
Identification of Fatty Acid Components and Key Genes for Synthesis during the Development of Pecan Fruit


Abstract
:1. Introduction
2. Materials and Methods
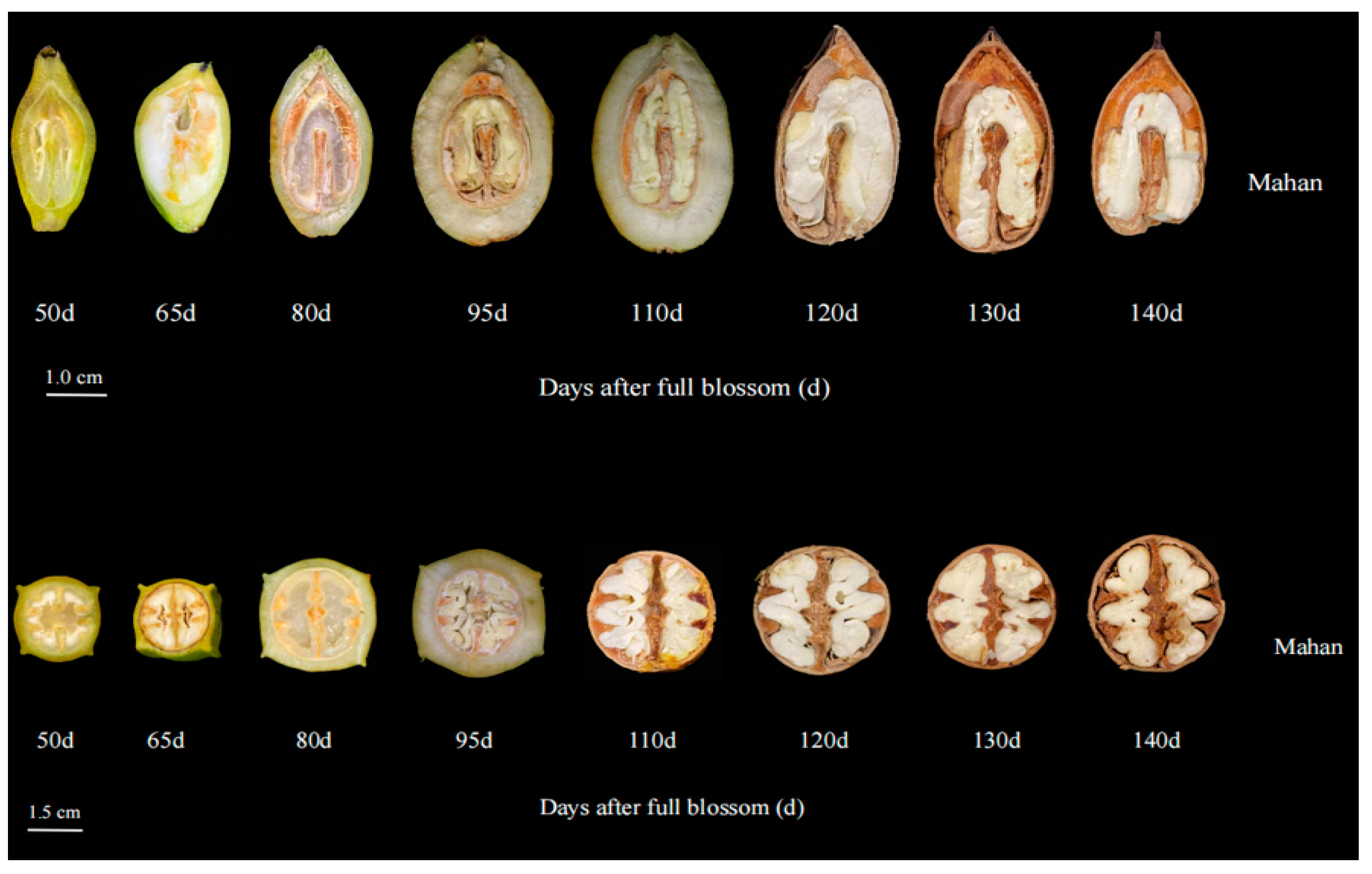
2.1. Plant Material and Treatment
2.2. Measurement of Biochemical Parameters
2.3. RNA Extraction, Library Construction, and Sequencing
2.4. Bioinformatics Analysis of RNA-Seq Data
2.5. Validation of RNA-Seq Data by qRT-PCR
3. Results and Discussion
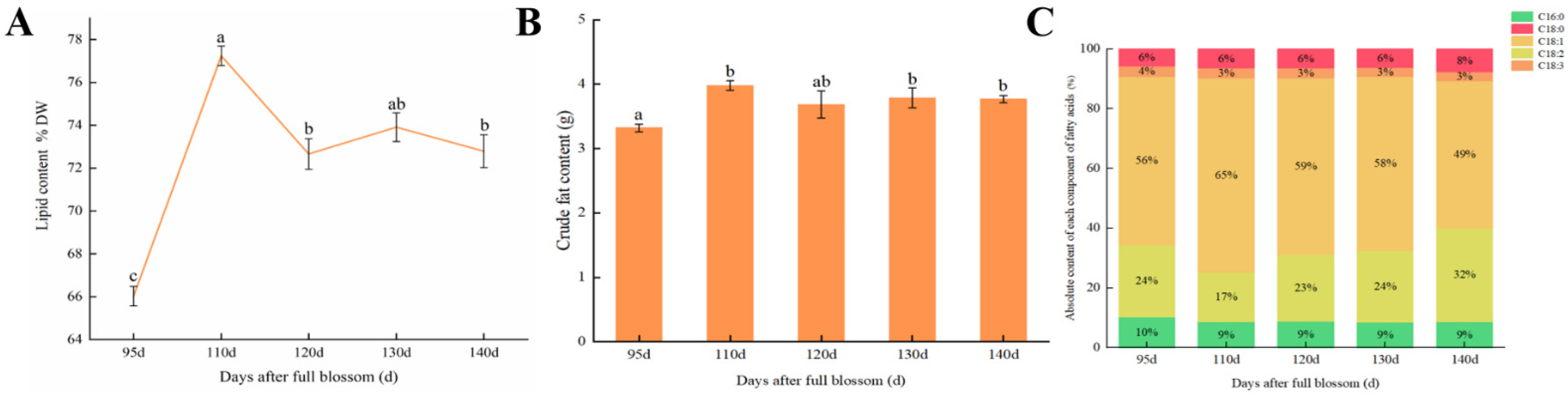
3.1. Biochemical Analysis of Lipid and Fatty Acid Content of Pecan Kernels
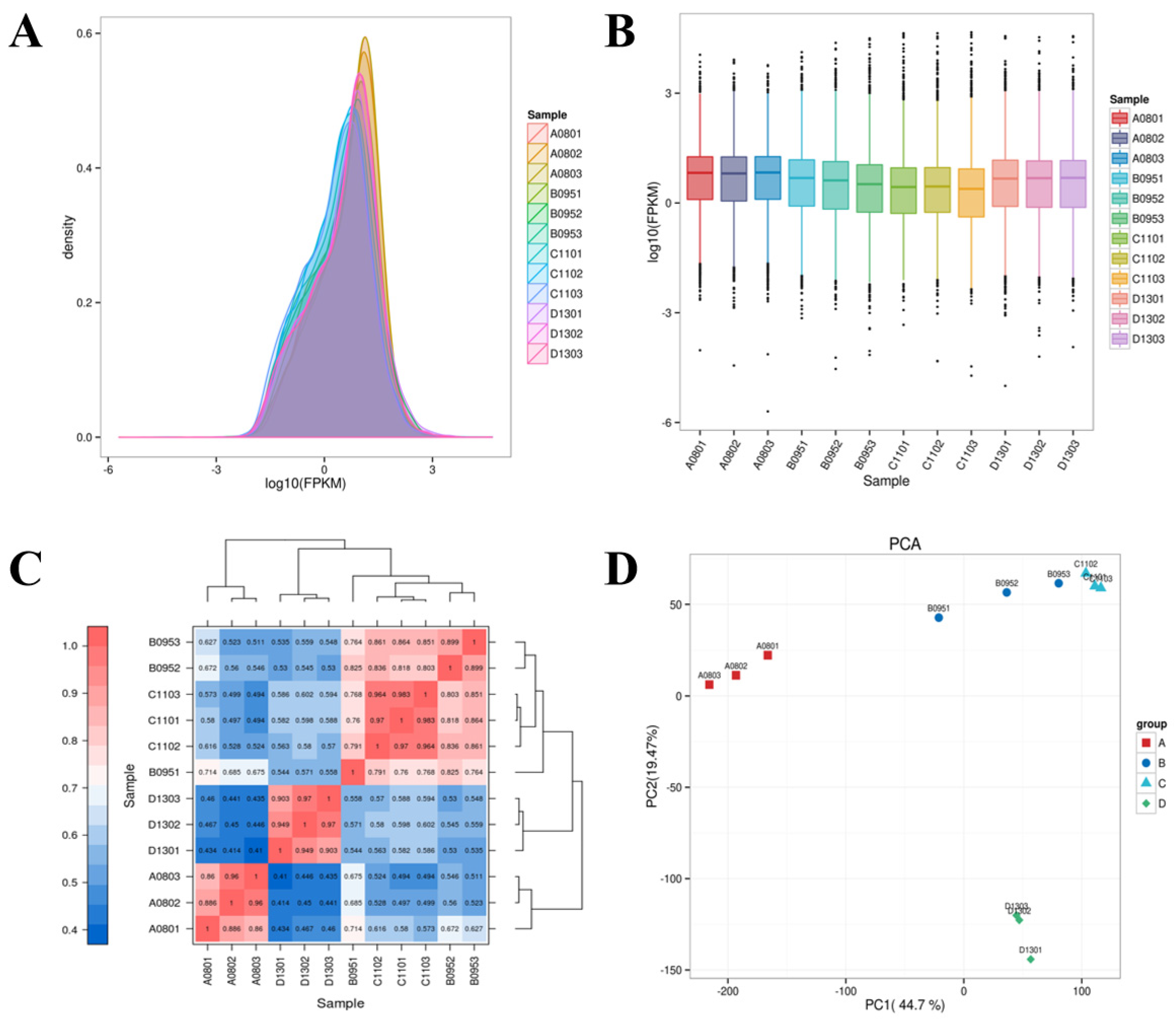
3.2. RNA-Seq Quality
3.3. Functional Annotation of Novel Genes
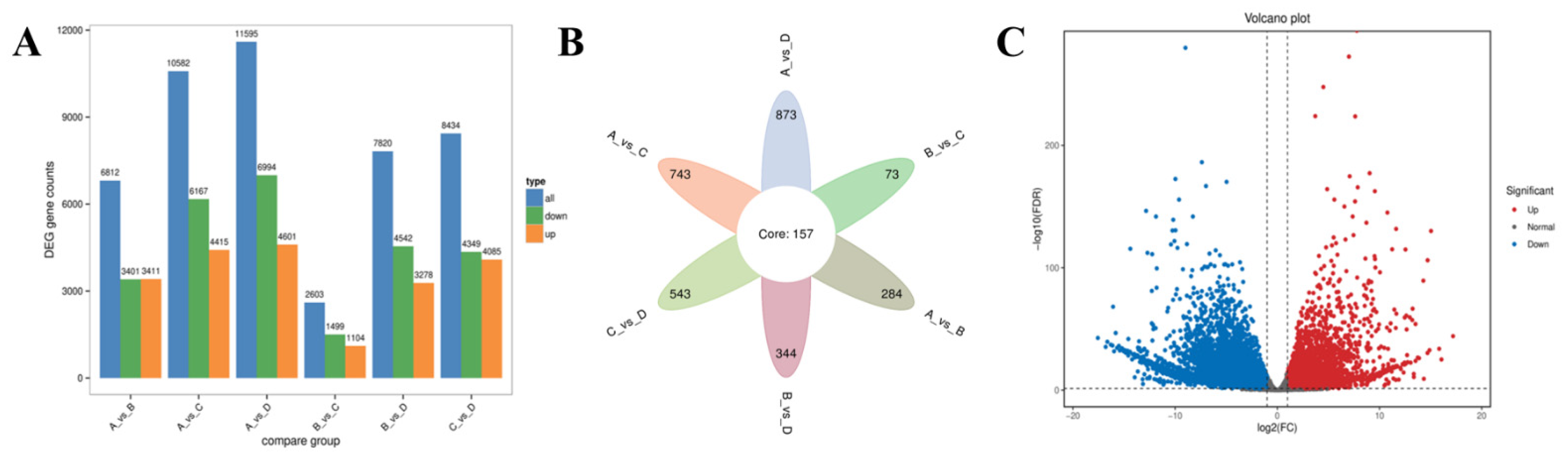
3.4. Differential Expression Analysis
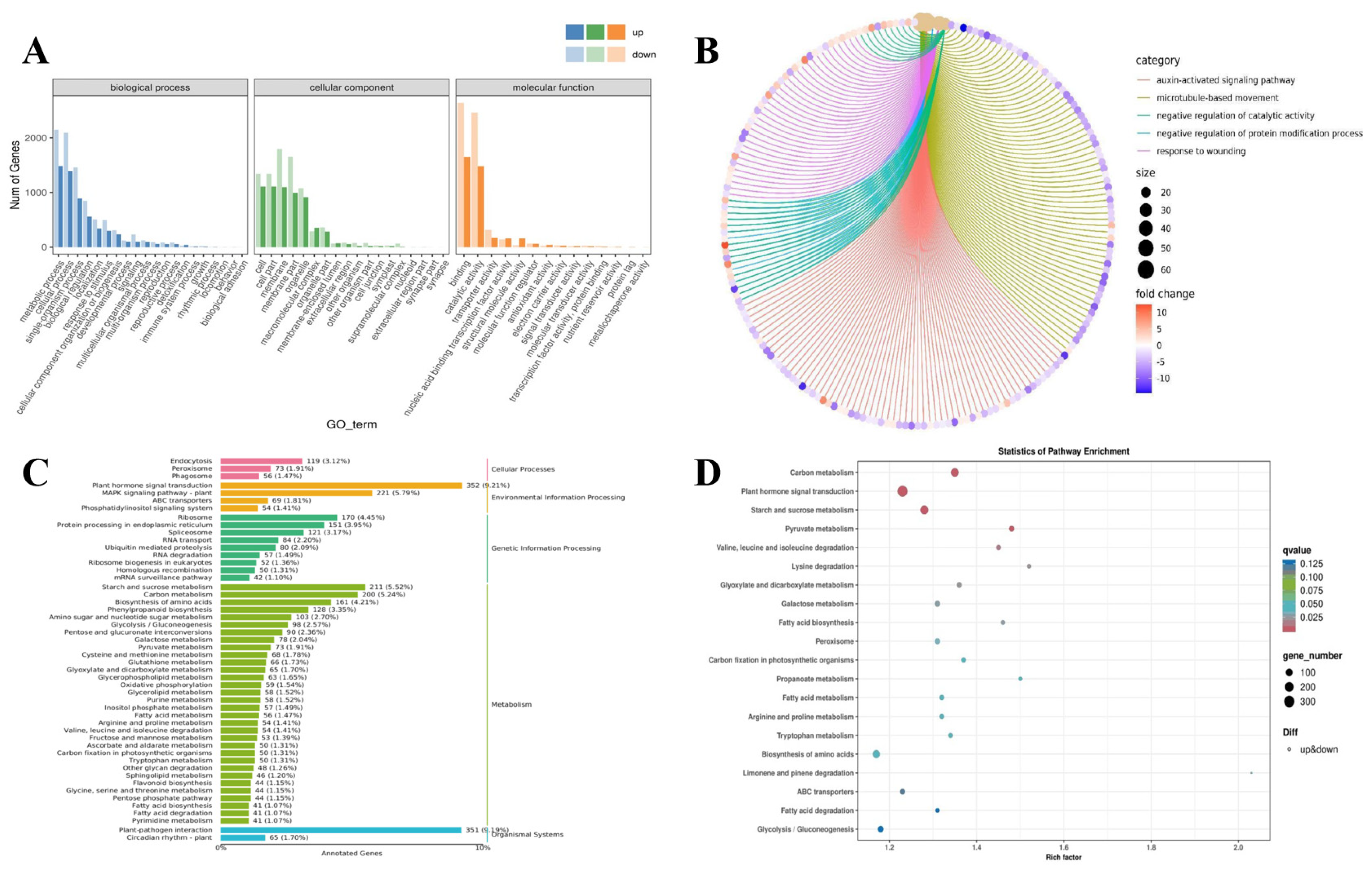
3.5. Enrichment Analysis of DEGs
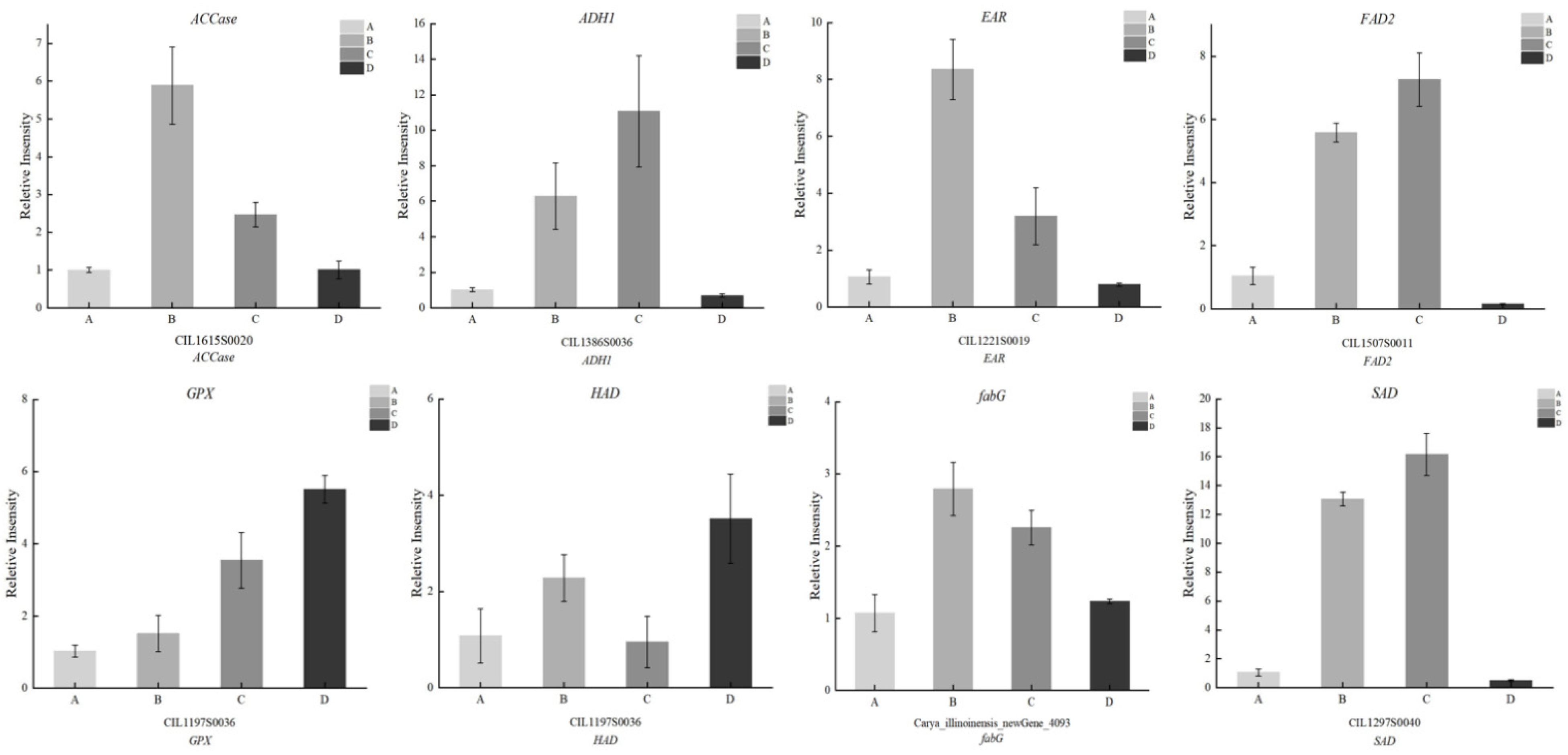
3.6. qPCR Validation of Gene Expression
3.7. Key Enzymes in the Fatty Acid Synthesis of Pecan
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Flack, J.R. The Spread and Domestication of the Pecan (Carya illinoinensis) in the United States. Ph.D. Thesis, University of Wisconsin, Madison, WI, USA, 1970. [Google Scholar]
- Hall, D.G. Pecan food potential in prehistoric North America. Econ. Bot. 2000, 54, 103–112. [Google Scholar] [CrossRef]
- Tanwar, B.; Modgil, R.; Goyal, A. Nutritional and phytochemical composition of pecan nut [Carya illinoinensis (Wangenh.) K. Koch] and its hypocholesterolemic effect in an animal model. Br. Food J. 2020, 123, 1433–1448. [Google Scholar] [CrossRef]
- Tong, X.; Szacilo, A.; Chen, H.T.; Tan, L.B.; Kong, L.Y. Using rich media to promote knowledge on nutrition and health benefits of pecans among young consumers. J. Agric. Food Res. 2022, 10, 100387. [Google Scholar] [CrossRef]
- Venkatachalam, M.; Sathe, S.K. Chemical composition of selected edible nut seeds. J. Agric. Food Chem. 2006, 54, 4705–4714. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Wang, F.; Hu, T.; Zhou, C.H. Lipidomic analyses of five Carya illinoinensis cultivars. Food Sci. Nutr. 2023, 11, 6336–6348. [Google Scholar] [CrossRef] [PubMed]
- Masoodi, L.; Gull, A.; Masoodi, F.A.; Gani, A.; Nissar, J.; Ahad, T.; Nayik, G.A.; Mukarram, S.A.; Kovács, B.; Prokisch, J.; et al. An Overview on traditional vs. green technology of extraction methods for producing high quality walnut oil. Agronomy 2022, 12, 2258. [Google Scholar] [CrossRef]
- Zou, K.Y.; Ying, M.; Sun, Z.J.; Xiong, Y.; Wu, B.; Yang, X.W. Research status and mechanism of polyunsaturated fatty acids in the treatment of alopecia. China Oils Fats 2023, 48, 69–72. [Google Scholar]
- Salas, J.J.; Sanchez, J.; Ramli, U.S.; Manaf, A.M.; Williams, M.; Harwood, J.L. Biochemistry of lipid metabolism in oliveand other oil fruits. Prog. Lipid Res. 2000, 39, 151–180. [Google Scholar] [CrossRef]
- Slocombe, S.P.; Cornah, J.; Pinfield, W.H.; Soady, K.; Zhang, Q.Y.; Gilday, A.; Dyer, J.M.; Graham, I.A. Oil accumulation in leaves directed by modification of fatty acid breakdown and lipid synthesis pathways. Plant Biotechnol. J. 2009, 7, 694–703. [Google Scholar] [CrossRef]
- Petrenko, V.; Sinturel, F.; Riezman, H.; Dibner, C. Lipid metabolism around the body clocks. Prog. Lipid Res. 2023, 91, 101235. [Google Scholar] [CrossRef]
- Ramesh, A.M.; Anuma, S.; Rahul, G.S.; Paul, T.S.; Peter, M.G.; Latha, R. Identification of two genes encoding microsomal oleate desaturases (FAD2) from the biodiesel plant Pongamia pinnata L. Trees 2016, 30, 1351–1360. [Google Scholar]
- Song, S.X. Cloning and Expression Analysis of Paeonia Ostii Fatty Acid Desaturase Gene PoFAD2. Ph.D. Thesis, Shandong Agricultural University, Tai’an, China, 2016. [Google Scholar]
- Wang, L.M.; Pei, M.H.; Xu, Y.J.; Chen, Y.M. Extraction of walnut oil body and its demulsification based on thin film drying-vacuum filtration technology. Trans. Chin. Soc. Agric. Eng. 2023, 39, 241–248. [Google Scholar]
- Geng, Q.N.; Chen, J.; Guo, R.; Zhang, L.Y.; Li, Q.; Yu, X.Z. Salt-assisted aqueous extraction combined with Span 20 allow the obtaining of a high-quality and yield walnut oil. LWT 2020, 121, 108956. [Google Scholar] [CrossRef]
- Jia, Z.; Wang, G.; Xuan, J.; Zhang, J.; Zhai, M.; Jia, X.; Guo, Z.; Li, M. Comparative Transcriptome Analysis of Pecan Female and Male Inflorescences. Russ. J. Plant Physiol. 2018, 15, 186–196. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. String Tie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.Y.; Li, J.Q.; Wu, S.F.; Zhu, Y.P.; Chen, Y.W.; He, F.C. Integrated nr database in protein annotation system and its localization. Comput. Eng. 2006, 32, 71–74. [Google Scholar]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.Z.; Lopez, R.; Magrane, M.; et al. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2004, 32, 115–119. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A. The COG database: A tool for genome scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef]
- Koonin, E.V.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Krylov, D.M.; Makarova, K.S.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; Rao, B.S.; et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004, 5, R7. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.Z.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, P.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Coin, L.; Durbin, R.; Finn, R.D.; Hollich, V.; Griffiths, J.S.; Khanna, A.; Marshall, M.; Moxon, S.; Sonnhammer, E.L.L.; et al. Pfam: The protein families database. Nucleic Acids Res. 2013, 1223, 276–280. [Google Scholar]
- Eddy, S.R. Profile hidden Markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome. Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Mo, Z.H.; Chen, Y.Q.; Lou, W.R.; Jia, X.D.; Zhai, M.; Xuan, J.P.; Guo, Z.R.; Li, Y.R. Identification of suitable reference genes for normalization of real-time quantitative PCR data in pecan (Carya illinoinensis). Trees 2020, 34, 1233–1241. [Google Scholar] [CrossRef]
- Özrenk, K.; Javidipour, I.; Yarilgac, T.; Balta, F.; Gündogdu, M. Fatty acids, tocopherols, selenium and total carotene of pistachios (P. vera L.) from Diyarbakir (Southestern Turkey) and walnuts (J. regia L.) from Erzincan (Eastern Turkey). Int. J. Food Sci. Technol. 2010, 18, 55–62. [Google Scholar] [CrossRef]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.S.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef]
- Sasaki, Y.; Nagano, Y. Plant acety-CoA carboxylase: Structure, biosynthesis, regulation, and gene manipulation for plant breeding. Biosci. Biotech. Bioch. 2004, 68, 1175–1184. [Google Scholar] [CrossRef]
- Wang, B.M.; Yan, S.H.; Tan, X.F. Study on the structure, function and expression regulation of ACCase genes in plants. Anhui Agric. Sci. Bulletin. 2021, 27, 17–24. [Google Scholar]
- Chen, S.L. Identification and Functional Study of Lipid Synthesis Related Genes in Peanut. Ph.D. Thesis, Chinese Academy of Agricultural Sciences, Beijing, China, 2012. [Google Scholar]
- Dong, H.J.; Cronan, J.E. Unsaturated fatty acid synthesis in Enterococcus faecalis requires a specific enoyl-ACP reductase. Mol. Microbiol. 2022, 118, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Shanklin, J.; Cahoon, E.B. Desaturation and related modifications of fatty acids. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1998, 49, 611–640. [Google Scholar] [CrossRef] [PubMed]
- Ben, A.R.; Ennouri, K.; Ercişli, S.; Hajer, B.H.; Mohsen, H.; Slim, S.; Ahmed Rebai, F.M. First study of correlation between oleic acid content and SAD gene poly-morphism in olive oil samples through statistical and bayesian modeling analyses. Lipids Health Dis. 2018, 17, 74. [Google Scholar]
- Dmitriev, A.A.; Kezimana, P.; Rozhmina, T.A.; Zhuchenko, A.A.; Povkhova, L.V.; Pushkova, E.N.; Novakovskiy, R.O.; Pavelek, M.; Vladimirov, G.N.; Nikolaev, E.N.; et al. Genetic diversity of SAD and FAD genes responsible for the fatty acid composition in flax cultivars and lines. BMC Plant Biol. 2020, 20 (Suppl. S1), 301. [Google Scholar] [CrossRef]
- Huang, R.M.; Zhou, Y.; Zhang, J.P.; Ji, F.Y.; Jin, F.; Fan, W.; Pei, D. Transcriptome Analysis of Walnut (Juglans regia L.) Embryos Reveals Key Developmental Stages and Genes Involved in Lipid Biosynthesis and Polyunsaturated Fatty Acid Metabolism. J. Agric. Food Chem. 2021, 69, 377–396. [Google Scholar] [CrossRef]
- Dhakal, K.H.; Jung, K.H.; Chae, J.H.; Shannon, J.G.; Lee, J.D. Variation of unsaturated fatty acids in soybean sprout of high oleic acid accessions. Food Chem. 2014, 164, 70–73. [Google Scholar] [CrossRef]
- Puttha, R.; Venkatachalam, K.; Hanpakdeesakul, S.; Wongsa, T.; Parametthanuwat, T.; Srean, P.; Pakeechai, K.; Charoenphun, N. Exploring the Potential of Sunflowers: Agronomy, Applications, and Opportunities within Bio-Circular-Green Economy. Horticulturae 2023, 9, 1079. [Google Scholar] [CrossRef]
- Dar, A.A.; Choudhury, A.R.; Kancharla, P.K.; Arumugam, N. The FAD2 Gene in Plants: Occurrence, Regulation, and Role. Front. Plant Sci. 2017, 8, 1789. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Sample Class | ||||
|---|---|---|---|---|---|
| 95 Days | 110 Days | 120 Days | 130 Days | 140 Days | |
| C16:0 | 7.810 ± 0.035 a | 6.430 ± 0.036 e | 6.513 ± 0.025 d | 6.677 ± 0.035 c | 6.937 ± 0.057 b |
| C18:0 | 2.143 ± 0.015 d | 2.327 ± 0.045 c | 2.320 ± 0.010 c | 2.420 ± 0.066 b | 3.010 ± 0.026 a |
| C18:1 | 70.423 ± 0.270 d | 78.080 ± 0.035 a | 72.057 ± 0.068 c | 72.970 ± 0.061 b | 64.107 ± 0.040 e |
| C18:2 | 18.307 ± 0.025 b | 11.970 ± 0.090 e | 17.970 ± 0.020 c | 16.747 ± 0.112 d | 24.803 ± 0.076 a |
| C18:3 | 1.320 ± 0.242 a | 1.187 ± 0.025 a | 1.143 ± 0.040 a | 1.187 ± 0.032 a | 1.140 ± 0.017 a |
| Sample | Total Reads | Mapped Reads | Uniq Mapped Reads | Multiple Map Reads | Reads Map to ‘+’ | Reads Map to ‘−’ |
|---|---|---|---|---|---|---|
| A801 | 44,611,820 | 41,188,894 (92.33%) | 40,148,548 (90.00%) | 1,040,346 (2.33%) | 21,217,825 (47.56%) | 21,251,742 (47.64%) |
| A802 | 48,505,208 | 45,161,914 (93.11%) | 44,064,795 (90.85%) | 1,097,11 (2.26%) | 23,229,480 (47.89%) | 23,269,975 (47.97%) |
| A803 | 52,534,948 | 48,558,039 (92.43%) | 47,362,292 (90.15%) | 1,195,747 (2.28%) | 24,990,755 (47.57%) | 25,024,059 (47.63%) |
| B951 | 45,958,908 | 42,897,631 (93.34%) | 41,749,011 (90.84%) | 1,148,620 (2.50%) | 22,133,775 (48.16%) | 22,172,300 (48.24%) |
| B952 | 46,090,540 | 43,103,232 (93.52%) | 41,304,339 (89.62%) | 1,798,893 (3.90%) | 22,673,701 (49.19%) | 22,695,095 (49.24%) |
| B953 | 47,187,014 | 44,972,673 (95.31%) | 43,449,342 (92.08%) | 11,523,331 (3.23%) | 23,431,052 (49.66%) | 23,453,666 (49.70%) |
| C1101 | 41,641,344 | 39,457,525 (94.76%) | 37,856,182 (90.91%) | 1,601,343 (3.85%) | 20,700,039 (49.71%) | 20,754,808 (49.84%) |
| C1102 | 41,792,266 | 39,992,132 (95.69%) | 38,311,525 (91.67%) | 1,680,580 (4.02%) | 21,042,988 (50.35%) | 21,053,642 (50.38%) |
| C1103 | 57,865,390 | 55,427,094 (95.79%) | 52,668,638 (91.02%) | 2,758,456 (4.77%) | 29,426,371 (50.85%) | 29,448.572 (50.89) |
| D1301 | 42,464,800 | 40,360,537 (95.04%) | 39,035,159 (91.92%) | 1,325,378 (3.12%) | 20,992,211 (49.43%) | 20,963,422 (49.37%) |
| D1302 | 43,473,704 | 41,497,574 (95.45%) | 40,109,711 (92.26%) | 1,387,863 (3.19%) | 21,618,944 (49.73%) | 21,622,341 (49.74%) |
| D1303 | 48,545,276 | 46,435,876 (95.65%) | 45,083,142 (92.87%) | 1,352,734 (2.79%) | 24,038,643 (49.52%) | 24,037,113 (49.51%) |
| DEG Set | Total | COG | GO | KEGG | KOG | NR | Pfam | Swiss-Prot | eggNOG |
|---|---|---|---|---|---|---|---|---|---|
| A_vs_B | 6365 | 2226 | 5248 | 4321 | 3262 | 6359 | 5291 | 4791 | 5481 |
| A_vs_C | 9863 | 3230 | 8079 | 6711 | 5116 | 9849 | 8132 | 7330 | 8394 |
| A_vs_D | 10,882 | 3608 | 8936 | 7465 | 5656 | 10,870 | 9009 | 8141 | 9346 |
| B_vs_C | 2437 | 829 | 1953 | 1631 | 1218 | 2435 | 2053 | 1796 | 2103 |
| B_vs_D | 7474 | 2621 | 6174 | 5230 | 4051 | 7470 | 6327 | 5697 | 6488 |
| C_vs_D | 7985 | 2806 | 6587 | 5622 | 4347 | 7979 | 6703 | 6052 | 6905 |
| Pathway Name | Pathways Number | DEGs Number |
|---|---|---|
| Fatty acid biosynthesis | ko00061 | 1 |
| Fatty acid elongation | ko00062 | 2 |
| Fatty acid degradation | ko00071 | 8 |
| Synthesis and degradation of ketone bodies | ko00072 | 2 |
| Cutin, suberine, and wax biosynthesis | ko00073 | 5 |
| Steroid biosynthesis | ko00100 | 8 |
| Glycerolipid metabolism | ko00561 | 14 |
| Glycerophospholipid metabolism | ko00564 | 7 |
| Ether lipid metabolism | ko00565 | 5 |
| Arachidonic acid metabolism | ko00590 | 3 |
| Linoleic acid metabolism | ko00591 | 4 |
| alpha-Linolenic acid metabolism | ko00592 | 9 |
| Sphingolipid metabolism | ko00600 | 14 |
| Fatty acid metabolism | ko01212 | 3 |
| Total | - | 85 |
| Annotation | 80 d | 95 d | 110 d | 130 d |
|---|---|---|---|---|
| 3-Hydroxyacyl-CoA dehydrogenase | 74.906 | 84.636 | 52.174 | 647.727 |
| Acyl-sn-glycerol-3-phosphate Acyltransferase | 74.060 | 92.385 | 55.007 | 120.418 |
| Acyl-[acyl-carrier-protein] desaturase | 221.144 | 1188.341 | 1787.679 | 0.977 |
| Acetyl-CoA carboxylase | 208.514 | 467.616 | 92.758 | 48.297 |
| Alcohol dehydrogenase class-P | 305.192 | 1659.110 | 1707.982 | 67.504 |
| Oxoacyl-[acyl-carrier protein] reductase | 93.442 | 868.771 | 707.290 | 99.485 |
| Glutathione peroxidase | 111.326 | 251.159 | 327.642 | 656.789 |
| Enoyl-[acyl-carrier protein] reductase I | 133.260 | 512.245 | 223.755 | 15.479 |
| Omega-6 fatty acid desaturase | 84.676 | 306.651 | 488.605 | 96.185 |
| Acetyl-CoA acyltransferase 1 | 144.337 | 69.065 | 60.953 | 237.704 |
| Gene ID | Gene Name | Protein Name | Gene Expression Patterns | |
|---|---|---|---|---|
| Upregulated | Downregulated | |||
| CIL1204S0021 | HAD, MFP2 | 3-hydroxyacyl-CoA dehydrogenase | 80–95 d, 110–130 d | 95–110 d |
| CIL1297S0040 | SAD, FAB2 | acyl-[acyl-carrier-protein] desaturase | 80–95 d, 95–110 d | 110–130 d |
| CIL1615S0020 | accC | acetyl-CoA carboxylase | 80–95 d | 95–110 d, 110–130 d |
| CIL1386S0036 | ADH1 | alcohol dehydrogenase class-P | 80–95 d, 95–110 d | 110–130 d |
| Carya_illinoinensis_newGene_4093 | fabG | 3-oxoacyl-[acyl-carrier protein] reductase | 80–95 d | 95–110 d, 110–130 d |
| CIL1197S0036 | gpx, btuE | glutathione peroxidase | 80–95 d, 95–110 d, 110–130 d | - |
| CIL1221S0019 | EAR, fabI | enoyl-[acyl-carrier protein] reductase I | 80–95 d, 95–110 d | 110–130 d |
| CIL1507S0011 | FAD2 | omega-6 fatty acid desaturase | 80–95 d, 95–110 d | 110–130 d |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, F.; Zhao, Z.; Hu, T.; Zhou, C. Identification of Fatty Acid Components and Key Genes for Synthesis during the Development of Pecan Fruit. Horticulturae 2023, 9, 1199. https://doi.org/10.3390/horticulturae9111199
Wang F, Zhao Z, Hu T, Zhou C. Identification of Fatty Acid Components and Key Genes for Synthesis during the Development of Pecan Fruit. Horticulturae. 2023; 9(11):1199. https://doi.org/10.3390/horticulturae9111199
Chicago/Turabian StyleWang, Fei, Zhe Zhao, Tian Hu, and Chunhua Zhou. 2023. "Identification of Fatty Acid Components and Key Genes for Synthesis during the Development of Pecan Fruit" Horticulturae 9, no. 11: 1199. https://doi.org/10.3390/horticulturae9111199
APA StyleWang, F., Zhao, Z., Hu, T., & Zhou, C. (2023). Identification of Fatty Acid Components and Key Genes for Synthesis during the Development of Pecan Fruit. Horticulturae, 9(11), 1199. https://doi.org/10.3390/horticulturae9111199