
Genome-Wide Association Study Detects Loci Involved in Scab Susceptibility in Japanese Apricot

 , and
, and
Abstract
1. Introduction
2. Results
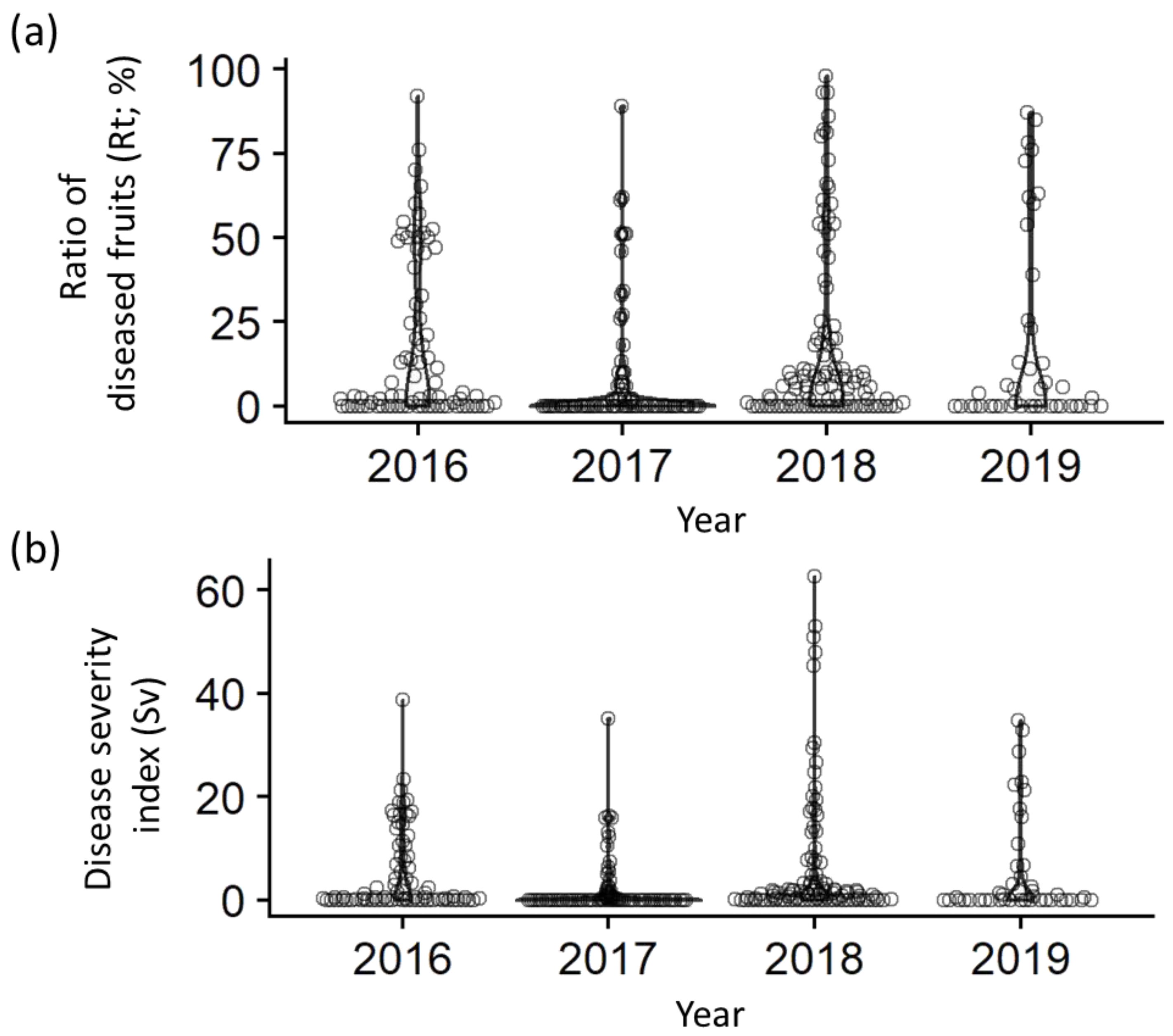
2.1. Phenotypic Variation of Scab Resistance
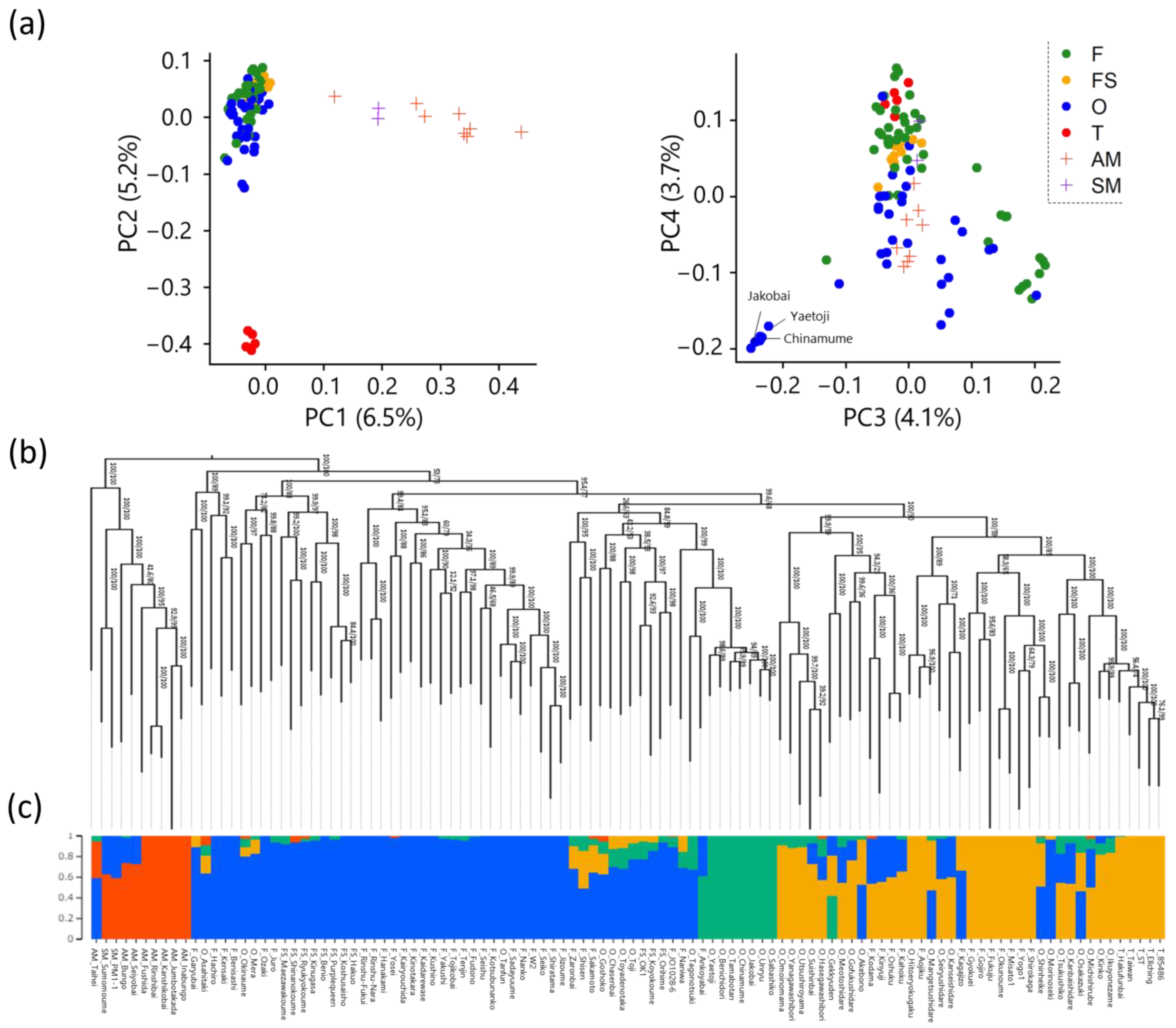
2.2. Population Structure of Japanese Apricot Accessions
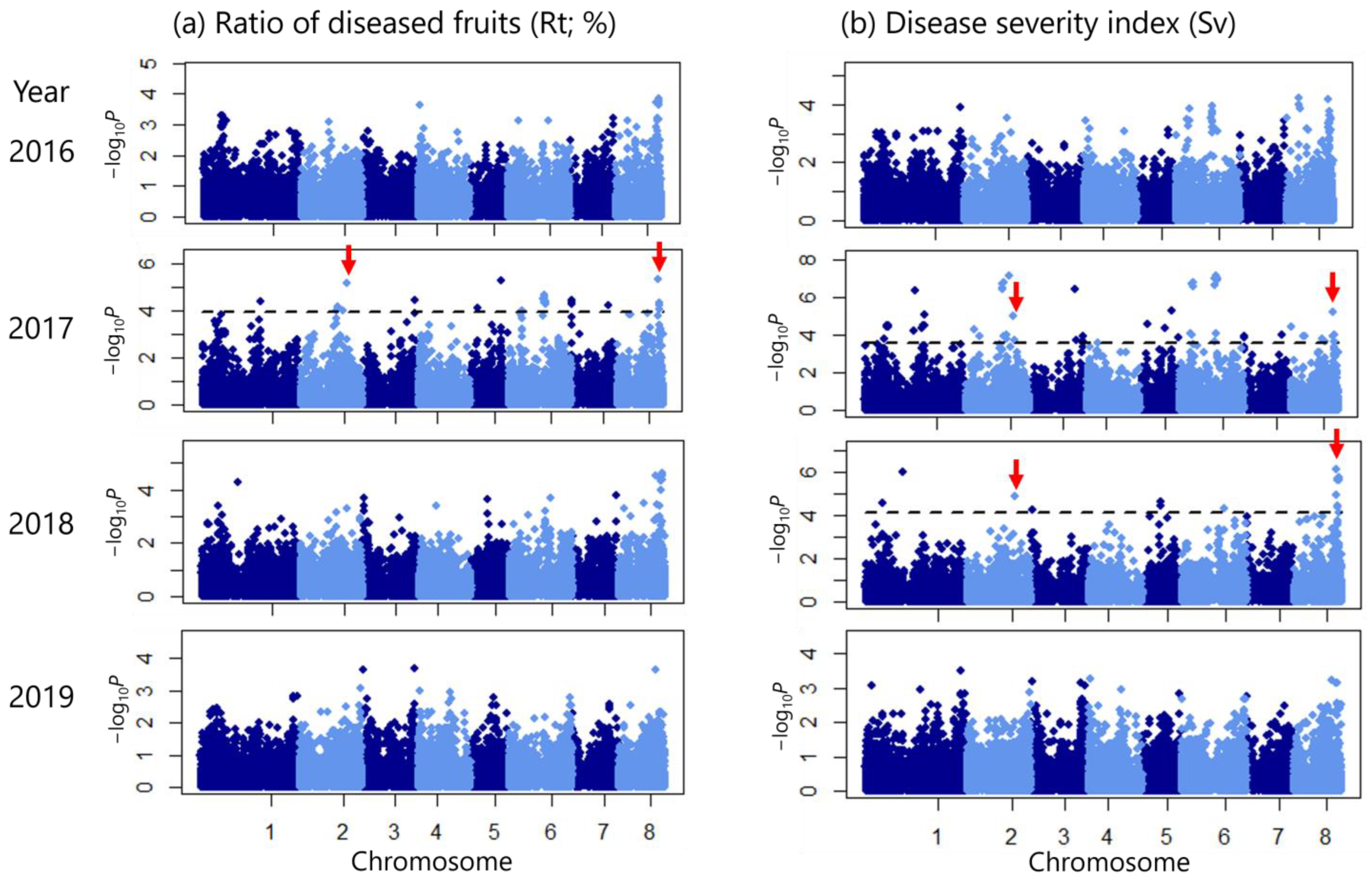
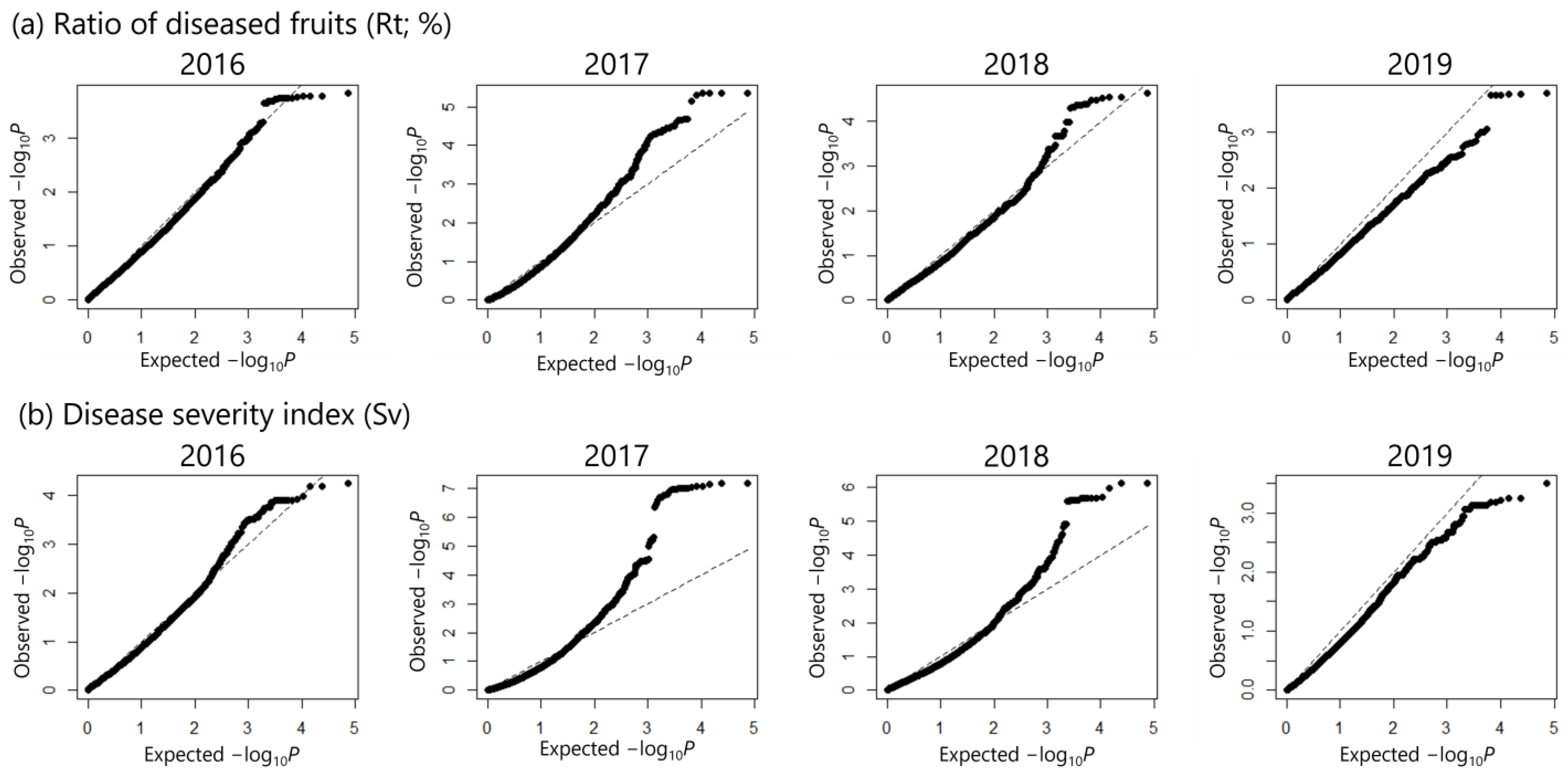
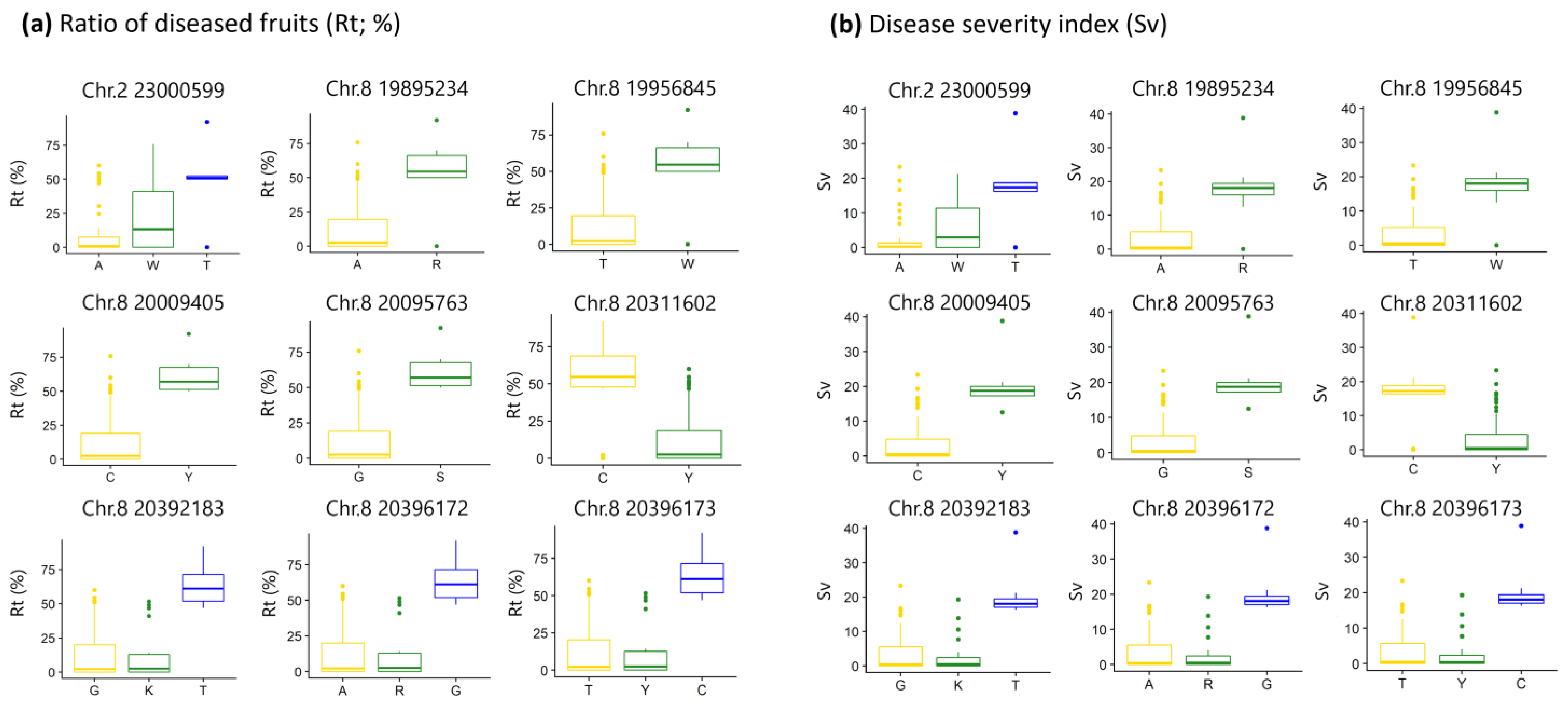
2.3. Multi-Year GWAS Identifies SNPs Associated with Disease Severity
2.4. Candidate Genes for Scab Severity
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Plant Materials
5.2. Evaluation of Scab Resistance
5.3. Genome-Wide Genotyping Using the Target Capture Method
5.4. Read Processing and SNP Calling
5.5. Population Structure Analysis
5.6. GWAS
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mega, K.; Tomita, E.; Kitamura, S.; Saito, S.; Mizukami, S. The Grand Dictionary of Horticulture; Aoba, T., Ed.; Shogakukan: Tokyo, Japan, 1988; ISBN 978-4-09-305111-8. [Google Scholar]
- Horiuchi, S.; Yoshida, M.; Kariya, H.; Nakamura, T.; Hasebe, H.; Suzaki, T.; Sakitani, T. Nihonnoume Sekainoume; Yokendo: Tokyo, Japan, 1996. [Google Scholar]
- Chen, J. China Mei Flower (Prunus Mume) Cultivars in Colour; China Forestry Publishing House: Beijing, China, 2017. [Google Scholar]
- Ministry of Agriculture, Forestry and Fisheries Japan the 96th Statistical Yearbook of Ministry of Agriculture, Forestry and Fisheries: MAFF. Available online: https://www.maff.go.jp/e/data/stat/96th/index.html (accessed on 5 May 2023).
- Ministry of Agriculture, Forestry and Fisheries Japan Survey on Production Dynamics of Specialty Fruit Trees (in Japanese). Available online: https://www.e-stat.go.jp/stat-search/file-download?statInfId=000040042773&fileKind=0 (accessed on 5 May 2023).
- Ikeda, K.; Nishide, M.; Tsujimoto, K.; Nagashima, S.; Kuwahara, T.; Mitani, T.; Koyama, A.H. Antiviral and Virucidal Activities of Umesu Phenolics on Influenza Viruses. Jpn. J. Infect. Dis. 2020, 73, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, S.; Yanaoka, K.; Utsunomiya, H.; Niwa, T.; Inada, K.; Deguchi, H.; Ueda, K.; Mukoubayashi, C.; Inoue, I.; Maekita, T.; et al. Inhibitory Effects of Japanese Apricot (Prunus Mume Siebold et Zucc.; Ume) on Helicobacter Pylori-Related Chronic Gastritis. Eur. J. Clin. Nutr. 2010, 64, 714–719. [Google Scholar] [CrossRef] [PubMed]
- Kono, R.; Nakamura, M.; Nomura, S.; Kitano, N.; Kagiya, T.; Okuno, Y.; Inada, K.; Tokuda, A.; Utsunomiya, H.; Ueno, M. Biological and Epidemiological Evidence of Anti-Allergic Effects of Traditional Japanese Food Ume (Prunus Mume). Sci. Rep. 2018, 8, 11638. [Google Scholar] [CrossRef] [PubMed]
- Bailly, C. Anticancer Properties of Prunus Mume Extracts (Chinese Plum, Japanese Apricot). J. Ethnopharmacol. 2020, 246, 112215. [Google Scholar] [CrossRef]
- Fisher, E.E. Venturia Carpophila Sp.Nov., the Ascigerous State of the Apricot Freckle Fungus. Trans. Br. Mycol. Soc. 1961, 44, 337-IN4. [Google Scholar] [CrossRef]
- Chen, C.; Bock, C.H.; Wood, B.W. Draft Genome Sequence of Venturia Carpophila, the Causal Agent of Peach Scab. Stand. Genom. Sci. 2017, 12, 68. [Google Scholar] [CrossRef]
- Takeda, T.; Hishiike, M.; Numaguchi, K. Occurrence of QoI-Resistant Strains of Cladosporium Carpophilum Causing Japanese Apricot Scab in Wakayama Prefecture. Ann. Rept. Kansai Pl. Prot. 2022, 64, 75–80. [Google Scholar] [CrossRef]
- Mori, M.; Yamana, T. Occurrence of DMI-Resistant Strains of Venturia Inaequalis Causing Apple Scab in Hokkaido. Ann. Rept. Soc. Pl. Prot. Nort. Jap. 2022, 76–80. [Google Scholar] [CrossRef]
- Shimada, T.; Haji, T.; Yamaguchi, M.; Takeda, T.; Nomura, K.; Yoshida, M. Classification of Mume (Prunus mume Sieb. et Zucc.) by RAPD Assay. J. Jpn. Soc. Hort. Sci. 1994, 63, 543–551. [Google Scholar] [CrossRef][Green Version]
- Numaguchi, K.; Ishio, S.; Kitamura, Y.; Nakamura, K.; Ishikawa, R.; Ishii, T. Microsatellite Marker Development and Population Structure Analysis in Japanese Apricot (Prunus mume Sieb. et Zucc.). Hort. J. 2019, 88, 222–231. [Google Scholar] [CrossRef]
- Hayashi, K.; Shimazu, K.; Yaegaki, H.; Yamaguchi, M.; Iketani, H.; Yamamoto, T. Genetic Diversity in Fruiting and Flower-Ornamental Japanese Apricot (Prunus mume) Germplasms Assessed by SSR Markers. Breed Sci. 2008, 58, 401–410. [Google Scholar] [CrossRef]
- Ohta, S.; Hayashi, K.; Yaegaki, H.; Mitsui, N.; Omura, M.; Nishitani, C.; Yamamoto, T. Genetic Relationship among Fruiting and Flower-Japanese Apricot Characterized by Chloroplast DNA Markers. DNA Polymorph. 2006, 14, 138–140. [Google Scholar]
- Numaguchi, K.; Akagi, T.; Kitamura, Y.; Ishikawa, R.; Ishii, T. Interspecific Introgression and Natural Selection in the Evolution of Japanese Apricot (Prunus mume). Plant J. 2020, 104, 1551–1567. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, H.; Sun, L.; Fan, G.; Ye, M.; Jiang, L.; Liu, X.; Ma, K.; Shi, C.; Bao, F.; et al. The Genetic Architecture of Floral Traits in the Woody Plant Prunus Mume. Nat. Commun. 2018, 9, 1702. [Google Scholar] [CrossRef]
- Tao, R.; Habu, T.; Yamane, H.; Sugiura, A.; Iwamoto, K. Molecular Markers for Self-Compatibility in Japanese Apricot (Prunus mume). HortScience 2000, 35, 1121–1123. [Google Scholar] [CrossRef]
- McClure, K.A.; Sawler, J.; Gardner, K.M.; Money, D.; Myles, S. Genomics: A Potential Panacea for the Perennial Problem. Am. J. Bot. 2014, 101, 1780–1790. [Google Scholar] [CrossRef]
- Groth, J.V.; Ozmon, E.A.; Busch, R.H. Repeatability and Relationship of Incidence and Severity Measures of Scab of Wheat Caused by Fusarium Graminearum in Inoculated Nurseries. Plant Dis. 1999, 83, 1033–1038. [Google Scholar] [CrossRef]
- Cardoso, J.E.; Santos, A.A.; Rossetti, A.G.; Vidal, J.C. Relationship between Incidence and Severity of Cashew Gummosis in Semiarid North-Eastern Brazil. Plant Pathol. 2004, 53, 363–367. [Google Scholar] [CrossRef]
- Verde, I.; Jenkins, J.; Dondini, L.; Micali, S.; Pagliarani, G.; Vendramin, E.; Paris, R.; Aramini, V.; Gazza, L.; Rossini, L.; et al. The Peach v2.0 Release: High-Resolution Linkage Mapping and Deep Resequencing Improve Chromosome-Scale Assembly and Contiguity. BMC Genom. 2017, 18, 225. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast Model-Based Estimation of Ancestry in Unrelated Individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Browning, B.L.; Zhou, Y.; Browning, S.R. A One-Penny Imputed Genome from Next-Generation Reference Panels. Am. J. Hum. Genet. 2018, 103, 338–348. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for Association Mapping of Complex Traits in Diverse Samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and Visualization of LD and Haplotype Maps. Bioinformatics 2005, 21, 263–265. [Google Scholar] [CrossRef]
- McDonald, B.A.; Stukenbrock, E.H. Rapid Emergence of Pathogens in Agro-Ecosystems: Global Threats to Agricultural Sustainability and Food Security. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2016, 371, 20160026. [Google Scholar] [CrossRef]
- Papp, D.; Gao, L.; Thapa, R.; Olmstead, D.; Khan, A. Field Apple Scab Susceptibility of a Diverse Malus Germplasm Collection Identifies Potential Sources of Resistance for Apple Breeding. CABI Agric. Biosci. 2020, 1, 16. [Google Scholar] [CrossRef]
- Kitamura, Y.; Takeda, T.; Numaguchi, K.; Tsuchida, Y.; Negoro, K.; Hayashi, K.; Iwamoto, K.; Hishiike, M.; Naka, K.; Shimazu, K. Breeding of scab-resistant Japanese apricot (Prunus mume Sieb. et Zucc.) “Seiko” and evaluation of possibility for fungicide-saving cultivation. Bull. Wakayama Prefect. Exp. Station. Agric. Forest. Fish. 2018, 6, 27–35. [Google Scholar]
- Numaguchi, K.; Kitamura, Y.; Takeda, T.; Shimomura, Y.; Tsunaki, K.; Kashiwamoto, T.; Shimazu, K.; Hishiike, M.; Iwamoto, K.; Negoro, K.; et al. Breeding of Japanese apricot (Prunus mume Sieb. et Zucc.) ‘Seishu’. Bull. Wakayama Prefect. Exp. Station. Agric. Forest. Fish. 2021, 9, 73–85. [Google Scholar]
- Korte, A.; Farlow, A. The Advantages and Limitations of Trait Analysis with GWAS: A Review. Plant Methods 2013, 9, 29. [Google Scholar] [CrossRef]
- Jones, D.A.; Jones, J.D.G. The Role of Leucine-Rich Repeat Proteins in Plant Defences. In Advances in Botanical Research; Andrews, J.H., Tommerup, I.C., Callow, J.A., Eds.; Academic Press: Cambridge, MA, USA, 1997; Volume 24, pp. 89–167. [Google Scholar]
- Weis, C.; Pfeilmeier, S.; Glawischnig, E.; Isono, E.; Pachl, F.; Hahne, H.; Kuster, B.; Eichmann, R.; Hückelhoven, R. Co-Immunoprecipitation-Based Identification of Putative BAX INHIBITOR-1-Interacting Proteins Involved in Cell Death Regulation and Plant–Powdery Mildew Interactions. Mol. Plant Pathol. 2013, 14, 791–802. [Google Scholar] [CrossRef]
- Van Damme, M.; Huibers, R.P.; Elberse, J.; Van den Ackerveken, G. Arabidopsis DMR6 Encodes a Putative 2OG-Fe(II) Oxygenase That Is Defense-Associated but Required for Susceptibility to Downy Mildew. Plant J. 2008, 54, 785–793. [Google Scholar] [CrossRef]
- Thatcher, L.F.; Powell, J.J.; Aitken, E.A.B.; Kazan, K.; Manners, J.M. The Lateral Organ Boundaries Domain Transcription Factor LBD20 Functions in Fusarium Wilt Susceptibility and Jasmonate Signaling in Arabidopsis. Plant Physiol. 2012, 160, 407–418. [Google Scholar] [CrossRef]
- Hu, Y.; Zhang, J.; Jia, H.; Sosso, D.; Li, T.; Frommer, W.B.; Yang, B.; White, F.F.; Wang, N.; Jones, J.B. Lateral Organ Boundaries 1 Is a Disease Susceptibility Gene for Citrus Bacterial Canker Disease. Proc. Natl. Acad. Sci. USA 2014, 111, E521–E529. [Google Scholar] [CrossRef] [PubMed]
- Rissel, D.; Peiter, E. Poly(ADP-Ribose) Polymerases in Plants and Their Human Counterparts: Parallels and Peculiarities. Int. J. Mol. Sci. 2019, 20, 1638. [Google Scholar] [CrossRef]
- Minamikawa, M.F.; Takada, N.; Terakami, S.; Saito, T.; Onogi, A.; Kajiya-Kanegae, H.; Hayashi, T.; Yamamoto, T.; Iwata, H. Genome-Wide Association Study and Genomic Prediction Using Parental and Breeding Populations of Japanese Pear (Pyrus Pyrifolia Nakai). Sci. Rep. 2018, 8, 11994. [Google Scholar] [CrossRef]
- Minamikawa, M.F.; Nonaka, K.; Kaminuma, E.; Kajiya-Kanegae, H.; Onogi, A.; Goto, S.; Yoshioka, T.; Imai, A.; Hamada, H.; Hayashi, T.; et al. Genome-Wide Association Study and Genomic Prediction in Citrus: Potential of Genomics-Assisted Breeding for Fruit Quality Traits. Sci. Rep. 2017, 7, 4721. [Google Scholar] [CrossRef] [PubMed]
- Roth, M.; Muranty, H.; Di Guardo, M.; Guerra, W.; Patocchi, A.; Costa, F. Genomic Prediction of Fruit Texture and Training Population Optimization towards the Application of Genomic Selection in Apple. Hortic. Res. 2020, 7, 148. [Google Scholar] [CrossRef]
- Li, Y.-L.; Weng, J.-C.; Hsiao, C.-C.; Chou, M.-T.; Tseng, C.-W.; Hung, J.-H. PEAT: An Intelligent and Efficient Paired-End Sequencing Adapter Trimming Algorithm. BMC Bioinform. 2015, 16, S2. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Auwera, G.A.; O’Connor, B.D. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra; O’Reilly Media: Sebastopol, CA, USA, 2020. [Google Scholar]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The Variant Call Format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Minh, B.Q.; Nguyen, M.A.T.; von Haeseler, A. Ultrafast Approximation for Phylogenetic Bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | 2016 | 2017 | 2018 | 2019 |
|---|---|---|---|---|
| 2016 | 0.62 a | 0.56 | 0.54 | |
| 2017 | 5.28 × 10−9 b | 0.52 | 0.59 | |
| 2018 | 2.83 × 10−7 | 1.03 × 10−6 | 0.76 | |
| 2019 | 6.28 × 10−4 | 1.18 × 10−4 | 2.24 × 10−9 |
| Year | 2016 | 2017 | 2018 | 2019 |
|---|---|---|---|---|
| 2016 | 0.61 a | 0.55 | 0.55 | |
| 2017 | 1.01 × 10−8 b | 0.56 | 0.63 | |
| 2018 | 4.02 × 10−7 | 5.98 × 10−8 | 0.79 | |
| 2019 | 4.95 × 10−4 | 3.07 × 10−4 | 1.92 × 10−10 |
| Chr. | Position | LD Block a | Candidate Gene | Gene Description b |
|---|---|---|---|---|
| 2 | 23000599 | NA | Prupe.2G190600 | NA |
| 8 | 19895234 | 19895155– 19903433 | Prupe.8G217900 | Protein kinase domain (Pkinase)//Leucine-rich repeat (LRR_1) //Leucine-rich repeat N-terminal domain (LRRNT_2) |
| Prupe.8G218000 | NA | |||
| 8 | 19956845 | NA | Prupe.8G219000 | Tetratricopeptide repeat protein, TPR |
| 8 | 20009405 | 20009216– 20009426 | Prupe.8G220100 | Leucine-rich repeat (LRR_1) |
| 8 | 20095763 | NA | Prupe.8G221600 | Ribophorin II |
| 8 | 20311602 | 20306531– 20311602 | Prupe.8G226500 | Oxidoreductase, 2OG-Fe II oxygenase family protein |
| Prupe.8G226600 | Oxidoreductase, 2OG-Fe II oxygenase family protein | |||
| Prupe.8G226700 | Oxidoreductase, 2OG-Fe II oxygenase family protein | |||
| 8 | 20392183 20396172 20396173 | 20377283– 20396173 | Prupe.8G227300 | Lob domain-containing protein 17-related |
| Prupe.8G227400 | Lob domain-containing protein 17-related | |||
| Prupe.8G227500 | Lob domain-containing protein 16 | |||
| Prupe.8G227600 | Poly [ADP-ribose] polymerase 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Numaguchi, K.; Kashiwamoto, T.; Ishikawa, R.; Ishii, T.; Kitamura, Y. Genome-Wide Association Study Detects Loci Involved in Scab Susceptibility in Japanese Apricot. Horticulturae 2023, 9, 872. https://doi.org/10.3390/horticulturae9080872
Numaguchi K, Kashiwamoto T, Ishikawa R, Ishii T, Kitamura Y. Genome-Wide Association Study Detects Loci Involved in Scab Susceptibility in Japanese Apricot. Horticulturae. 2023; 9(8):872. https://doi.org/10.3390/horticulturae9080872
Chicago/Turabian StyleNumaguchi, Koji, Tomoaki Kashiwamoto, Ryo Ishikawa, Takashige Ishii, and Yuto Kitamura. 2023. "Genome-Wide Association Study Detects Loci Involved in Scab Susceptibility in Japanese Apricot" Horticulturae 9, no. 8: 872. https://doi.org/10.3390/horticulturae9080872
APA StyleNumaguchi, K., Kashiwamoto, T., Ishikawa, R., Ishii, T., & Kitamura, Y. (2023). Genome-Wide Association Study Detects Loci Involved in Scab Susceptibility in Japanese Apricot. Horticulturae, 9(8), 872. https://doi.org/10.3390/horticulturae9080872