
Improving Lithium-Ion Battery Performance: Nano Al2O3 Coatings on High-Mass Loading LiFePO4 Cathodes via Atomic Layer Deposition

 ,
,  , ,
, ,
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Section
2.1. Al2O3 Coating of LFP Electrode by ALD
2.2. Electrochemical Measurements
2.3. Characterization of the LFP Electrodes
3. Results and Discussion
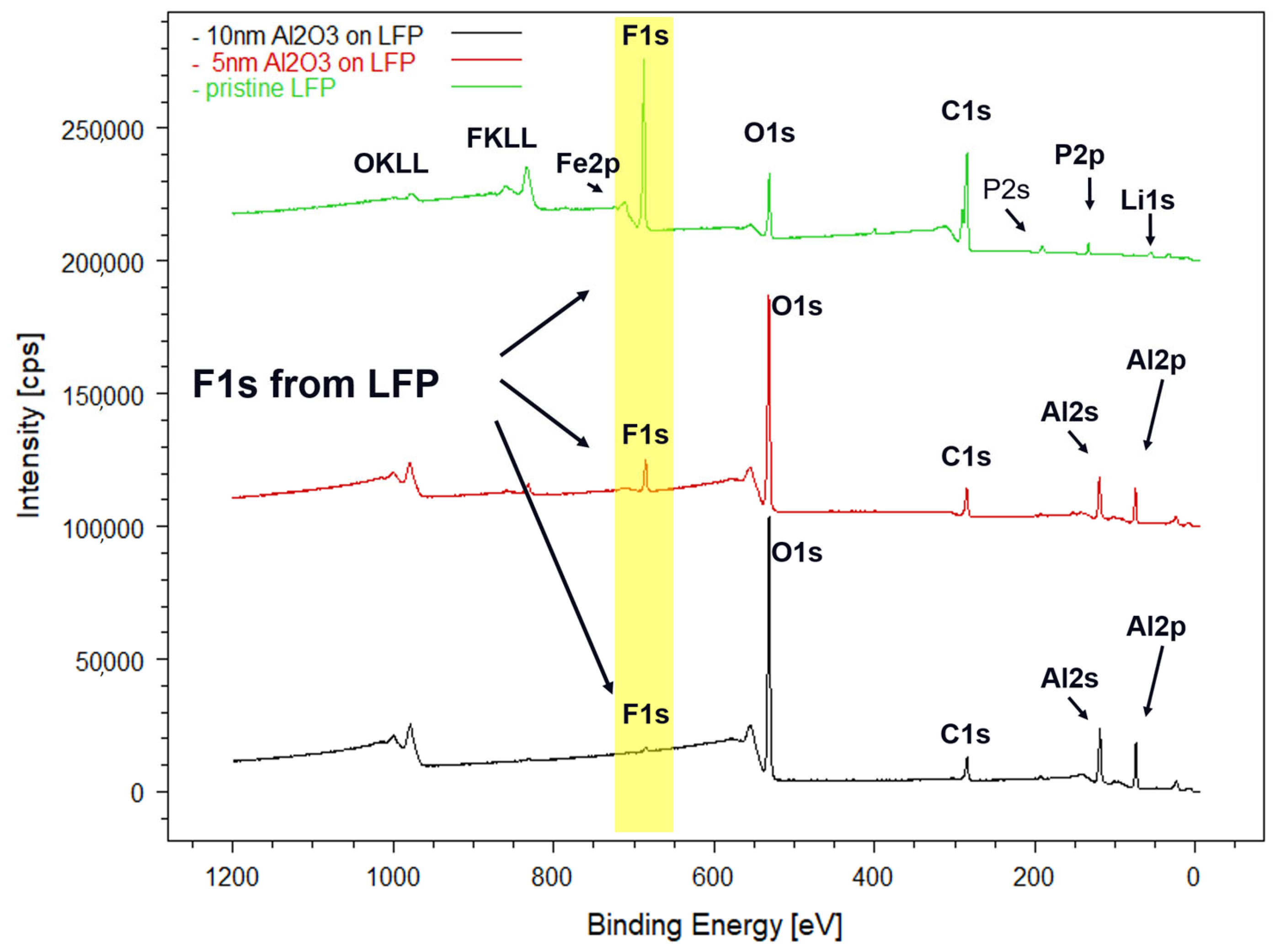
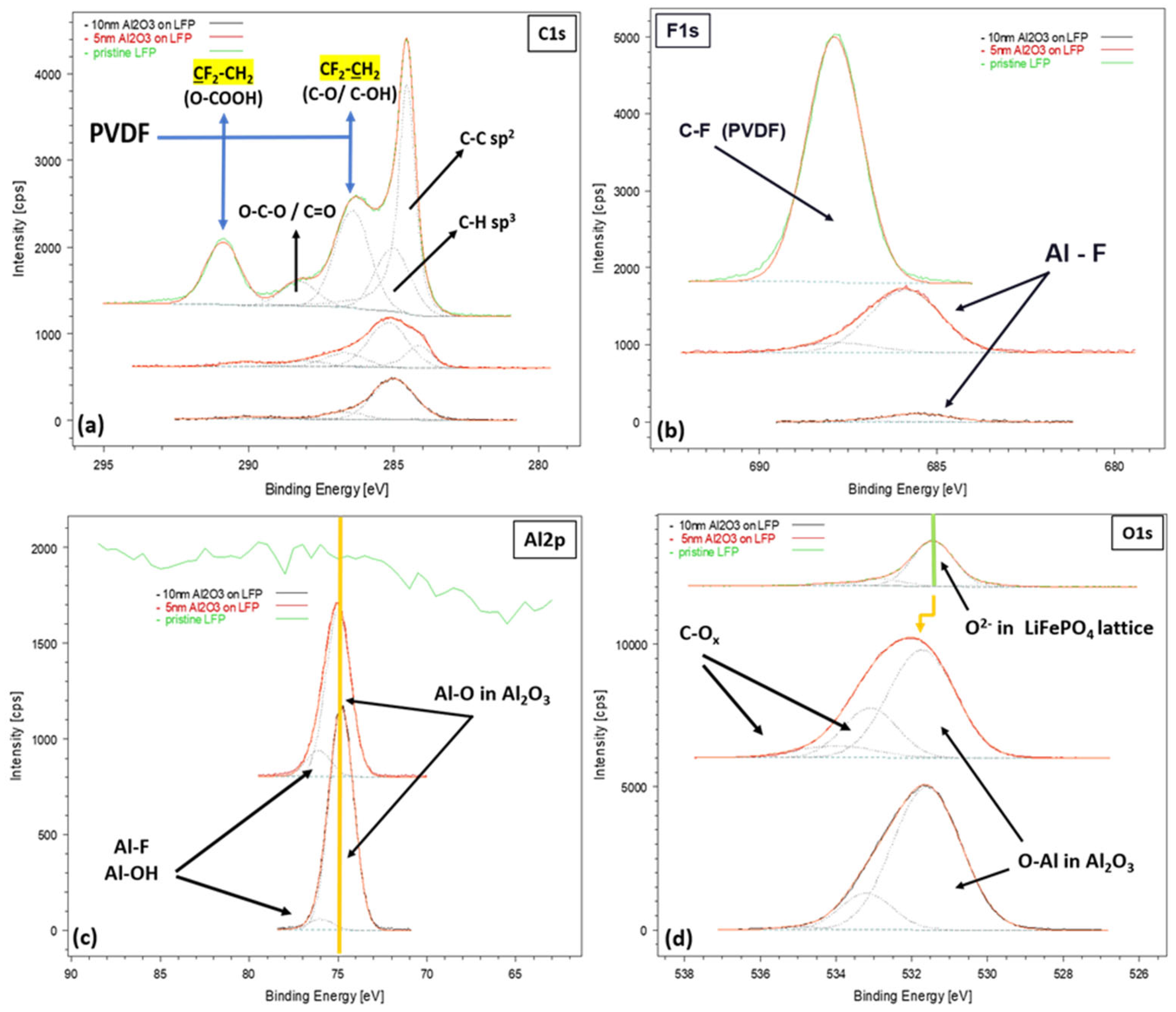
3.1. Structural Characterization of the Pristine LFP Electrode and of the Al2O3-Coated Electrodes
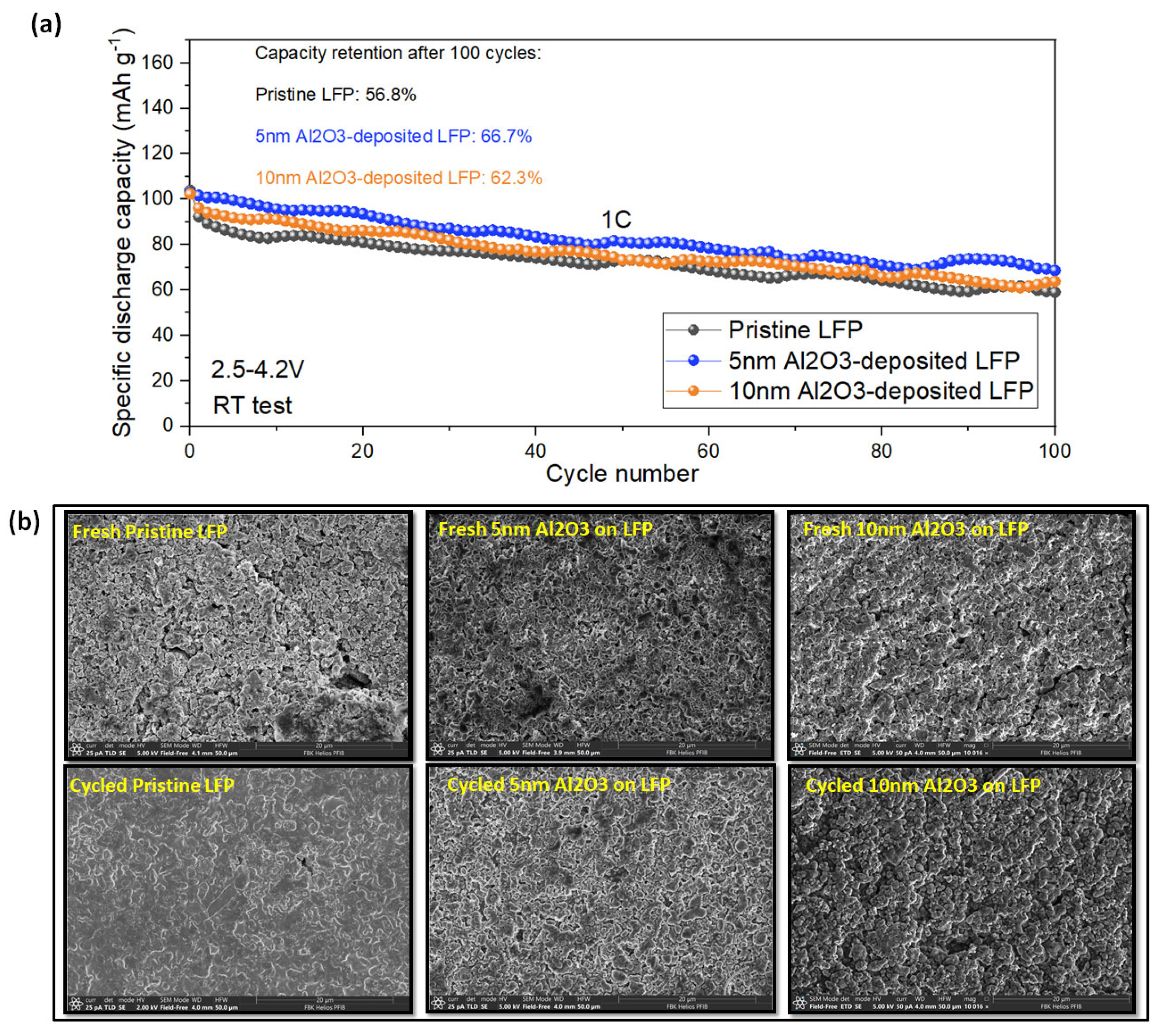
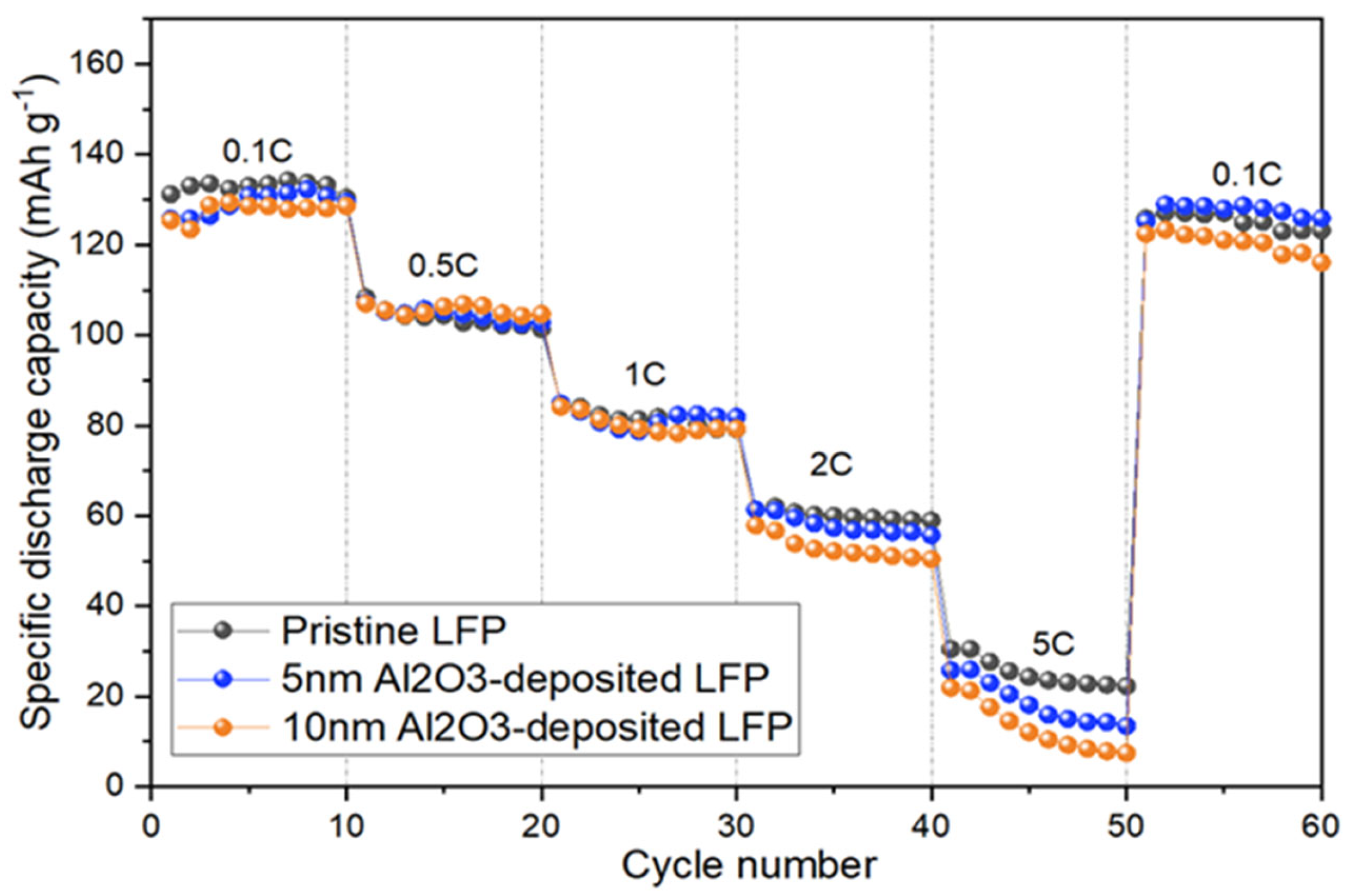
3.2. Electrochemical and Post-Mortem Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xu, C.; Steubing, B.; Hu, M.; Harpprecht, C.; van der Meide, M.; Tukker, A. Future Greenhouse Gas Emissions of Automotive Lithium-Ion Battery Cell Production. Resour. Conserv. Recycl. 2022, 187, 106606. [Google Scholar] [CrossRef]
- Gonçalves, R.; Lanceros-Méndez, S.; Costa, C.M. Electrode Fabrication Process and Its Influence in Lithium-Ion Battery Performance: State of the Art and Future Trends. Electrochem. Commun. 2022, 135, 107210. [Google Scholar] [CrossRef]
- Lithium-Ion Battery Market to Exceed $120 Billion by 2028: Electric Vehicles and Renewable Energy Drive Global Expansion. Available online: https://finance.yahoo.com/news/lithium-ion-battery-market-exceed-121800187.html?guccounter=1&guce_referrer=aHR0cHM6Ly93d3cuZ29vZ2xlLmNvbS8&guce_referrer_sig=AQAAANlnQLaH5owzBNcTLsFa9nCpN610I1L0fIQV_Huuhev0AKDIiV_-R2GSUCG7S7rQALIiGC5mzs9U3VylcBQrvJ8medgdzentpdMCrTHstCfaSOuqh-BfrIibVxZY9ykQGw6MgAD4FZOuyiPY6tYz-QW15QFGSaCJxyjR_1Ji9kUA (accessed on 26 April 2024).
- Huang, Y. The Discovery of Cathode Materials for Lithium-Ion Batteries from the View of Interdisciplinarity. Interdiscip. Mater. 2022, 1, 323–329. [Google Scholar] [CrossRef]
- Stenina, I.; Minakova, P.; Kulova, T.; Yaroslavtsev, A. Electrochemical Properties of LiFePO4 Cathodes: The Effect of Carbon Additives. Batteries 2022, 8, 111. [Google Scholar] [CrossRef]
- Salimi, P.; Tieuli, S.; Taghavi, S.; Venezia, E.; Fugattini, S.; Lauciello, S.; Prato, M.; Marras, S.; Li, T.; Signoretto, M.; et al. Sustainable Lithium-Ion Batteries Based on Metals-Free Tannery Waste Biochar. Green Chem. 2022, 24, 4119–4129. [Google Scholar] [CrossRef]
- Parviz, Z.; Salimi, P.; Javadian, S.; Gharibi, H.; Morsali, A.; Bayat, E.; Leoncino, L.; Lauciello, S.; Zaccaria, R.P. Fabrication of Sustainable Hybrid MOF/Silica Electrodes for Current Lithium-Ion Batteries and Beyond. ACS Appl. Energy Mater. 2022, 5, 15155–15165. [Google Scholar] [CrossRef]
- Huang, X.; Chen, K.; Liu, Y. Interfacial Effect of Nano Al2O3 Modifying LiFePO4 to Improve Capacity Retention and Rate Capability of Lithium Ion Batteries. Mater. Res. Express 2018, 6, 015511. [Google Scholar] [CrossRef]
- Beletskii, E.V.; Alekseeva, E.V.; Spiridonova, D.V.; Yankin, A.N.; Levin, O.V. Overcharge Cycling Effect on the Surface Layers and Crystalline Structure of LiFePO4 Cathodes of Li-Ion Batteries. Energies 2019, 12, 4652. [Google Scholar] [CrossRef]
- Wang, L.; Qiu, J.; Wang, X.; Chen, L.; Cao, G.; Wang, J.; Zhang, H.; He, X. Insights for Understanding Multiscale Degradation of LiFePO4 Cathodes. eScience 2022, 2, 125–137. [Google Scholar] [CrossRef]
- Jiang, J.; Shi, W.; Zheng, J.; Zuo, P.; Xiao, J.; Chen, X.; Xu, W.; Zhang, J.-G. Optimized Operating Range for Large-Format LiFePO4/Graphite Batteries. J. Electrochem. Soc. 2014, 161, A336–A341. [Google Scholar] [CrossRef]
- David, L.A.; Dahlberg, K.; Mohanty, D.; Ruther, R.E.; Huq, A.; An, S.J.; Mao, C.; King, D.; Stevenson, L.; Wood, D.L. Unveiling the Role of Al2O3 in Preventing Surface Reconstruction During High-Voltage Cycling of Lithium-Ion. Batteries 2019, 2, 1308–1313. [Google Scholar] [CrossRef]
- Prakasha, K.R.; Sathish, M.; Bera, P.; Prakash, A.S. Mitigating the Surface Degradation and Voltage Decay of Li1.2Ni0.13Mn0.54Co0.13O2 Cathode Material through Surface Modification Using Li2ZrO3. ACS Omega 2017, 2, 2308–2316. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Wang, L.; Xu, J.; Wang, Y.; Zhou, A.; Li, J. Improved Electrochemical Performance of LiCoO2 Electrodes with ZnO Coating by Radio Frequency Magnetron Sputtering. ACS Appl. Mater. Interfaces 2014, 6, 15853–15859. [Google Scholar] [CrossRef] [PubMed]
- Etinger-Geller, Y.; Polishchuk, I.; Seknazi, E.; Livne, A.; Ciatto, G.; Pokroy, B. Surface Reconstruction Causes Structural Variations in Nanometric Amorphous Al2O3. Phys. Chem. Chem. Phys. 2019, 21, 14887–14891. [Google Scholar] [CrossRef]
- Yoo, J.E.; Zazpe, R.; Cha, G.; Prikryl, J.; Hwang, I.; Macak, J.M.; Schmuki, P. Uniform ALD Deposition of Pt Nanoparticles within 1D Anodic TiO2 Nanotubes for Photocatalytic H2 Generation. Electrochem. Commun. 2018, 86, 6–11. [Google Scholar] [CrossRef]
- Yasmeen, S.; Ryu, S.W.; Lee, S.H.; Lee, H.B.R. Atomic Layer Deposition Beyond Thin Film Deposition Technology. Adv. Mater. Technol. 2023, 8, 202200876. [Google Scholar] [CrossRef]
- Pan, H.; Zhou, L.; Zheng, W.; Liu, X.; Zhang, J.; Pinna, N. Atomic Layer Deposition to Heterostructures for Application in Gas Sensors. Int. J. Extrem. Manuf. 2023, 5, 022008. [Google Scholar] [CrossRef]
- Ansari, M.Z.; Hussain, I.; Mohapatra, D.; Ansari, S.A.; Rahighi, R.; Nandi, D.K.; Song, W.; Kim, S.H. Atomic Layer Deposition—A Versatile Toolbox for Designing/Engineering Electrodes for Advanced Supercapacitors. Adv. Sci. 2023, 11, e202303055. [Google Scholar] [CrossRef]
- George, S.M. Atomic Layer Deposition: An Overview. Chem. Rev. 2010, 110, 111–131. [Google Scholar] [CrossRef]
- Cao, Y.Q.; Wang, S.S.; Liu, C.; Wu, D.; Li, A.D. Atomic Layer Deposition of ZnO/TiO2 Nanolaminates as Ultra-Long Life Anode Material for Lithium-Ion Batteries. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef]
- Mohanty, D.; Dahlberg, K.; King, D.M.; David, L.A.; Sefat, A.S.; Wood, D.L.; Daniel, C.; Dhar, S.; Mahajan, V.; Lee, M.; et al. Modification of Ni-Rich FCG NMC and NCA Cathodes by Atomic Layer Deposition: Preventing Surface Phase Transitions for High-Voltage Lithium-Ion Batteries. Sci. Rep. 2016, 6, 26532. [Google Scholar] [CrossRef]
- Kazyak, E.; Chen, K.H.; Wood, K.N.; Davis, A.L.; Thompson, T.; Bielinski, A.R.; Sanchez, A.J.; Wang, X.; Wang, C.; Sakamoto, J.; et al. Atomic Layer Deposition of the Solid Electrolyte Garnet Li7La3Zr2O12. Chem. Mater. 2017, 29, 3785–3792. [Google Scholar] [CrossRef]
- Han, B.; Key, B.; Lipton, A.S.; Vaughey, J.T.; Trevey, J.; Dogan, F.; Hughes, B. Influence of Coating Protocols on Alumina-Coated Cathode Material: Atomic Layer Deposition versus Wet-Chemical Coating. J. Electrochem. Soc. 2019, 166, A3679–A3684. [Google Scholar] [CrossRef]
- Jung, Y.S.; Cavanagh, A.S.; Dillon, A.C.; Groner, M.D.; George, S.M.; Lee, S.-H. Enhanced Stability of LiCoO2 Cathodes in Lithium-Ion Batteries Using Surface Modification by Atomic Layer Deposition. J. Electrochem. Soc. 2010, 157, A75. [Google Scholar] [CrossRef]
- Shi, C.; Hamann, T.; Takeuchi, S.; Alexander, G.V.; Nolan, A.M.; Limpert, M.; Fu, Z.; O’Neill, J.; Godbey, G.; Dura, J.A.; et al. 3D Asymmetric Bilayer Garnet-Hybridized High-Energy-Density Lithium−Sulfur Batteries. ACS Appl. Mater. Interfaces 2023, 15, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Koshtyal, Y.; Olkhovskii, D.; Rumyantsev, A.; Maximov, M. Applications and Advantages of Atomic Layer Deposition for Lithium-Ion Batteries Cathodes: Review. Batter. 2022, 8, 184. [Google Scholar] [CrossRef]
- Sahoo, P.P.; Güneren, A.; Hudec, B.; Mičušík, M.; Lenčéš, Z.; Siffalovic, P.; Frohlich, K. Improved Properties of Li-Ion Battery Electrodes Protected By Al2O3 and ZnO Ultrathin Layers Prepared By Atomic Layer Deposition. ECS Meet. Abstr. 2022, MA2022-02, 1136. [Google Scholar] [CrossRef]
- Li, X.; Liu, J.; Banis, M.N.; Lushington, A.; Li, R.; Cai, M.; Sun, X. Atomic Layer Deposition of Solid-State Electrolyte Coated Cathode Materials with Superior High-Voltage Cycling Behavior for Lithium Ion Battery Application. Energy Environ. Sci. 2014, 7, 768–778. [Google Scholar] [CrossRef]
- Jung, Y.S.; Cavanagh, A.S.; Riley, L.A.; Kang, S.H.; Dillon, A.C.; Groner, M.D.; George, S.M.; Lee, S.H. Ultrathin Direct Atomic Layer Deposition on Composite Electrodes for Highly Durable and Safe Li-Ion Batteries. Adv. Mater. 2010, 22, 2172–2176. [Google Scholar] [CrossRef] [PubMed]
- Khotimah, C.; Cheng, H.M.; Wang, F.M. Enhanced Cyclability of Lithium-Ion Batteries Resulting from Atomic Layer Deposition of Al2O3 on LiFePO4 Electrodes. J. Solid State Electrochem. 2024. [Google Scholar] [CrossRef]
- Hu, C.; Geng, M.; Yang, H.; Fan, M.; Sun, Z.; Yu, R.; Wei, B. A Review of Capacity Fade Mechanism and Promotion Strategies for Lithium Iron Phosphate Batteries. Coatings 2024, 14, 832. [Google Scholar] [CrossRef]
- Speranza, G.; Canteri, R. RxpsG a New Open Project for Photoelectron and Electron Spectroscopy Data Processing. SoftwareX 2019, 10, 100282. [Google Scholar] [CrossRef]
- Moulder, J.; Stickle, W.; Sobol, W.; Bomben, K.D. Handbook of X-Ray Photoelectron Spectroscopy. Perkin-Elmer Corp. 1992, 40, 221. [Google Scholar]
- Beamson, G.; Briggs, D. High Resolution Monochromated X-Ray Photoelectron Spectroscopy of Organic Polymers: A Comparison between Solid State Data for Organic Polymers and Gas Phase Data for Small Molecules. Mol. Phys. 1992, 76, 919–936. [Google Scholar] [CrossRef]
- Evshchik, E.Y.; Sanginov, E.A.; Kayumov, R.R.; Zhuravlev, V.D.; Bushkova, O.V.; Dobrovolsky, Y.A. Li4Ti5O12/LiFePO4 Solid-State Lithium-Ion Full Cell with LithiatedNafion Membrane. Int. J. Electrochem. Sci. 2020, 15, 2216–2225. [Google Scholar] [CrossRef]
- Buqa, H.; Goers, D.; Holzapfel, M.; Spahr, M.E.; Novák, P. High Rate Capability of Graphite Negative Electrodes for Lithium-Ion Batteries. J. Electrochem. Soc. 2005, 152, A474. [Google Scholar] [CrossRef]
- Dokko, K.; Nakata, N.; Kanamura, K. High Rate Discharge Capability of Single Particle Electrode of LiCoO2. J. Power Sources 2009, 189, 783–785. [Google Scholar] [CrossRef]
- Yuan, L.X.; Wang, Z.H.; Zhang, W.X.; Hu, X.L.; Chen, J.T.; Huang, Y.H.; Goodenough, J.B. Development and Challenges of LiFePO4 Cathode Material for Lithium-Ion Batteries. Energy Environ. Sci. 2011, 4, 269–284. [Google Scholar] [CrossRef]
- Peled, E.; Menkin, S. Review—SEI: Past, Present and Future. J. Electrochem. Soc. 2017, 164, A1703–A1719. [Google Scholar] [CrossRef]
- Barsoukov, E.; Macdonald, J.R. (Eds.) Impedance Spectroscopy; John Wiley & Sons: Hoboken, NJ, USA, 2018. [Google Scholar] [CrossRef]
- Brug, G.J.; van den Eeden, A.L.G.; Sluyters-Rehbach, M.; Sluyters, J.H. The Analysis of Electrode Impedances Complicated by the Presence of a Constant Phase Element. J. Electroanal. Chem. Interfacial Electrochem. 1984, 176, 275–295. [Google Scholar] [CrossRef]
- Bal, B.; Ozdogru, B.; Nguyen, D.T.; Li, Z.; Murugesan, V.; Çapraz, Ö.Ö. Probing the Formation of Cathode-Electrolyte Interphase on Lithium Iron Phosphate Cathodes via Operando Mechanical Measurements. ACS Appl. Mater. Interfaces 2023, 15, 42449–42459. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Zhou, J.; Liu, G.; Wang, L. Lithium-Ions Diffusion Kinetic in LiFePO4/Carbon Nanoparticles Synthesized by Microwave Plasma Chemical Vapor Deposition for Lithium-Ion Batteries. Appl. Surf. Sci. 2018, 433, 35–44. [Google Scholar] [CrossRef]
- Wang, J.; Koenig, G.M. Comparison of Lithium Diffusion Coefficient Measurements in Tellurium Electrodes via Different Electrochemical Techniques. J. Electrochem. Soc. 2023, 170, 050534. [Google Scholar] [CrossRef]
- Ho, C.; Raistrick, I.D.; Huggins, R.A. Application of A-C Techniques to the Study of Lithium Diffusion in Tungsten Trioxide Thin Films. J. Electrochem. Soc. 1980, 127, 343–350. [Google Scholar] [CrossRef]
- Ahsan, Z.; Ding, B.; Cai, Z.; Wen, C.; Yang, W.; Ma, Y.; Zhang, S. Recent Progress in Capacity Enhancement of LiFePO4cathode for Li-Ion Batteries. J. Electrochem. Energy Convers. Storage 2021, 18, 010801. [Google Scholar] [CrossRef]
- Baboo, J.P.; Yatoo, M.A.; Dent, M.; Hojaji Najafabadi, E.; Lekakou, C.; Slade, R.; Hinder, S.J.; Watts, J.F. Exploring Different Binders for a LiFePO4 Battery, Battery Testing, Modeling and Simulations. Energies 2022, 15, 2332. [Google Scholar] [CrossRef]
- Kroff, M.; Hevia, S.A.; O’Shea, J.N.; de Muro, I.G.; Palomares, V.; Rojo, T.; del Río, R. Lithium Iron Phosphate/Carbon (LFP/C) Composite Using Nanocellulose as a Reducing Agent and Carbon Source. Polymers 2023, 15, 2628. [Google Scholar] [CrossRef]
- Lee, H.; Lim, H.S.; Ren, X.; Yu, L.; Engelhard, M.H.; Han, K.S.; Lee, J.; Kim, H.T.; Xiao, J.; Liu, J.; et al. Detrimental Effects of Chemical Crossover from the Lithium Anode to Cathode in Rechargeable Lithium Metal Batteries. ACS Energy Lett. 2018, 3, 2921–2930. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salimi, P.; Gottardi, G.; Morais, W.G.; Bartali, R.; Laidani, N.; Macchi, E.G. Improving Lithium-Ion Battery Performance: Nano Al2O3 Coatings on High-Mass Loading LiFePO4 Cathodes via Atomic Layer Deposition. Batteries 2024, 10, 304. https://doi.org/10.3390/batteries10090304
Salimi P, Gottardi G, Morais WG, Bartali R, Laidani N, Macchi EG. Improving Lithium-Ion Battery Performance: Nano Al2O3 Coatings on High-Mass Loading LiFePO4 Cathodes via Atomic Layer Deposition. Batteries. 2024; 10(9):304. https://doi.org/10.3390/batteries10090304
Chicago/Turabian StyleSalimi, Pejman, Gloria Gottardi, William G. Morais, Ruben Bartali, Nadhira Laidani, and Edoardo Gino Macchi. 2024. "Improving Lithium-Ion Battery Performance: Nano Al2O3 Coatings on High-Mass Loading LiFePO4 Cathodes via Atomic Layer Deposition" Batteries 10, no. 9: 304. https://doi.org/10.3390/batteries10090304
APA StyleSalimi, P., Gottardi, G., Morais, W. G., Bartali, R., Laidani, N., & Macchi, E. G. (2024). Improving Lithium-Ion Battery Performance: Nano Al2O3 Coatings on High-Mass Loading LiFePO4 Cathodes via Atomic Layer Deposition. Batteries, 10(9), 304. https://doi.org/10.3390/batteries10090304