
Predicting Pt-195 NMR Chemical Shift and 1J(195Pt-31P) Coupling Constant for Pt(0) Complexes Using the NMR-DKH Basis Sets




Abstract
:
1. Introduction
2. Theoretical Methodology
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Dilruba, S.; Kalayda, G.V. Platinum-Based Drugs: Past, Present and Future. Cancer Chemother. Pharmacol. 2016, 77, 1103–1124. [Google Scholar] [CrossRef]
- Deo, K.M.; Ang, D.L.; McGhie, B.; Rajamanickam, A.; Dhiman, A.; Khoury, A.; Holland, J.; Bjelosevic, A.; Pages, B.; Gordon, C.; et al. Platinum Coordination Compounds with Potent Anticancer Activity. Coord. Chem. Rev. 2018, 375, 148–163. [Google Scholar] [CrossRef]
- Dedieu, A. Theoretical Studies in Palladium and Platinum Molecular Chemistry. Chem. Rev. 2000, 100, 543–600. [Google Scholar] [CrossRef]
- Li, C.-J.; Chen, L. Organic Chemistry in Water. Chem. Soc. Rev. 2006, 35, 68–82. [Google Scholar] [CrossRef]
- Li, C.-J. Organic Reactions in Aqueous Media with a Focus on Carbon−Carbon Bond Formations: A Decade Update. Chem. Rev. 2005, 105, 3095–3166. [Google Scholar] [CrossRef] [PubMed]
- Silbestri, G.F.; Flores, J.C.; de Jesús, E. Water-Soluble N-Heterocyclic Carbene Platinum(0) Complexes: Recyclable Catalysts for the Hydrosilylation of Alkynes in Water at Room Temperature. Organometallics 2012, 31, 3355–3360. [Google Scholar] [CrossRef]
- Nakajima, Y.; Shimada, S. Hydrosilylation Reaction of Olefins: Recent Advances and Perspectives. RSC Adv. 2015, 5, 20603–20616. [Google Scholar] [CrossRef]
- Sprengers, J.W.; de Greef, M.; Duin, M.A.; Elsevier, C.J. Stable Platinum(0) Catalysts for Catalytic Hydrosilylation of Styrene and Synthesis of [Pt(Ar-Bian)(H2-Alkene)] Complexes. Eur. J. Inorg. Chem. 2003, 2003, 3811–3819. [Google Scholar] [CrossRef]
- Troegel, D.; Stohrer, J. Recent Advances and Actual Challenges in Late Transition Metal Catalyzed Hydrosilylation of Olefins from an Industrial Point of View. Coord. Chem. Rev. 2011, 255, 1440–1459. [Google Scholar] [CrossRef]
- Markó, I.E.; Stérin, S.; Buisine, O.; Mignani, G.; Branlard, P.; Tinant, B.; Declercq, J.-P. Selective and Efficient Platinum(0)-Carbene Complexes As Hydrosilylation Catalysts. Science 2002, 298, 204–206. [Google Scholar] [CrossRef]
- Pregosin, P.S. Platinum NMR Spectroscopy. Annu. Rep. NMR Spectrosc. 1986, 17, 285–349. [Google Scholar] [CrossRef]
- Georgii, I.; Mann, B.E.; Taylor, B.F.; Musco, A. 195Pt n.m.r. Spectra of Some [Pt(PR3)n] Complexes. Inorg. Chim. Acta 1984, 86, L81–L82. [Google Scholar] [CrossRef]
- Still, B.M.; Kumar, P.G.A.; Aldrich-Wright, J.R.; Price, W.S. 195Pt NMR—Theory and Application. Chem. Soc. Rev. 2007, 36, 665–686. [Google Scholar] [CrossRef]
- Pregosin, P.S. Platinum-195 Nuclear Magnetic Resonance. Coord. Chem. Rev. 1982, 44, 247–291. [Google Scholar] [CrossRef]
- Priqueler, J.R.L.; Butler, I.S.; Rochon, F.D. An Overview of 195Pt Nuclear Magnetic Resonance Spectroscopy. Appl. Spectrosc. Rev. 2006, 41, 185–226. [Google Scholar] [CrossRef]
- Vaara, J. Theory and Computation of Nuclear Magnetic Resonance Parameters. Phys. Chem. Chem. Phys. 2007, 9, 5399. [Google Scholar] [CrossRef] [PubMed]
- Bühl, M.; Kaupp, M.; Malkina, O.L.; Malkin, V.G. The DFT Route to NMR Chemical Shifts. J. Comput. Chem. 1999, 20, 91–105. [Google Scholar] [CrossRef]
- Paschoal, D.; Guerra, C.F.; de Oliveira, M.A.L.; Ramalho, T.C.; dos Santos, H.F. Predicting Pt-195 NMR Chemical Shift Using New Relativistic All-Electron Basis Set. J. Comput. Chem. 2016, 37, 2360–2373. [Google Scholar] [CrossRef]
- Carvalho, J.; Paschoal, D.; Fonseca Guerra, C.; dos Santos, H.F. Nonrelativistic Protocol for Calculating the 1J(195Pt-15N) Coupling Constant in Pt(II)-Complexes Using All-Electron Gaussian Basis-Set. Chem. Phys. Lett. 2020, 745, 137279. [Google Scholar] [CrossRef]
- da Silva Paschoal, D.F.; da Silva Gomes, M.; Machado, L.P.N.; dos Santos, H.F. Basis Sets for Heavy Atoms. In Basis Sets in Computational Chemistry. Lecture Notes in Chemistry; Perlt, E., Ed.; Springer: Cham, Switzerland, 2021; Volume 107, pp. 183–214. [Google Scholar]
- Semenov, V.A.; Samultsev, D.O.; Rusakova, I.L.; Krivdin, L.B. Computational Multinuclear NMR of Platinum Complexes: A Relativistic Four-Component Study. J. Phys. Chem. A 2019, 123, 4908–4920. [Google Scholar] [CrossRef]
- Sterzel, M.; Autschbach, J. Toward an Accurate Determination of 195Pt Chemical Shifts by Density Functional Computations: The Importance of Unspecific Solvent Effects and the Dependence of Pt Magnetic Shielding Constants on Structural Parameters. Inorg. Chem. 2006, 45, 3316–3324. [Google Scholar] [CrossRef] [PubMed]
- Semenov, V.A.; Rusakov, Y.Y.; Samultsev, D.O.; Krivdin, L.B. Geometries and NMR Properties of Cisplatin and Transplatin Revisited at the Four-Component Relativistic Level. Mendeleev Commun. 2019, 29, 315–317. [Google Scholar] [CrossRef]
- Sutter, K.; Truflandier, L.A.; Autschbach, J. NMR J-Coupling Constants in Cisplatin Derivatives Studied by Molecular Dynamics and Relativistic DFT. ChemPhysChem 2011, 12, 1448–1455. [Google Scholar] [CrossRef] [PubMed]
- Tsipis, A.C.; Karapetsas, I.N. Accurate Prediction of 195Pt NMR Chemical Shifts for a Series of Pt(II) and Pt(IV) Antitumor Agents by a Non-Relativistic DFT Computational Protocol. Dalton Trans. 2014, 43, 5409–5426. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, T.M.; Ziegler, T. Prediction of 195 Pt NMR Chemical Shifts by Density Functional Theory Computations: The Importance of Magnetic Coupling and Relativistic Effects in Explaining Trends. J. Phys. Chem. A 1999, 103, 7535–7543. [Google Scholar] [CrossRef]
- Berkefeld, A.; Reimann, M.; Hörner, G.; Kaupp, M.; Schubert, H. C–P vs C–H Bond Cleavage of Triphenylphosphine at Platinum(0): Mechanism of Formation, Reactivity, Redox Chemistry, and NMR Chemical Shift Calculations of a μ-Phosphanido Diplatinum(II) Platform. Organometallics 2020, 39, 443–452. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for the Transition Metal Atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for K to Au Including the Outermost Core Orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297. [Google Scholar] [CrossRef] [PubMed]
- Scalmani, G.; Frisch, M.J. Continuous Surface Charge Polarizable Continuum Models of Solvation. I. General Formalism. J. Chem. Phys. 2010, 132, 114110. [Google Scholar] [CrossRef] [PubMed]
- Cheeseman, J.R.; Trucks, G.W.; Keith, T.A.; Frisch, M.J. A Comparison of Models for Calculating Nuclear Magnetic Resonance Shielding Tensors. J. Chem. Phys. 1996, 104, 5497–5509. [Google Scholar] [CrossRef]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient Implementation of the Gauge-Independent Atomic Orbital Method for NMR Chemical Shift Calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett 1997, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian ∼09 Revision D.01. Available online: https://www.scienceopen.com/document?vid=839f33cc-9114-4a55-8f1a-3f1520324ef5 (accessed on 20 September 2021).
- Mann, B.E.; Musco, A. A 31P Nuclear Magnetic Resonance Investigation of the Structure, Equilibria, and Kinetics of [Pt(PR3)n] in Solution. J. Chem. Soc. Dalton Trans. 1980, 776–785. [Google Scholar] [CrossRef]
- Benn, R.; Michael Büch, H.; Reinhardt, R.-D. Heavy Metal Spin- ½ Nuclei. Platinum-195 Relaxation Times in Phosphorus-Platinum(0) Compounds and Their Dependence on the Geometry of the Complex. Magn. Reson. Chem. 1985, 23, 559–564. [Google Scholar] [CrossRef]
- Koie, Y.; Shinoda, S.; Saito, Y. Platinum-195 Nuclear Magnetic Resonance Study of Platinum(0) Complexes Containing a Series of Acetylenes. J. Chem. Soc. Dalton Trans. 1981, 1082–1088. [Google Scholar] [CrossRef]
- Kennedy, J.D.; McFarlane, W.; Puddephatt, R.J.; Thompson, P.J. Magnetic Double-Resonance Studies of Platinum-195 Chemical Shifts in Organoplatinum Compounds. J. Chem. Soc. Dalton Trans. 1976, 874–879. [Google Scholar] [CrossRef]
- Wrackmeyer, B.; Klimkina, E.V.; Schmalz, T.; Milius, W. Synthesis, NMR Spectroscopic Characterization and Structure of a Divinyldisilazane-(Triphenylphosphine)Platinum(0) Complex: Observation of Isotope-Induced Chemical Shifts 1Δ12/13C(195Pt). Magn. Reson. Chem. 2013, 51, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Varilla, A.M.; Baquero, E.A.; Silbestri, G.F.; Gonzalez-Arellano, C.; de Jesús, E.; Flores, J.C. Synthesis and Behavior of Novel Sulfonated Water-Soluble N-Heterocyclic Carbene (η4-Diene) Platinum(0) Complexes. Dalton Trans. 2015, 44, 18360–18369. [Google Scholar] [CrossRef] [PubMed]
- Buchner, M.R.; Bechlars, B.; Wahl, B.; Ruhland, K. Influence of Electronically and Sterically Tunable Cinnamate Ligands on the Spectroscopic Properties and Reactivity of Bis(Triphenylphosphine)Platinum(0) Olefin Complexes. Organometallics 2013, 32, 1643–1653. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
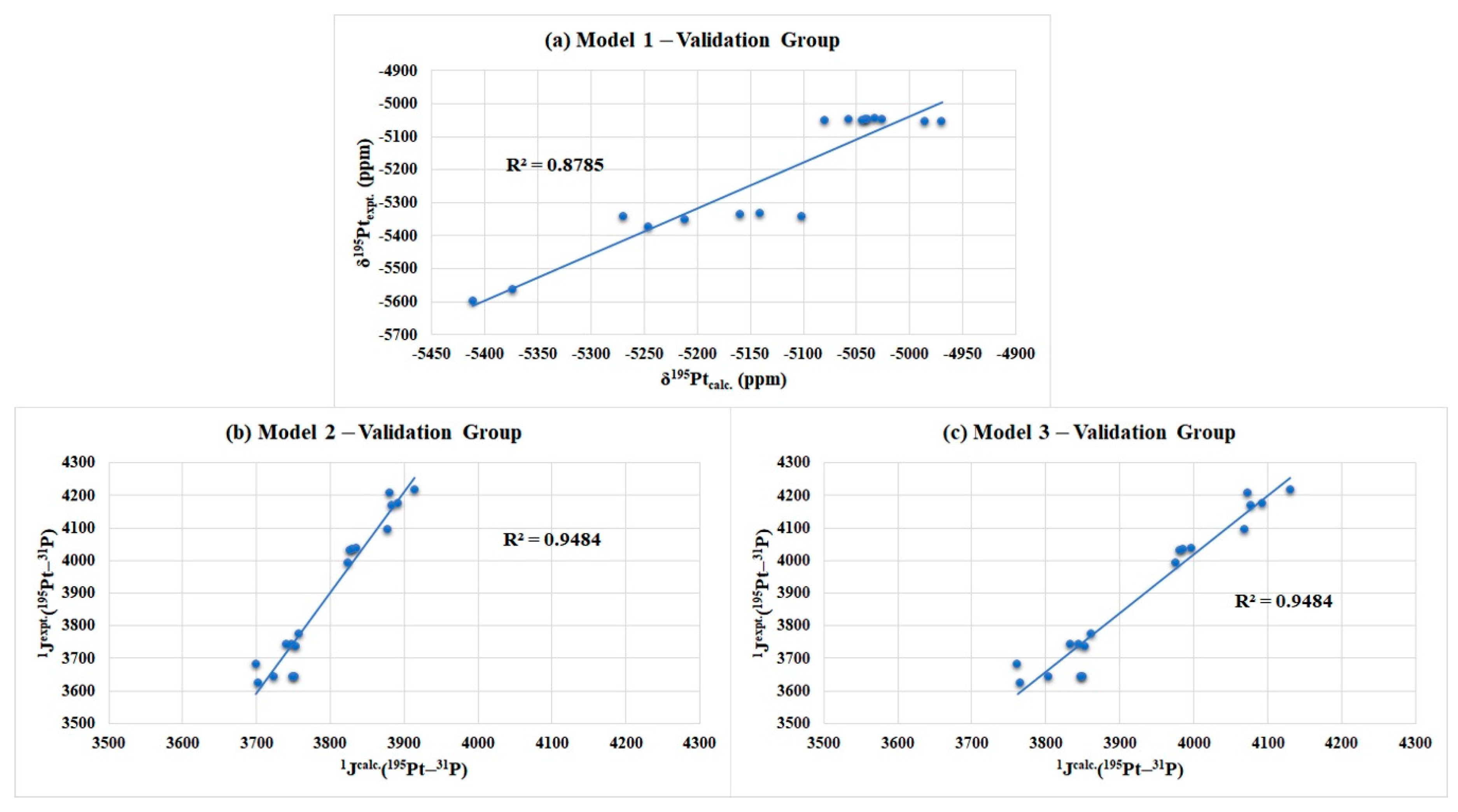
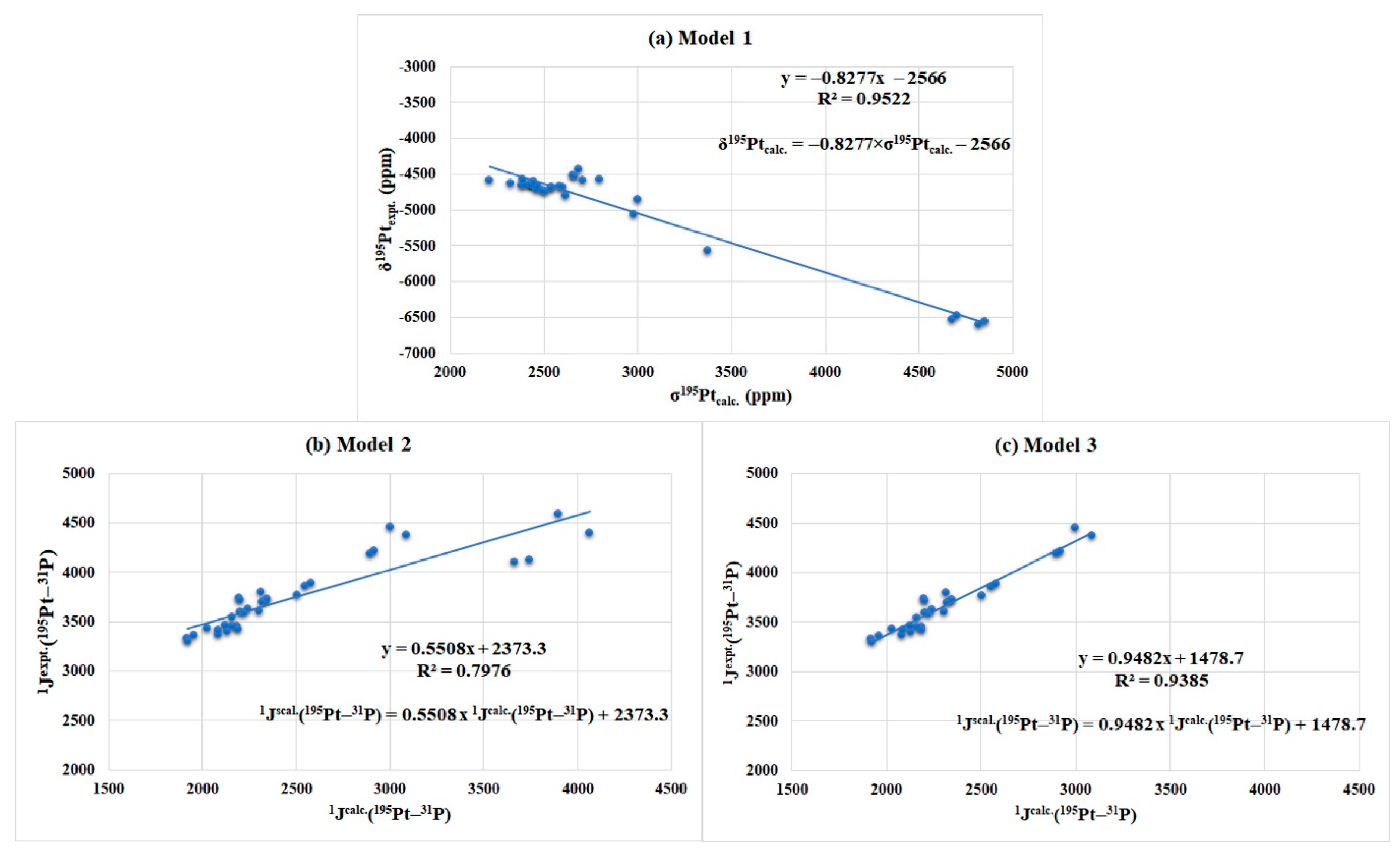
| Models | Linear Regression Models | R2 |
|---|---|---|
| Model 1 a | 0.9522 | |
| Model 2 b | 0.7976 | |
| Model 3 c | 0.9385 |
| δ195Pt (ppm) | 1J(195Pt–31P) (Hz) | |||||||
|---|---|---|---|---|---|---|---|---|
| Pt(0) Complexes | Solvent | Model 1 | Expt. | Model 0 | Model 2 | Model 3 | Expt. | |
| Linear Geometry (C.N. = 2) | ||||||||
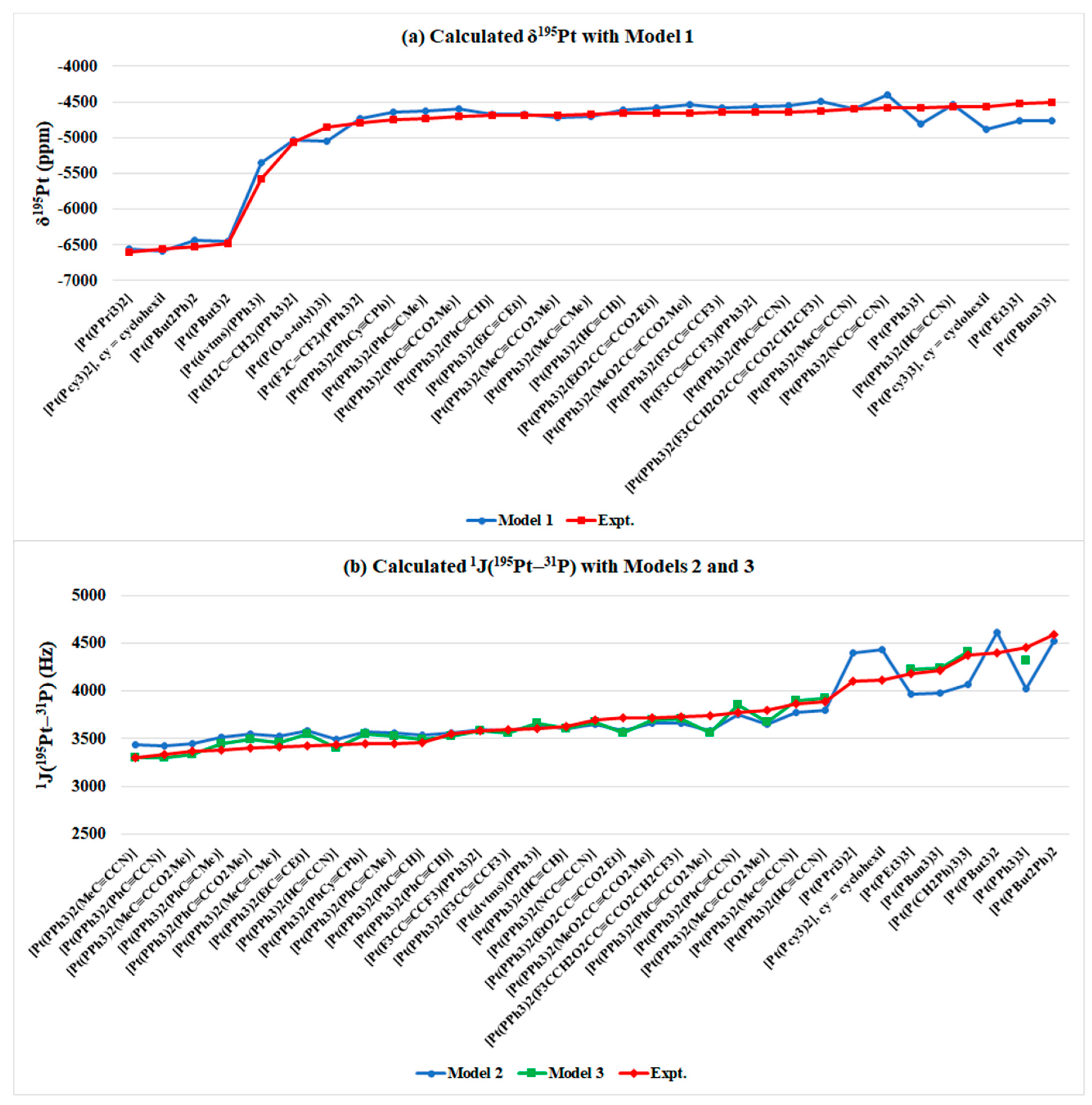
| 1 | [Pt(PPri3)2] | Toluene | −6557 | −6607 a | 3665 | 4392 | - | 4104 b |
| 2 | [Pt(Pcy3)2], cy = cyclohexil | Toluene | −6583 | −6555 a | 3742 | 4435 | - | 4120 b |
| 3 | [Pt(PBut2Ph)2 | Toluene | −6434 | −6526 a | 3898 | 4520 | - | 4592 b |
| 4 | [Pt(PBut3)2 | THF-d8 | −6457 | −6479 c | 4065 | 4612 | - | 4399 c |
| MAD/MRD (C.N. = 2) | 48/0.7% | 461/11% | 222/5.3% | - | ||||
| Trigonal Geometry (C.N. = 3) | ||||||||
| 5 | [Pt(PEt3)3] | Toluene | −4768 | −4526 a | 2897 | 3969 | 4226 | 4188 b |
| 6 | [Pt(PBun3)3] | Toluene | −4761 | −4511 a | 2916 | 3980 | 4244 | 4211 b |
| 7 | [Pt(P(CH2Ph)3)3] | Toluene | −4790 | −4439 a | 3086 | 4073 | 4405 | 4377 b |
| 8 | [Pt(Pcy3)3], cy = cyclohexil | Toluene | −4878 | −4567 a | - | - | - | - |
| 9 | [Pt(PPh3)2(PhCy≡CPh)] | CD2Cl2 | −4642 | −4741 d | 2186 | 3577 | 3551 | 3452 d |
| 10 | [Pt(PPh3)2(PhC≡CMe)] | CD2Cl2 | −4630 | −4727 d | 2083 | 3520 | 3454 | 3377 d |
| - | - | 2159 | 3562 | 3526 | 3454 d | |||
| 11 | [Pt(PPh3)2(PhC≡CCO2Me)] | CD2Cl2 | −4602 | −4710 d | 2130 | 3547 | 3499 | 3403 d |
| - | - | 2196 | 3583 | 3561 | 3741 d | |||
| 12 | [Pt(PPh3)2(PhC≡CH)] | CD2Cl2 | −4668 | −4690 d | 2121 | 3541 | 3490 | 3464 d |
| - | - | 2158 | 3562 | 3525 | 3547 d | |||
| 13 | [Pt(PPh3)2(EtC≡CEt)] | CD2Cl2 | −4669 | −4689 d | 2189 | 3579 | 3554 | 3425 d |
| 14 | [Pt(PPh3)2(MeC≡CCO2Me)] | CD2Cl2 | −4719 | −4682 d | 1957 | 3451 | 3334 | 3366 d |
| - | - | 2312 | 3647 | 3671 | 3803 d | |||
| 15 | [Pt(PPh3)2(MeC≡CMe)] | CD2Cl2 | −4702 | −4674 d | 2086 | 3522 | 3457 | 3420 d |
| 16 | [Pt(PPh3)2(HC≡CH)] | CD2Cl2 | −4607 | −4658 d | 2242 | 3608 | 3604 | 3626 d |
| 17 | [Pt(PPh3)2(EtO2CC≡CCO2Et)] | CD2Cl2 | −4586 | −4655 d | 2203 | 3587 | 3567 | 3722 d |
| 18 | [Pt(PPh3)2(MeO2CC≡CCO2Me)] | CD2Cl2 | −4537 | −4653 d | 2342 | 3664 | 3700 | 3722 d |
| 19 | [Pt(PPh3)2(F3CC≡CCF3)] | CD2Cl2 | −4576 | −4645 d | 2202 | 3586 | 3566 | 3595 d |
| 20 | [Pt(PPh3)2(PhC≡CCN)] | CD2Cl2 | −4549 | −4640 d | 1919 | 3430 | 3298 | 3336 d |
| - | - | 2506 | 3754 | 3855 | 3772 d | |||
| 21 | [Pt(PPh3)2(F3CH2CO2CC≡CCO2CH2CF3)] | CD2Cl2 | −4488 | −4626 d | 2346 | 3665 | 3703 | 3726 d |
| 22 | [Pt(PPh3)2(MeC≡CCN)] | CD2Cl2 | −4590 | −4598 d | 1923 | 3433 | 3302 | 3303 d |
| - | - | 2550 | 3778 | 3896 | 3864 d | |||
| 23 | [Pt(PPh3)2(HC≡CCN)] | CD2Cl2 | −4539 | −4573 d | 2026 | 3489 | 3400 | 3434 d |
| - | - | 2581 | 3795 | 3926 | 3887 d | |||
| 24 | [Pt(PPh3)2(NCC≡CCN)] | CD2Cl2 | −4395 | −4586 d | 2320 | 3651 | 3678 | 3696 d |
| 25 | [Pt(F3CC≡CCF3)(PPh3)2] | CDCl3 | −4570 | −4645 e | 2222 | 3597 | 3585 | 3590 e |
| 26 | [Pt(F2C=CF2)(PPh3)2] | CDCl3 | −4728 | −4791 e | - | - | - | - |
| 27 | [Pt(H2C=CH2)(PPh3)2] | CDCl3 | −5033 | −5065e | - | - | - | - |
| 28 | [Pt(P(O-o-tolyl)3)] | CD2Cl2 | −5049 | −4858 c | - | - | - | - |
| 29 | [Pt(PPh3)3] | THF-d8 | −4804 | −4583 c | 2999 | 4025 | 4322 | 4455 c |
| 30 | [Pt(dvtms)(PPh3)] | CDCl3 | −5358 | −5572 f | 2304 | 3643 | 3664 | 3609 f |
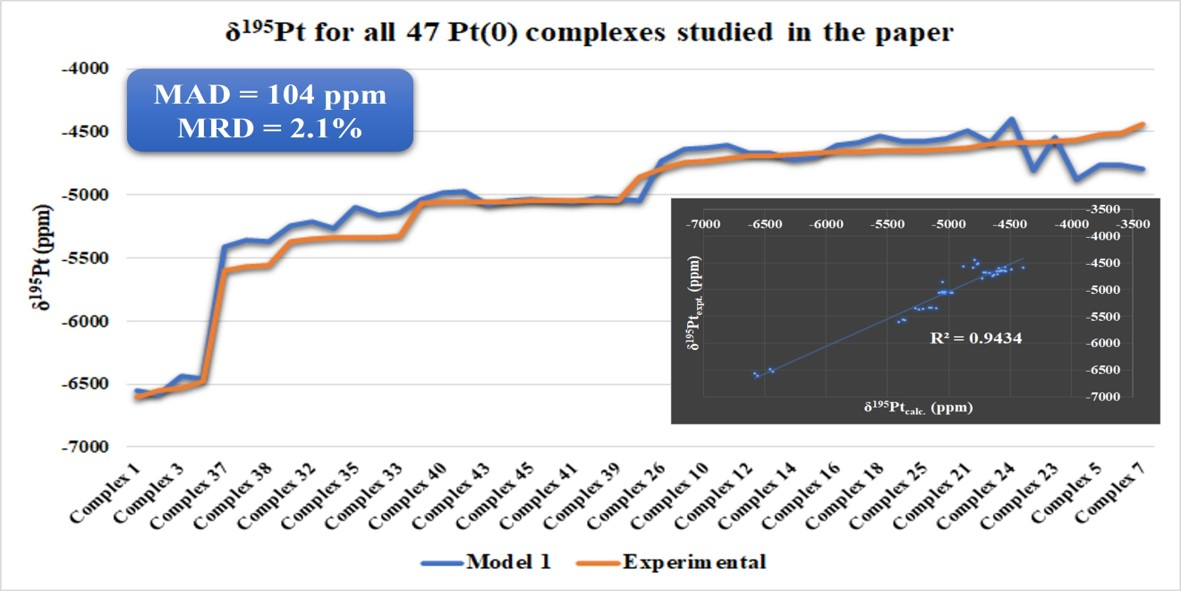
| MAD/MRD (C.N. = 3) | - | 120/2.6% | - | 1359/37% | 114/3.0% | 58/1.6% | - | |
| MAD/MRD (All Pt(0) complexes studied) | - | 107/2.3% | - | 1250/34% | 127/3.3% | 58/1.6% | - | |
| δ195Pt (ppm) | 1J(195Pt–31P) (Hz) | |||||||
|---|---|---|---|---|---|---|---|---|
| Pt(0) Complexes | Solvent | Model 1 | Expt. | Model 0 | Model 2 | Model 3 | Expt. | |
| Trigonal Geometry (C.N. = 3) | ||||||||
| 31 | [Pt(ICy)(dvtms)] | CDCl3 | −5270 | −5343 a | - | - | - | - |
| 32 | [Pt(Mes-NHC-Prn-SO3Na)(dvtms)] | DMSO | −5212 | −5352 b | - | - | - | - |
| 33 | [Pt(IPr-4-SO3Na)(dvtms)] | DMSO | −5141 | −5332 b | - | - | - | - |
| 34 | [Pt(IXy-4-SO3Na)(dvtms)] | D2O | −5160 | −5336 c | - | - | - | - |
| 35 | anti-[Pt(IMes-4-SO3Na)(dvtms)] | D2O | −5102 | −5342 c | - | - | - | - |
| 36 | syn-[Pt(SIMes-4-SO3Na)(dvtms)] | D2O | −5246 | −5372 c | - | - | - | - |
| 37 | [Pt(Mes-NHC-Prn-SO3Na)(AE)] | DMSO | −5411 | −5597 c | - | - | - | - |
| 38 | [Pt(IXy-4,4-SO3Na)(AE)] | DMSO | −5374 | −5562 c | - | - | - | - |
| 39 | [Pt(PPh3)2(MeOPhHC=CHCOOPhOMe)] | Benzene | −5033 | −5044 d | 2412 | 3702 | 3766 | 3625 d |
| trans/cis COOR | - | - | 2797 | 3914 | 4131 | 4218 d | ||
| 40 | [Pt(PPh3)2(PhHC=CHCOOPhMe)] | Benzene | −4985 | −5053 d | 2502 | 3751 | 3851 | 3645 d |
| trans/cis COOR | - | - | 2741 | 3883 | 4077 | 4170 d | ||
| 41 | [Pt(PPh3)2(NO2PhHC=CHCOOPhNO2)] | Benzene | −5057 | −5047 d | 2408 | 3700 | 3762 | 3682 d |
| trans/cis COOR | - | - | 2731 | 3878 | 4068 | 4095 d | ||
| 42 | [Pt(PPh3)2(NO2PhHC=CHCOOPh)] | Benzene | −5026 | −5047 d | 2505 | 3753 | 3854 | 3737 d |
| trans/cis COOR | - | - | 2656 | 3836 | 3997 | 4039 d | ||
| 43 | [Pt(PPh3)2(MePhHC=CHCOOPh)] | Benzene | −5080 | −5052 d | 2453 | 3724 | 3804 | 3644 d |
| trans/cis COOR | - | - | 2736 | 3880 | 4073 | 4207 d | ||
| 44 | [Pt(PPh3)2(PhHC=CHCOOPh)] | Benzene | −4970 | −5053 d | 2499 | 3750 | 3848 | 3642 d |
| trans/cis COOR | - | - | 2757 | 3892 | 4093 | 4176 d | ||
| 45 | [Pt(PPh3)2(NO2PhHC=CHCOOPhOMe)] | Benzene | −5040 | −5049 d | 2495 | 3748 | 3845 | 3742 d |
| trans/cis COOR | - | - | 2639 | 3827 | 3981 | 4032 d | ||
| 46 | [Pt(PPh3)2(NO2PhHC=CHCOOPhMe)] | Benzene | −5041 | −5048 d | 2483 | 3741 | 3833 | 3742 d |
| trans/cis COOR | - | - | 2644 | 3830 | 3986 | 4033 d | ||
| 47 | [Pt(PPh3)2(NO2PhHC=CHCOOPri)] | Benzene | −5045 | −5051 d | 2513 | 3757 | 3861 | 3774 d |
| trans/cis COOR | - | - | 2633 | 3824 | 3975 | 3993 d | ||
| MAD/MRD (Pt(0) complexes studied) | - | 92/1.7% | - | 1311/34% | 146/3.6% | 98/2.6% | - | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
e Silva, J.H.C.; Dos Santos, H.F.; Paschoal, D.F.S. Predicting Pt-195 NMR Chemical Shift and 1J(195Pt-31P) Coupling Constant for Pt(0) Complexes Using the NMR-DKH Basis Sets. Magnetochemistry 2021, 7, 148. https://doi.org/10.3390/magnetochemistry7110148
e Silva JHC, Dos Santos HF, Paschoal DFS. Predicting Pt-195 NMR Chemical Shift and 1J(195Pt-31P) Coupling Constant for Pt(0) Complexes Using the NMR-DKH Basis Sets. Magnetochemistry. 2021; 7(11):148. https://doi.org/10.3390/magnetochemistry7110148
Chicago/Turabian Stylee Silva, Joyce H. C., Hélio F. Dos Santos, and Diego F. S. Paschoal. 2021. "Predicting Pt-195 NMR Chemical Shift and 1J(195Pt-31P) Coupling Constant for Pt(0) Complexes Using the NMR-DKH Basis Sets" Magnetochemistry 7, no. 11: 148. https://doi.org/10.3390/magnetochemistry7110148
APA Stylee Silva, J. H. C., Dos Santos, H. F., & Paschoal, D. F. S. (2021). Predicting Pt-195 NMR Chemical Shift and 1J(195Pt-31P) Coupling Constant for Pt(0) Complexes Using the NMR-DKH Basis Sets. Magnetochemistry, 7(11), 148. https://doi.org/10.3390/magnetochemistry7110148