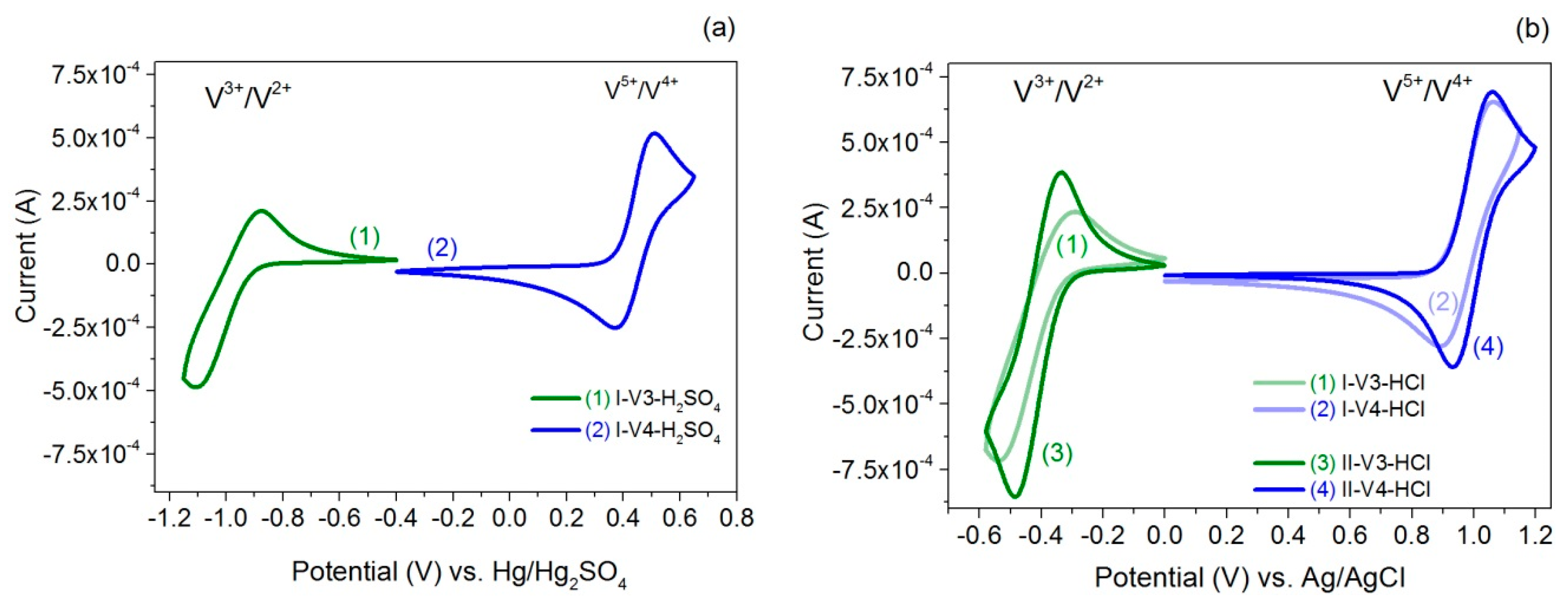
2.3. Cyclic Voltammetry
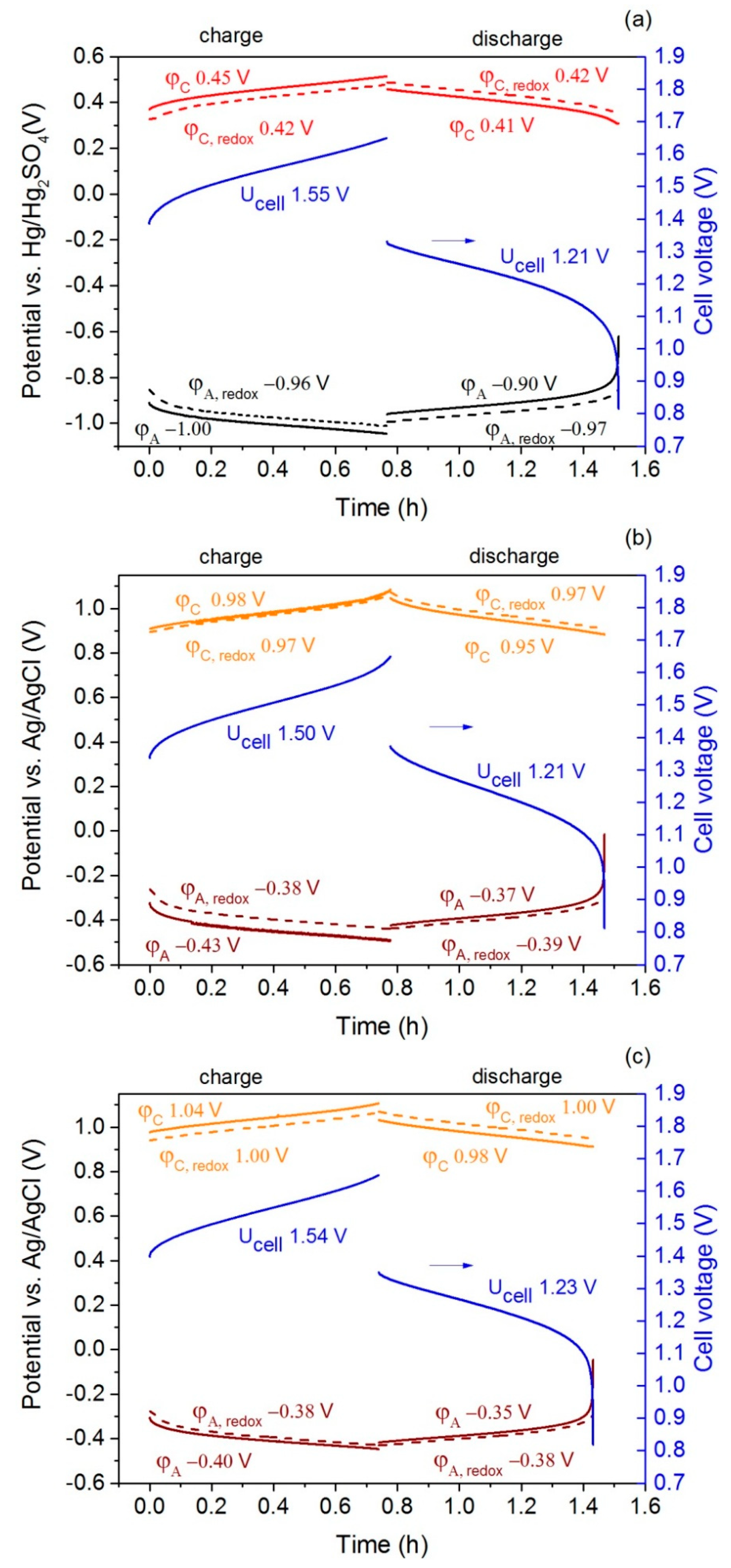
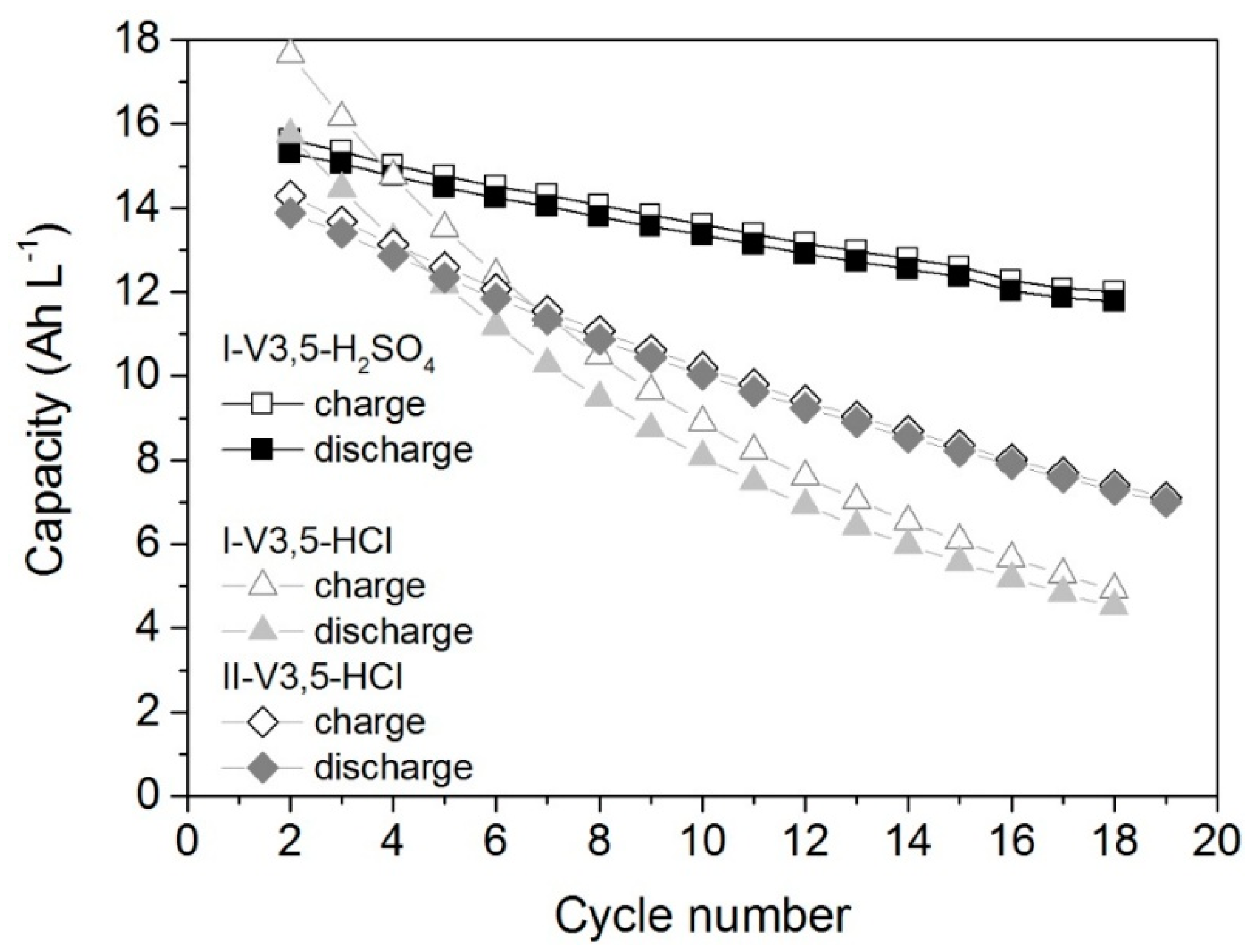
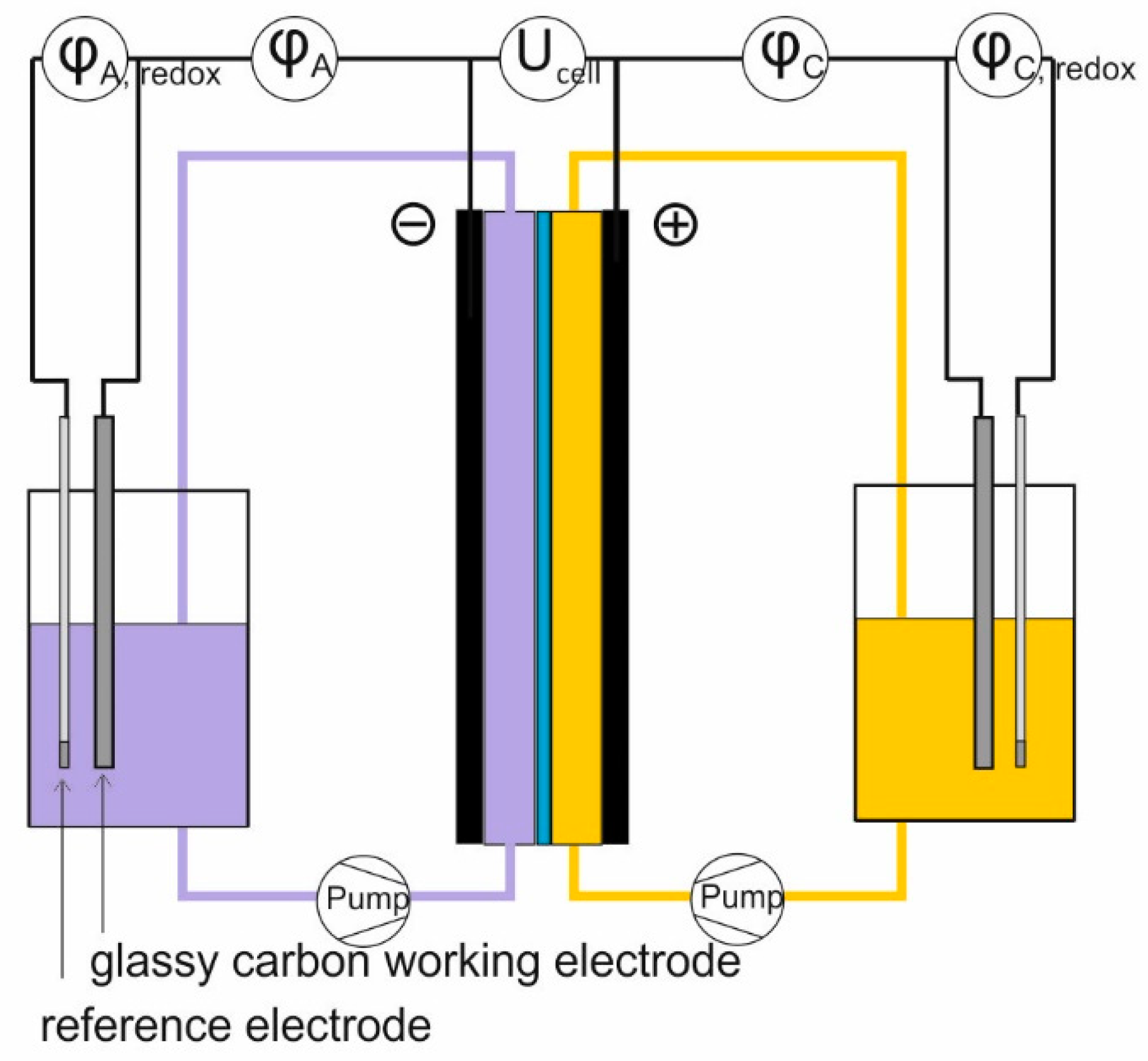
2.4. Cell Test
2.5. Thermal Stability
3. Materials and Methods
3.1. Chemicals
3.2. Electrolyte Preparation
3.3. Electrolyte Characterization
3.3.1. Titration and Gravimetric Analysis
3.3.2. Conductivity Measurement
3.3.3. Optical Absorption Spectra (UV-Vis)
3.3.4. Cyclic Voltammetry (CV)
3.3.5. Thermal Stability
3.4. Cell Test
4. Conclusions
- (1)
- The electrolyte preparation procedures should be considered to differentiate between the proton effect of an acidic matrix and the complexing effect or nature of the counter ions on the electrochemical properties of V(III)/V(II) and V(V)/V(IV) couples, and on the stability of the fully charged V(V) electrolyte at elevated temperatures.
- (2)
- Provided that the electrolyte samples have the same conductivity and total vanadium concentration, the characteristics of the cell operated with these electrolytes (capacity, energy efficiency, and activation losses) are similar. The cyclability of the cell with sulfuric acid electrolyte is better compared to the cells with hydrochloric acid.
- (3)
- V(V) in catholyte at an SoC of 98% in hydrochloric acid undergoes reduction by chloride ions, which can proceed slower or faster depending on the amount of free acid in the electrolyte and the temperature.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kausar, N.; Howe, R.; Skyllas-Kazacos, M. Raman spectroscopy studies of concentrated vanadium redox battery positive electrolytes. J. Appl. Electrochem. 2001, 31, 1327–1332. [Google Scholar] [CrossRef]
- Rahman, F.; Skyllas-Kazacos, M. Solubility of vanadyl sulfate in concentrated sulfuric acid solutions. J. Power Sources 1998, 72, 105–110. [Google Scholar] [CrossRef]
- Vijayakumar, M.; Li, L.; Nie, Z.; Yang, Z.; Hu, J. Structure and stability of hexa-aqua V(iii) cations in vanadium redox flow battery electrolytes. Phys. Chem. Chem. Phys. 2012, 14, 10233–10242. [Google Scholar] [CrossRef] [PubMed]
- Roznyatovskaya, N.V.; Roznyatovsky, V.A.; Höhne, C.-C.; Fühl, M.; Gerber, T.; Küttinger, M.; Noack, J.; Fischer, P.; Pinkwart, K.; Tübke, J. The role of phosphate additive in stabilization of sulphuric-acid-based vanadium(V) electrolyte for all-vanadium redox-flow batteries. J. Power Sources 2017, 363, 234–243. [Google Scholar] [CrossRef]
- Skyllas-Kazacos, M.; Cao, L.; Kazacos, M.; Kausar, N.; Mousa, A. Vanadium Electrolyte Studies for the Vanadium Redox Battery-A Review. ChemSusChem 2016, 9, 1521–1543. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.; Kim, S.; Kim, R.; Choi, Y.; Kim, S.; Jung, H.-Y.; Yang, J.H.; Kim, H.-T. A review of vanadium electrolytes for vanadium redox flow batteries. Renew. Sustain. Energy Rev. 2017, 69, 263–274. [Google Scholar] [CrossRef]
- Lu, W.; Li, X.; Zhang, H. The next generation vanadium flow batteries with high power density—A perspective. Phys. Chem. Chem. Phys. 2018, 20, 23–35. [Google Scholar] [CrossRef]
- Lee, J.G.; Park, S.J.; Cho, Y.I.; Shul, Y.G. A novel cathodic electrolyte based on H2C2O4 for a stable vanadium redox flow battery with high charge-discharge capacities. RSC Adv. 2013, 3, 21347–21351. [Google Scholar] [CrossRef]
- Kim, S.; Vijayakumar, M.; Wang, W.; Zhang, J.; Chen, B.; Nie, Z.; Chen, F.; Hu, J.; Li, L.; Yang, Z. Chloride supporting electrolytes for all-vanadium redox flow batteries. Phys. Chem. Chem. Phys. 2011, 13, 18186–18193. [Google Scholar] [CrossRef]
- Li, L.; Kim, S.; Wang, W.; Vijayakumar, M.; Nie, Z.; Chen, B.; Zhang, J.; Xia, G.; Hu, J.; Graff, G.; et al. A Stable Vanadium Redox-Flow Battery with High Energy Density for Large-Scale Energy Storage. Adv. Energy Mater. 2011, 1, 394–400. [Google Scholar] [CrossRef]
- Cao, L.; Skyllas-Kazacos, M.; Menictas, C.; Noack, J. A review of electrolyte additives and impurities in vanadium redox flow batteries. J. Energy Chem. 2018, 27, 1269–1291. [Google Scholar] [CrossRef]
- Sum, E.; Rychcik, M.; Skyllas-Kazacos, M. Investigation of the V(V)/V(IV) system for use in the positive half-cell of a redox battery. J. Power Sources 1985, 16, 85–95. [Google Scholar] [CrossRef]
- Rahman, F.; Skyllas-Kazacos, M. Vanadium redox battery: Positive half-cell electrolyte studies. J. Power Sources 2009, 189, 1212–1219. [Google Scholar] [CrossRef]
- He, Z.; Li, Z.; Zhou, Z.; Tu, F.; Jiang, Y.; Pan, C.; Liu, S. Improved performance of vanadium redox battery using methylsulfonic acid solution as supporting electrolyte. J. Renew. Sustain. Energy 2013, 5, 023130. [Google Scholar] [CrossRef]
- Skyllas-Kazacos, M.; Kazacos, M. State of charge monitoring methods for vanadium redox flow battery control. J. Power Sources 2011, 196, 8822–8827. [Google Scholar] [CrossRef]
- Fraenkel, D. Electrolytic Nature of Aqueous Sulfuric Acid. 2. Acidity. J. Phys. Chem. B 2012, 116, 11678–11686. [Google Scholar] [CrossRef] [PubMed]
- Crans, D.C.; Tracey, A.S. The Chemistry of Vanadium in Aqueous and Nonaqueous Solution. In Vanadium Compounds, Chemistry, Biochemistry, and Therapeutic Applications; Tracey, A.S., Crans, D.C., Eds.; American Chemical Society: Washington, DC, USA, 1998; Volume 711, pp. 2–29. [Google Scholar]
- Martin, E.L.; Bentley, K.E. Spectrophotometric Investigation of Vanadium(II), Vanadium(III), and Vanadium(IV) in Various Media. Anal. Chem. 1962, 34, 354–358. [Google Scholar] [CrossRef]
- Pajdowski, L. A spectrophotometric study of the hydrolysis of vanadium (III) ion. J. Inorg. Nuclear Chem. 1966, 28, 433–442. [Google Scholar] [CrossRef]
- Ballhausen, C.J.; Gray, H.B. The Electronic Structure of the Vanadyl Ion. Inorg. Chem. 1962, 1, 111–122. [Google Scholar] [CrossRef]
- Vijayakumar, M.; Burton, S.D.; Huang, C.; Li, L.; Yang, Z.; Graff, G.L.; Liu, J.; Hu, J.; Skyllas-Kazacos, M. Nuclear magnetic resonance studies on vanadium(IV) electrolyte solutions for vanadium redox flow battery. J. Power Sources 2010, 195, 7709–7717. [Google Scholar] [CrossRef]
- Roznyatovskaya, N.; Noack, J.; Fuehl, M.; Pinkwart, K.; Tuebke, J. Towards an all-vanadium redox-flow battery electrolyte: Electrooxidation of V(III) in V(IV)/V(III) redox couple. Electrochim. Acta 2016, 211, 926–932. [Google Scholar] [CrossRef]
- Skyllas-Kazacos, M. Novel vanadium chloride/polyhalide redox flow battery. J. Power Sources 2003, 124, 299–302. [Google Scholar] [CrossRef]
- Noack, J.; Roznyatovskaya, N.; Kunzendorf, J.; Skyllas-Kazacos, M.; Menictas, C.; Tübke, J. The influence of electrochemical treatment on electrode reactions for vanadium redox-flow batteries. J. Energy Chem. 2018, 27, 1341–1352. [Google Scholar] [CrossRef]
- Compton, R.G.; Banks, G.E. Understanding Voltammetry, 2nd ed.; Imperial College Press: London, UK, 2011; pp. 2–444. ISBN 9781848165861. [Google Scholar]
- Tang, A.; Bao, J.; Skyllas-Kazacos, M. Studies on pressure losses and flow rate optimization in vanadium redox flow battery. J. Power Sources 2014, 248, 154–162. [Google Scholar] [CrossRef]
- Noack, J.; Vorhauser, L.; Pinkwart, K.; Tübke, J. Aging Studies of Vanadium Redox Flow Batteries. ECS Trans. 2011, 33, 3–9. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Total V Conc. (M) | Molar Content (%) | Total Counter Ion Conc. (M) | Conductivity (mS∙cm−1) | Comment | ||
|---|---|---|---|---|---|---|---|
| V(III) | V(IV) | V(V) | |||||
| I-V4-H2SO4 ** | 1.66 | 100 | 4.7 | 330 | 3 M free H2SO4 | ||
| I-V3-H2SO4 | 1.63 | 100 | 230 | ||||
| I-V5-H2SO4 | 1.61 | 1.5 | 98.5 | 450 | |||
| I-V3.5-H2SO4 | 1.65 | 45.9 | 54.1 | 270 | |||
| I-V4-HCl ** | 1.63 | 95.6 | 4.4 | 6.1 | 330 | targeted 3 M free HCl | |
| I-V3-HCl | 1.67 | 98.8 | 1.2 | 160 | |||
| I-V5-HCl | 1.59 | 1.7 | 98.3 | 480 | freshly prepared | ||
| I-V3.5-HCl | 1.66 | 47.2 | 52.8 | 240 | |||
| II-V4-HCl | 1.57 | 100 | 365 | ||||
| II-V3-HCl | 1.68 | 93.1 | 6.9 | 240 | |||
| II-V5-HCl | 1.54 | 1.7 | 98.3 | 490 | freshly prepared | ||
| II-V3.5-HCl ** | 1.60 | 47.5 | 52.4 | 7.6 | 320 | ||
| 3 M H2SO4 | 3 | 810 | |||||
| 3 M HCl | 3 | 710 | |||||
| Sample | V(III) | V(IV) | ||
|---|---|---|---|---|
| 3T1g(F) → 3T1g(P) | 3T1g(F) → 3T2g | 2B2 → 2B1 | 2B2 → 2E(I) | |
| I-V3-H2SO4, I-V4-H2SO4 | 403 nm | 610 nm | 633 nm | 760 nm |
| I-V3-HCl, I-V4-HCl | 413 nm | 605 nm | 633 nm | 760 nm |
| II-V3-HCl, II-V4-HCl | 414 nm | 620 nm | 633 nm | 760 nm |
| Sample | Redox Couple | E1/2 (V) vs. Ag/AgCl | E1/2 (V) vs. Hg/Hg2SO4 | ∆E (V) | Ucell, theor. (V) |
|---|---|---|---|---|---|
| I-V3-H2SO4 | V(III)/V(II) | −0.99 | 0.23 | 1.43 | |
| I-V4-H2SO4 | V(V)/V(IV) | 0.44 | 0.13 | ||
| I-V3-HCl | V(III)/V(II) | −0.42 | 0.25 | 1.39 | |
| I-V4-HCl | V(V)/V(IV) | 0.97 | 0.16 | ||
| II-V3-HCl | V(III)/V(II) | −0.41 | 0.14 | 1.41 | |
| II-V4-HCl | V(V)/V(IV) | 1.00 | 0.13 |
| Parameter | I-V3.5-H2SO4 | I-V3.5-HCl | II-V3.5-HCl | |||
|---|---|---|---|---|---|---|
| Charge | Discharge | Charge | Discharge | Charge | Discharge | |
| φA − φA, redox (V) ** | −0.04 ± 0.01 | 0.06 ± 0.02 | −0.05 ± 0.01 | 0.03 ± 0.01 | −0.02 ± 0.01 | 0.02 ± 0.01 |
| φC − φC, redox (V) ** | 0.03 ± 0.01 | −0.02 ± 0.01 | 0.02 ± 0.01 | −0.02 ± 0.01 | 0.03 ± 0.01 | −0.03 ± 0.01 |
| Ucell − (φC − φA) (V) ** | 0.09 ± 0.01 | −0.10 ± 0.01 | 0.080 ± 0.01 | −0.10 ± 0.01 | 0.10 ± 0.01 | −0.11 ± 0.01 |
| Capacity (Ah∙L−1) * | 15.7 | 15.3 | 17.5 | 15.7 | 14.8 | 13.9 |
| Energy (Wh∙L−1) * | 24.1 | 18.5 | 26.5 | 19.0 | 22.7 | 17.1 |
| Coulombic efficiency (%) | 97 | 90 | 94 | |||
| Energy efficiency (%) | 77 | 72 | 75 | |||
| Sample | Conditions | Total V Concentration (M) | Molar Content (%) | Comment | |
|---|---|---|---|---|---|
| V(IV) | V(V) | ||||
| I-V5-H2SO4 | initial | 1.61 | 1.5 | 98.5 | |
| after 3 weeks at RT | 1.62 | 1.4 | 98.6 | unchanged | |
| after 6 weeks at RT | 1.62 | 1.6 | 98.4 | unchanged | |
| after 1 day at 45 °C | 1.62 | 2 | 98.0 | traces of precipitate | |
| after 5 weeks at RT and 2 days at 45 °C | 1.40 | 1.8 | 98.2 | precipitation | |
| I-V5-HCl | initial | 1.59 | 1.7 | 98.3 | |
| after 3 weeks at RT | 1.60 | 3.1 | 97.0 | SoC shift | |
| after 6 weeks at RT | 1.59 | 4.0 | 96.0 | SoC shift | |
| after 1 day at 45 °C | 1.55 | 4.1 | 95.9 | precipitation | |
| after 5 weeks at RT and 2 days at 45 °C | 1.40 | 6.0 | 94.0 | precipitation | |
| II-V5-HCl | initial | 1.54 | 1.7 | 98.3 | |
| after 3 weeks at RT | 1.52 | 5.4 | 94.6 | SoC shift | |
| after 5 weeks at RT | 1.53 | 7.3 | 92.7 | SoC shift | |
| after 1 day at 45 °C | 1.54 | 9.0 | 91.0 | SoC shift | |
| after 4 weeks at RT and 2 days at 45 °C | 1.53 | 11.4 | 88.6 | SoC shift | |
| Sample | VCl3 | V2O5 | HCl | Targeted Concentration | |
| HCl 3 M | HCl 32% w/w | Vanadium | |||
| I-V4-HCl | 0.4 moles | 0.2 moles | 0.5 L (1.5 moles) | 37 mL (0.4 moles) | 1.6 M |
| II-V3.5-HCl stock * | 0.85 moles | 0.13 moles | 0.5 L (1.5 moles) | 25 mL (0.26 moles) | 2.2 M |
| Sample | VOSO4∙5.1H2O | H2SO4 95% w/w | H2O | Targeted concentration | |
| Vanadium | |||||
| I-V4-H2SO4 | 204 g (0.8 moles) | 84 ml (1.5 moles) | to 0.5 L | 1.6 M | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roznyatovskaya, N.; Noack, J.; Mild, H.; Fühl, M.; Fischer, P.; Pinkwart, K.; Tübke, J.; Skyllas-Kazacos, M. Vanadium Electrolyte for All-Vanadium Redox-Flow Batteries: The Effect of the Counter Ion. Batteries 2019, 5, 13. https://doi.org/10.3390/batteries5010013
Roznyatovskaya N, Noack J, Mild H, Fühl M, Fischer P, Pinkwart K, Tübke J, Skyllas-Kazacos M. Vanadium Electrolyte for All-Vanadium Redox-Flow Batteries: The Effect of the Counter Ion. Batteries. 2019; 5(1):13. https://doi.org/10.3390/batteries5010013
Chicago/Turabian StyleRoznyatovskaya, Nataliya, Jens Noack, Heiko Mild, Matthias Fühl, Peter Fischer, Karsten Pinkwart, Jens Tübke, and Maria Skyllas-Kazacos. 2019. "Vanadium Electrolyte for All-Vanadium Redox-Flow Batteries: The Effect of the Counter Ion" Batteries 5, no. 1: 13. https://doi.org/10.3390/batteries5010013
APA StyleRoznyatovskaya, N., Noack, J., Mild, H., Fühl, M., Fischer, P., Pinkwart, K., Tübke, J., & Skyllas-Kazacos, M. (2019). Vanadium Electrolyte for All-Vanadium Redox-Flow Batteries: The Effect of the Counter Ion. Batteries, 5(1), 13. https://doi.org/10.3390/batteries5010013