

Identification and Analysis of Reference and Tissue-Specific Genes in Bitter Gourd Based on Transcriptome Data
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Tissue Collection
2.2. RNA-Seq and Data Analysis
2.3. Selection of Reference Genes and Primer Design
2.4. RNA Isolation and Reverse Transcription
2.5. qRT-PCR Analysis
2.6. Stability Analysis
2.7. Identification of Tissue-Specific Genes
2.8. Statistical Analysis
3. Results
3.1. Identification of Candidate Reference Genes in Bitter Gourd Based on an RNA-Seq Dataset
3.2. Verification of Primer Specificity and PCR Amplification Efficiency
3.3. Expression Profiles and Cycle Quantification Values of the Reference Genes
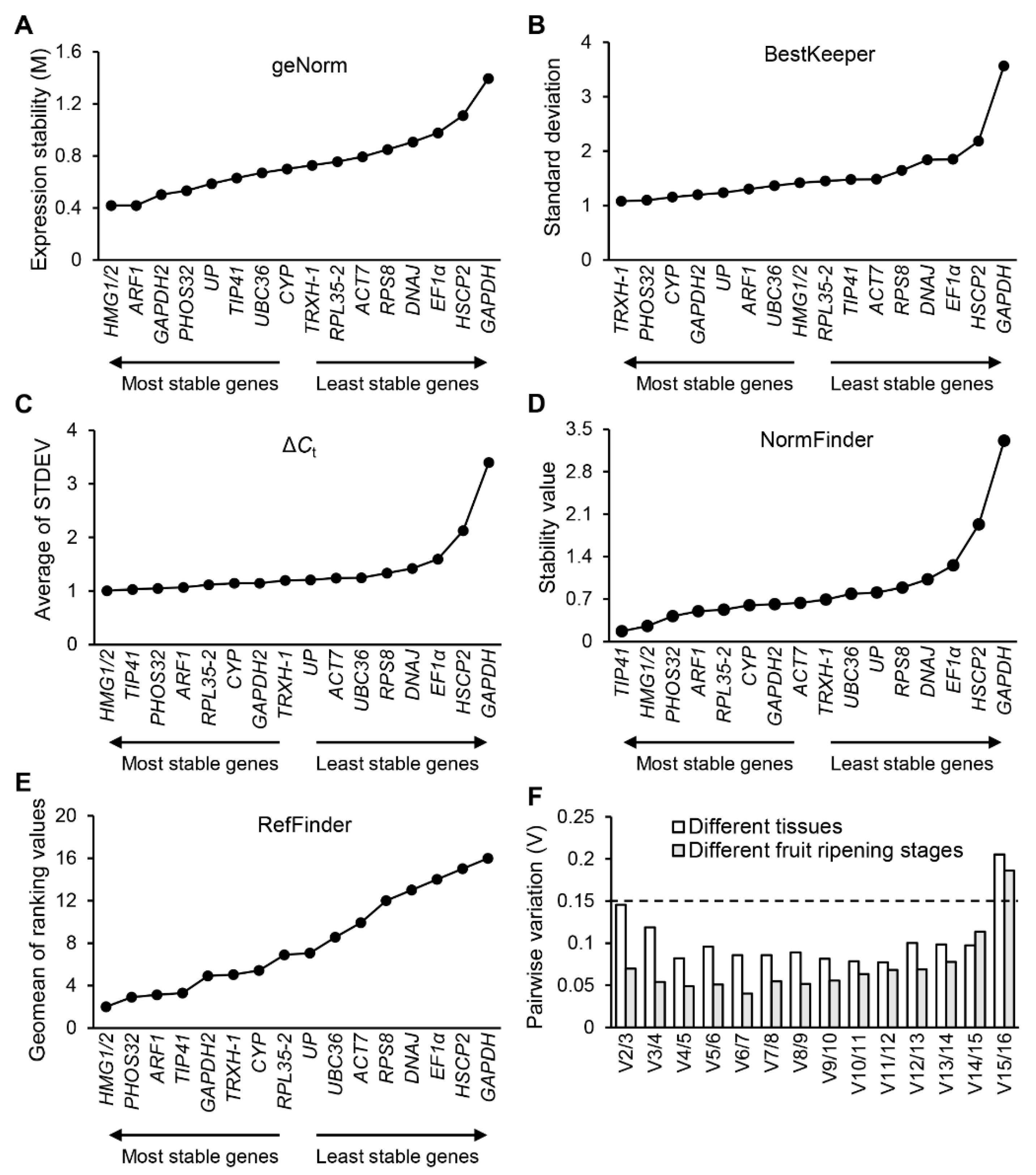
3.4. Expression Stability Analysis of TRGs and NRGs
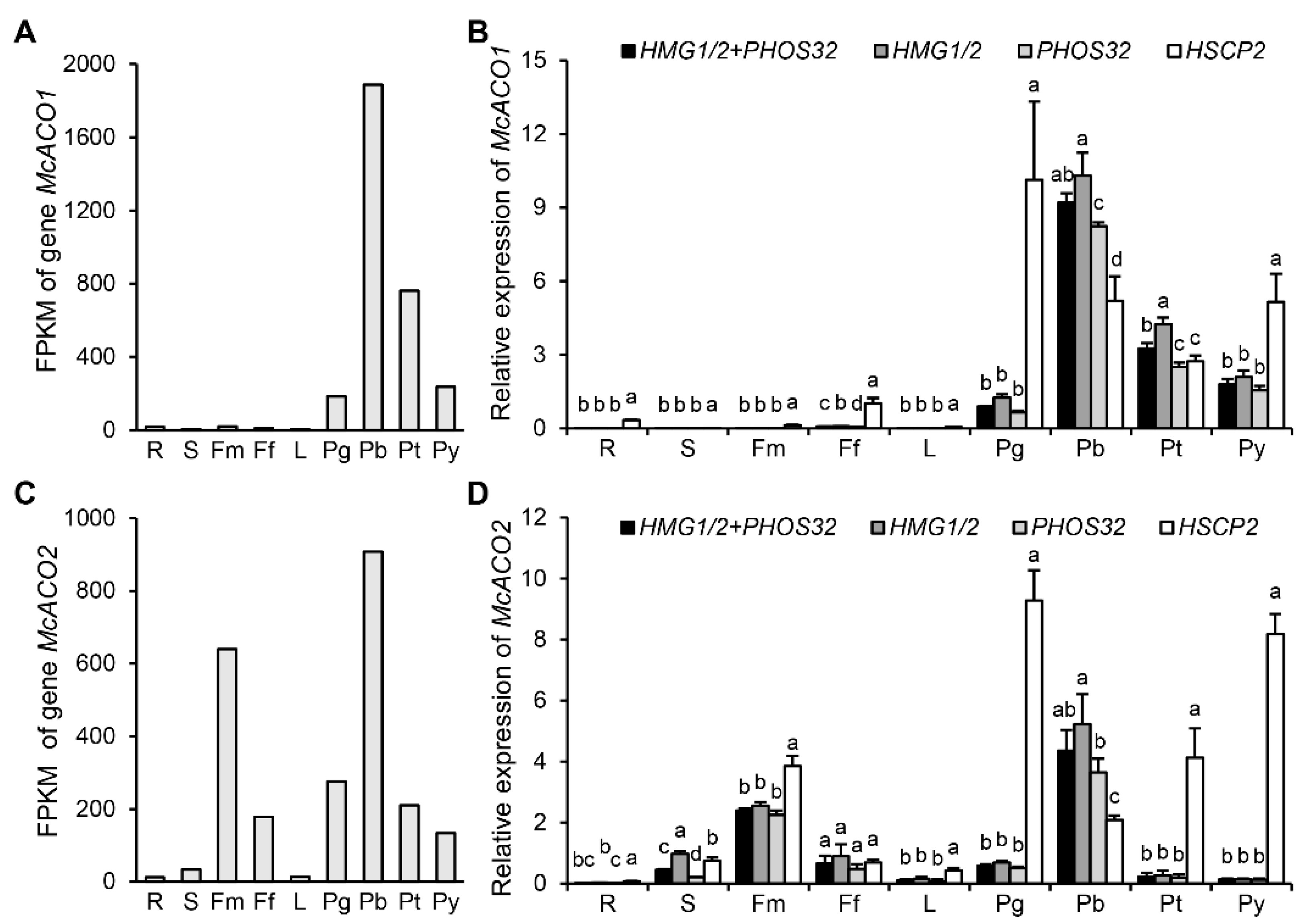
3.5. Validation of HMG1/2 and PHOS32 as Optimal NRGs
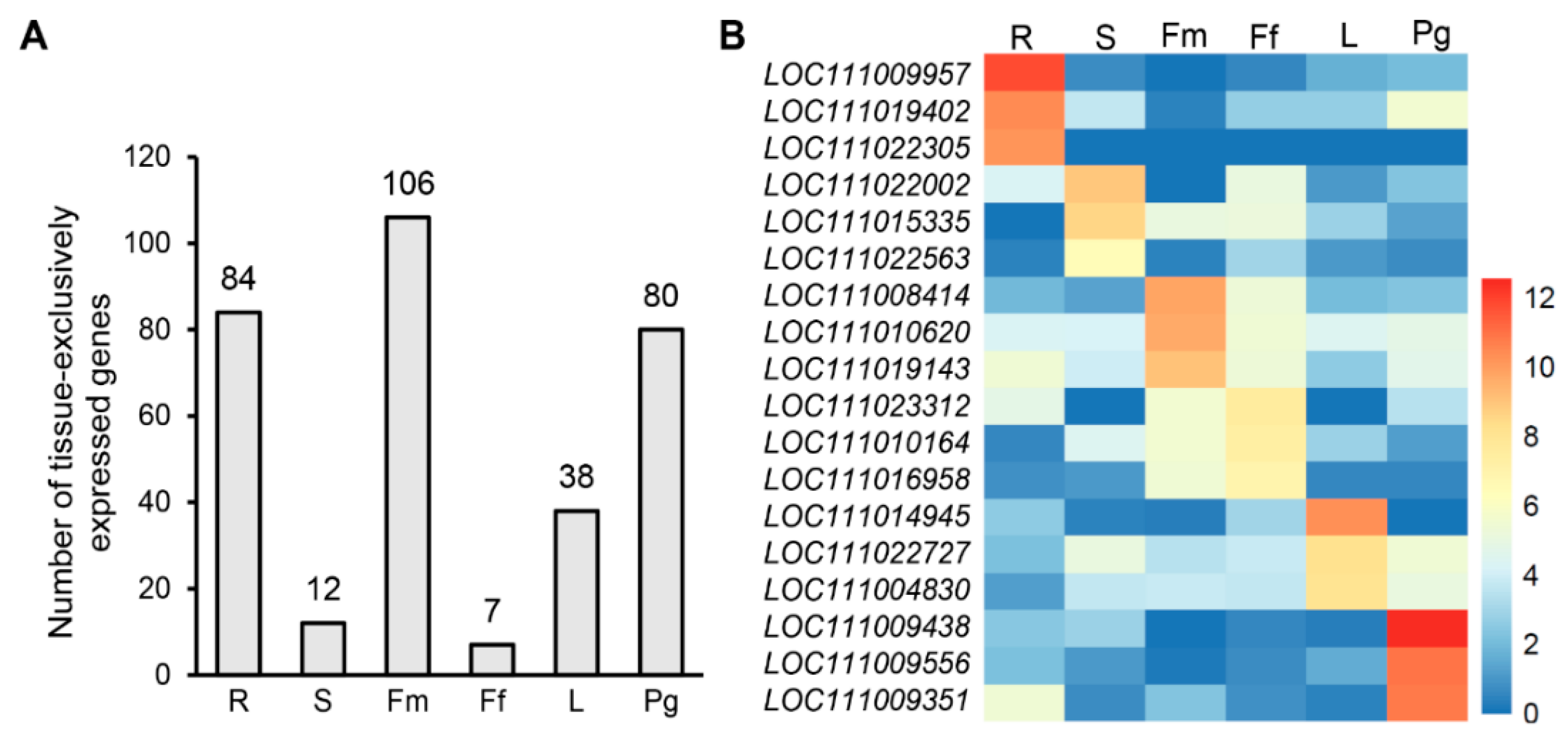
3.6. Identification of Candidate Tissue-Specific Genes Based on an RNA-Seq Dataset
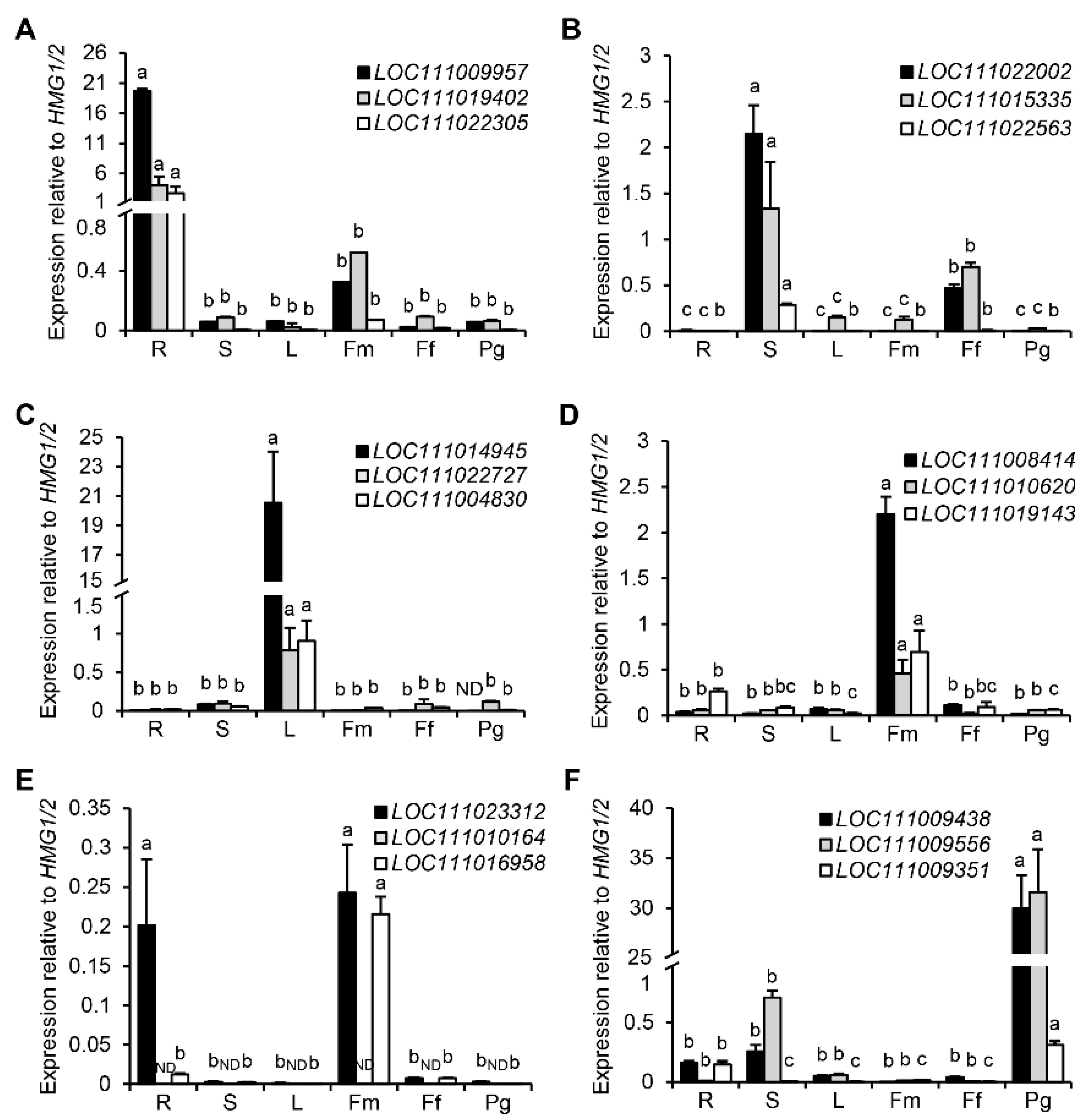
3.7. Validation of 18 Tissue-Specific Genes Using HMG1/2
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Andersen, C.L.; Jensen, J.L.; Orntoft, T.F. Normalization of real-time quantitative reverse transcription-PCR data: A model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004, 64, 5245–5250. [Google Scholar] [CrossRef] [PubMed]
- Sgamma, T.; Pape, J.; Massiah, A.; Jackson, S. Selection of reference genes for diurnal and developmental time-course real-time PCR expression analyses in lettuce. Plant Methods 2016, 12, 21. [Google Scholar] [CrossRef] [PubMed]
- Yi, N.; Kim, Y.S.; Jeong, M.H.; Oh, S.J.; Jeong, J.S.; Park, S.H.; Jung, H.; Choi, Y.D.; Kim, J.K. Functional analysis of six drought-inducible promoters in transgenic rice plants throughout all stages of plant growth. Planta 2010, 232, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Lu, L.; Zhang, L.; Wang, A.; Wang, N.; Liang, Z.; Lu, X.; Tang, K. Molecular evolution of the E8 promoter in tomato and some of its relative wild species. J. Biosci. 2009, 34, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.K.; Drossard, J.; Lewis, D.; Altmann, F.; Boyle, J.; Christou, P.; Cole, T.; Dale, P.; van Dolleweerd, C.J.; Isitt, V.; et al. Regulatory approval and a first-in-human phase I clinical trial of a monoclonal antibody produced in transgenic tobacco plants. Plant Biotechnol. J. 2015, 13, 1106–1120. [Google Scholar] [CrossRef] [PubMed]
- Hu, A.; Yang, X.; Zhu, J.; Wang, X.; Liu, J.; Wang, J.; Wu, H.; Zhang, H.; Zhang, H. Selection and validation of appropriate reference genes for RT-qPCR analysis of Nitraria sibirica under various abiotic stresses. BMC Plant Biol. 2022, 22, 592. [Google Scholar] [CrossRef]
- Wang, Z.; Xu, J.; Liu, Y.; Chen, J.; Lin, H.; Huang, Y.; Bian, X.; Zhao, Y. Selection and validation of appropriate reference genes for real-time quantitative PCR analysis in Momordica charantia. Phytochemistry 2019, 164, 1–11. [Google Scholar] [CrossRef]
- Lin, F.; Bao, H.; Yang, J.; Liu, Y.; Dai, H.; Zhao, H. Genome-Wide Discovery of Tissue-Specific Genes in Maize. Plant Mol. Biol. Rep. 2016, 34, 1204–1214. [Google Scholar] [CrossRef]
- Chen, L.; Li, Y.; Wang, Y.; Li, W.; Feng, X.; Zhao, L. Use of High Resolution Spatiotemporal Gene Expression Data to Uncover Novel Tissue-Specific Promoters in Tomato. Agriculture 2021, 11, 1195. [Google Scholar] [CrossRef]
- Xun, H.; Zhang, X.; Yu, J.; Pang, J.; Wang, S.; Liu, B.; Dong, Y.; Jiang, L.; Guo, D. Analysis of expression characteristics of soybean leaf and root tissue-specific promoters in Arabidopsis and soybean. Transgenic Res. 2021, 30, 799–810. [Google Scholar] [CrossRef] [PubMed]
- Kryuchkova-Mostacci, N.; Robinson-Rechavi, M. A benchmark of gene expression tissue-specificity metrics. Brief. Bioinform. 2017, 18, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Yanai, I.; Benjamin, H.; Shmoish, M.; Chalifa-Caspi, V.; Shklar, M.; Ophir, R.; Bar-Even, A.; Horn-Saban, S.; Safran, M.; Domany, E.; et al. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics 2005, 21, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Jia, S.; Shen, M.; Zhang, F.; Xie, J. Recent Advances in Momordica charantia: Functional Components and Biological Activities. Int. J. Mol. Sci. 2017, 18, 2555. [Google Scholar] [CrossRef]
- Guo, J.; Xu, W.; Hu, Y.; Huang, J.; Zhao, Y.; Zhang, L.; Huang, C.H.; Ma, H. Phylotranscriptomics in Cucurbitaceae Reveal Multiple Whole-Genome Duplications and Key Morphological and Molecular Innovations. Mol. Plant 2020, 13, 1117–1133. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Ji, M.; Liu, W.; Chen, L.; Cai, Z.; Zhao, Y.; Bi, X. Antidiabetic activities of a cucurbitane–type triterpenoid compound from Momordica charantia in alloxan–induced diabetic mice. Mol. Med. Rep. 2016, 14, 4865–4872. [Google Scholar] [CrossRef]
- Sun, Y.; Sun, F.; Li, J.; Wu, M.; Fan, X.; Meng, Y.; Meng, Y. Mono-PEGylation of Alpha-MMC and MAP30 from Momordica charantia L.: Production, Identification and Anti-Tumor Activity. Molecules 2016, 21, 1457. [Google Scholar] [CrossRef]
- Zhang, C.; Chen, H.; Bai, W. Characterization of Momordica charantia L. polysaccharide and its protective effect on pancreatic cells injury in STZ-induced diabetic mice. Int. J. Biol. Macromol. 2018, 115, 45–52. [Google Scholar] [CrossRef]
- Deshaware, S.; Gupta, S.; Singhal, R.S.; Joshi, M.; Variyar, P.S. Debittering of bitter gourd juice using beta-cyclodextrin: Mechanism and effect on antidiabetic potential. Food Chem. 2018, 262, 78–85. [Google Scholar] [CrossRef]
- Yan, J.K.; Wu, L.X.; Qiao, Z.R.; Cai, W.D.; Ma, H. Effect of different drying methods on the product quality and bioactive polysaccharides of bitter gourd (Momordica charantia L.) slices. Food Chem. 2019, 271, 588–596. [Google Scholar] [CrossRef]
- Prajapati, U.; Asrey, R.; Varghese, E.; Singh, A.K.; Pal Singh, M. Effects of postharvest ultraviolet-C treatment on shelf-life and quality of bitter gourd fruit during storage. Food Packag. Shelf Life 2021, 28, 100665. [Google Scholar] [CrossRef]
- Takase, S.; Kera, K.; Hirao, Y.; Hosouchi, T.; Kotake, Y.; Nagashima, Y.; Mannen, K.; Suzuki, H.; Kushiro, T. Identification of triterpene biosynthetic genes from Momordica charantia using RNA-seq analysis. Biosci. Biotechnol. Biochem. 2019, 83, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Cheng, J.; Cui, J.; Hu, F.; Dong, J.; Liu, J.; Zou, Y.; Hu, K. MC03g0810, an Important Candidate Gene Controlling Black Seed Coat Color in Bitter Gourd (Momordica spp.). Front. Plant Sci. 2022, 13, 875631. [Google Scholar] [CrossRef]
- Zhong, J.; Cui, J.; Liu, J.; Zhong, C.; Hu, F.; Dong, J.; Cheng, J.; Hu, K. Fine-mapping and candidate gene analysis of the Mcgy1 locus responsible for gynoecy in bitter gourd (Momordica spp.). Theor. Appl. Genet 2023, 136, 81. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, J.; Liu, Z.; Xia, P.; Yuan, X.; Ning, Y. Genome-wide identification and expression analysis of the MLO gene family reveal a candidate gene associated with powdery mildew susceptibility in bitter gourd (Momordica charantia). Eur. J. Plant Pathol. 2020, 159, 163–178. [Google Scholar] [CrossRef]
- Tuan, P.A.; Kim, J.K.; Park, N.I.; Lee, S.Y.; Park, S.U. Carotenoid content and expression of phytoene synthase and phytoene desaturase genes in bitter melon (Momordica charantia). Food Chem. 2011, 126, 1686–1692. [Google Scholar] [CrossRef] [PubMed]
- Rajaee Behbahani, S.; Iranbakhsh, A.; Ebadi, M.; Majd, A.; Ardebili, Z.O. Red elemental selenium nanoparticles mediated substantial variations in growth, tissue differentiation, metabolism, gene transcription, epigenetic cytosine DNA methylation, and callogenesis in bittermelon (Momordica charantia); an in vitro experiment. PLoS ONE 2020, 15, e0235556. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Tu, P.; Wang, K.; Gao, F.; Yang, W.; Zhu, Y.; Li, S. Defining reference genes for quantitative real-time PCR analysis of anther development in rice. Acta Biochim. Biophys. Sin. 2014, 46, 305–312. [Google Scholar] [CrossRef]
- Tang, X.; Zhang, N.; Si, H.; Calderon-Urrea, A. Selection and validation of reference genes for RT-qPCR analysis in potato under abiotic stress. Plant Methods 2017, 13, 85. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, Z.; Ding, Z.; Meng, H.; Shen, R.; Tang, H.; Liu, Y.G.; Chen, L. Public-transcriptome-database-assisted selection and validation of reliable reference genes for qRT-PCR in rice. Sci. China Life Sci. 2020, 63, 92–101. [Google Scholar] [CrossRef]
- Zhao, G.; Wang, M.; Gan, Y.; Gong, H.; Li, J.; Zheng, X.; Liu, X.; Zhao, S.; Luo, J.; Wu, H. Identification of suitable reference genes for quantitative reverse transcription PCR in Luffa (Luffa cylindrica). Physiol. Mol. Biol. Plants 2022, 28, 737–747. [Google Scholar] [CrossRef]
- Chen, F.Z.; You, L.J.; Yang, F.; Wang, L.N.; Guo, X.Q.; Gao, F.; Hua, C.; Tan, C.; Fang, L.; Shan, R.Q.; et al. CNGBdb: China National GeneBank DataBase. Yi Chuan 2020, 42, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Chen, F.; Gao, F.; Li, L.; Liu, K.; You, L.; Hua, C.; Yang, F.; Liu, W.; Peng, C.; et al. CNSA: A data repository for archiving omics data. Database 2020, 2020, baaa055. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, research0034.1. [Google Scholar] [CrossRef]
- Pfaffl, M.W.; Tichopad, A.; Prgomet, C.; Neuvians, T.P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper-Excel-based tool using pair-wise correlations. Biotechnol. Lett. 2004, 26, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Silver, N.; Best, S.; Jiang, J.; Thein, S.L. Selection of housekeeping genes for gene expression studies in human reticulocytes using real-time PCR. BMC Mol. Biol. 2006, 7, 33. [Google Scholar] [CrossRef]
- Xie, F.; Xiao, P.; Chen, D.; Xu, L.; Zhang, B. miRDeepFinder: A miRNA analysis tool for deep sequencing of plant small RNAs. Plant Mol. Biol. 2012, 80, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Wang, J.; Zhang, B. RefFinder: A web-based tool for comprehensively analyzing and identifying reference genes. Funct Integr Genomics 2023, 23, 125. [Google Scholar] [CrossRef]
- Wan, H.; Zhao, Z.; Qian, C.; Sui, Y.; Malik, A.A.; Chen, J. Selection of appropriate reference genes for gene expression studies by quantitative real-time polymerase chain reaction in cucumber. Anal. Biochem. 2010, 399, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Die, J.V.; Roman, B.; Nadal, S.; Gonzalez-Verdejo, C.I. Evaluation of candidate reference genes for expression studies in Pisum sativum under different experimental conditions. Planta 2010, 232, 145–153. [Google Scholar] [CrossRef]
- Wang, H.L.; Li, L.; Tang, S.; Yuan, C.; Tian, Q.; Su, Y.; Li, H.G.; Zhao, L.; Yin, W.; Zhao, R.; et al. Evaluation of Appropriate Reference Genes for Reverse Transcription-Quantitative PCR Studies in Different Tissues of a Desert Poplar via Comparision of Different Algorithms. Int. J. Mol. Sci. 2015, 16, 20468–20491. [Google Scholar] [CrossRef]
- Thomas, J.O.; Travers, A.A. HMG1 and 2, and related ‘architectural’ DNA-binding proteins. Trends Biochem. Sci. 2001, 26, 167–174. [Google Scholar] [CrossRef]
- Luo, D.; Wu, Z.; Bai, Q.; Zhang, Y.; Huang, M.; Huang, Y.; Li, X. Universal Stress Proteins: From Gene to Function. Int. J. Mol. Sci. 2023, 24, 4725. [Google Scholar] [CrossRef]
- Chi, Y.H.; Koo, S.S.; Oh, H.T.; Lee, E.S.; Park, J.H.; Phan, K.A.T.; Wi, S.D.; Bae, S.B.; Paeng, S.K.; Chae, H.B.; et al. The Physiological Functions of Universal Stress Proteins and Their Molecular Mechanism to Protect Plants From Environmental Stresses. Front. Plant Sci. 2019, 10, 750. [Google Scholar] [CrossRef] [PubMed]
- Feng, K.; Liu, J.X.; Xing, G.M.; Sun, S.; Li, S.; Duan, A.Q.; Wang, F.; Li, M.Y.; Xu, Z.S.; Xiong, A.S. Selection of appropriate reference genes for RT-qPCR analysis under abiotic stress and hormone treatment in celery. PeerJ 2019, 7, e7925. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhang, R.; Zhou, Z. Identification and Validation of Reference Genes for Gene Expression Analysis in Schima superba. Genes 2021, 12, 732. [Google Scholar] [CrossRef]
- Atkinson, R.G.; Gunaseelan, K.; Wang, M.Y.; Luo, L.; Wang, T.; Norling, C.L.; Johnston, S.L.; Maddumage, R.; Schroder, R.; Schaffer, R.J. Dissecting the role of climacteric ethylene in kiwifruit (Actinidia chinensis) ripening using a 1-aminocyclopropane-1-carboxylic acid oxidase knockdown line. J. Exp. Bot. 2011, 62, 3821–3835. [Google Scholar] [CrossRef]
- Houben, M.; Van de Poel, B. 1-Aminocyclopropane-1-Carboxylic Acid Oxidase (ACO): The Enzyme That Makes the Plant Hormone Ethylene. Front. Plant Sci. 2019, 10, 695. [Google Scholar] [CrossRef]
- Odell, J.T.; Nagy, F.; Chua, N.H. Identification of DNA sequences required for activity of the cauliflower mosaic virus 35S promoter. Nature 1985, 313, 810–812. [Google Scholar] [CrossRef]
- Christensen, A.H.; Quail, P.H. Ubiquitin promoter-based vectors for high-level expression of selectable and/or screenable marker genes in monocotyledonous plants. Transgenic Res. 1996, 5, 213–218. [Google Scholar] [CrossRef]
- Chen, H.; Tang, W.; Xu, C.; Li, X.; Lin, Y.; Zhang, Q. Transgenic indica rice plants harboring a synthetic cry2A* gene of Bacillus thuringiensis exhibit enhanced resistance against lepidopteran rice pests. Theor. Appl. Genet 2005, 111, 1330–1337. [Google Scholar] [CrossRef]
- Werner, T.; Nehnevajova, E.; Kollmer, I.; Novak, O.; Strnad, M.; Kramer, U.; Schmulling, T. Root-specific reduction of cytokinin causes enhanced root growth, drought tolerance, and leaf mineral enrichment in Arabidopsis and tobacco. Plant Cell. 2010, 22, 3905–3920. [Google Scholar] [CrossRef] [PubMed]
- Hirai, T.; Kim, Y.W.; Kato, K.; Hiwasa-Tanase, K.; Ezura, H. Uniform accumulation of recombinant miraculin protein in transgenic tomato fruit using a fruit-ripening-specific E8 promoter. Transgenic Res. 2011, 20, 1285–1292. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| cGene Symbol (Gene ID) | Gene Description | Primer Sequence (5′–3′) Forward/Reverse | Amplicon Length (bp) | Amplification Efficiency (%) | R2 |
|---|---|---|---|---|---|
| CYP(LOC111007901) | Peptidyl-prolyl cis-trans isomerase | CCAAATTGTTGACGGCATGG/ GTAGAGCCAAGGCATCAATC | 128 | 99.3 | 0.999 |
| EF1α(LOC111011110) | Elongation factor 1-alpha | CTGTCGCAGTTGGTGTTATC/ CTTGTAAACCTCAGACGGAG | 131 | 103.0 | 0.989 |
| TIP41(LOC111017412) | TIP41-like protein | GGACACTCGTATGCATTGCG/ AGATGACGCTGGGATCGTTG | 146 | 103.8 | 0.998 |
| ACT7(LOC111005604) | Actin-7 | CAAGGTTGTTGCTCCACCAG/ GCACTTCCTGTGGACAATGG | 142 | 105.6 | 0.997 |
| GAPDH(LOC111016929) | Glyceraldehyde-3-phosphate dehydrogenase | AGTCCTCGACCAGAAGTTCG/ GTTGAGTGCAGCAGCTCTTG | 127 | 94.4 | 0.995 |
| DNAJ(LOC111018672) | DnaJ protein | GACTCATTTGGATAGCCGTC/ GAATGGCCTCTGGTACATTG | 115 | 107.8 | 0.998 |
| HSCP2(LOC111013580) | Heat shock cognate protein 2 | CTTATGGTGCTGCAGTTCAG/ CAACACTGTCATGACACCAC | 131 | 108.4 | 0.996 |
| ARF1(LOC111006269) | ADP-ribosylation factor 1 | CCTAATGCGATGAATGCTGC/ CTAAACCCTCGTATAGACCC | 122 | 108.3 | 0.998 |
| UP(LOC111009092) | Uncharacterized protein | CCGCAACATCTGCATCAATC/ CTTGTTGCGGACGAATTTCC | 127 | 109.7 | 0.998 |
| HMG1/2(LOC111012664) | HMG1/2-like protein | GCACCTTACATTGCTAAGGC/ CTCAGACATGGACTTCTCAG | 129 | 105.6 | 0.999 |
| TRXH-1(LOC111013893) | Thioredoxin H-type 1-like protein | GAAAGTGGACGTGGATGAAG/ TCCACCTTATCTGCACCAAC | 129 | 108.8 | 0.998 |
| PHOS32(LOC111009491) | Universal stress protein PHOS32 | CGTGAGAAGTTATGTGAGGC/ CACCACATAGTTGCTGACAC | 117 | 107.1 | 0.996 |
| GAPDH2(LOC111008959) | Glyceraldehyde-3-phosphate dehydrogenase 2 | GAAGACGATGTTGTGTCCTC/ TCATTGTCGTACCACGAGAC | 119 | 103.9 | 0.998 |
| RPL35-2(LOC111010277) | 60S ribosomal protein L35-2 | CGCTTAGGGAAGCTTACAAG/ TCGCTCGGTCTTTAGAGATG | 116 | 100.9 | 0.993 |
| UBC36(LOC111012841) | Ubiquitin-conjugating enzyme E2 36 | CAAATGGAGTCCTGCTCTAC/ GCAATGTTCTCAGAAAGCGG | 99 | 106.5 | 0.997 |
| RPS8(LOC111011823) | 40S ribosomal protein S8 | CTGCAGCATCTGCTAAGAAG/ TGTGGGTCAAGCTTACGATC | 118 | 95.9 | 0.998 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, Y.; Ma, Y.; Luo, J.; Li, J.; Zheng, X.; Gong, H.; Deng, L.; Zhao, G.; Luo, C.; Liu, X.; et al. Identification and Analysis of Reference and Tissue-Specific Genes in Bitter Gourd Based on Transcriptome Data. Horticulturae 2023, 9, 1262. https://doi.org/10.3390/horticulturae9121262
Zheng Y, Ma Y, Luo J, Li J, Zheng X, Gong H, Deng L, Zhao G, Luo C, Liu X, et al. Identification and Analysis of Reference and Tissue-Specific Genes in Bitter Gourd Based on Transcriptome Data. Horticulturae. 2023; 9(12):1262. https://doi.org/10.3390/horticulturae9121262
Chicago/Turabian StyleZheng, Yangyi, Yao Ma, Jianning Luo, Junxing Li, Xiaoming Zheng, Hao Gong, Liting Deng, Gangjun Zhao, Caixia Luo, Xiaoxi Liu, and et al. 2023. "Identification and Analysis of Reference and Tissue-Specific Genes in Bitter Gourd Based on Transcriptome Data" Horticulturae 9, no. 12: 1262. https://doi.org/10.3390/horticulturae9121262
APA StyleZheng, Y., Ma, Y., Luo, J., Li, J., Zheng, X., Gong, H., Deng, L., Zhao, G., Luo, C., Liu, X., & Wu, H. (2023). Identification and Analysis of Reference and Tissue-Specific Genes in Bitter Gourd Based on Transcriptome Data. Horticulturae, 9(12), 1262. https://doi.org/10.3390/horticulturae9121262