
Characterization and Expression Profiling of Orphan Genes in Rapeseed (Brassica napus) Provide Insights into Tissue Development and Cold Stress Adaptation


Abstract
1. Introduction
2. Materials and Methods
2.1. Database Retrieval
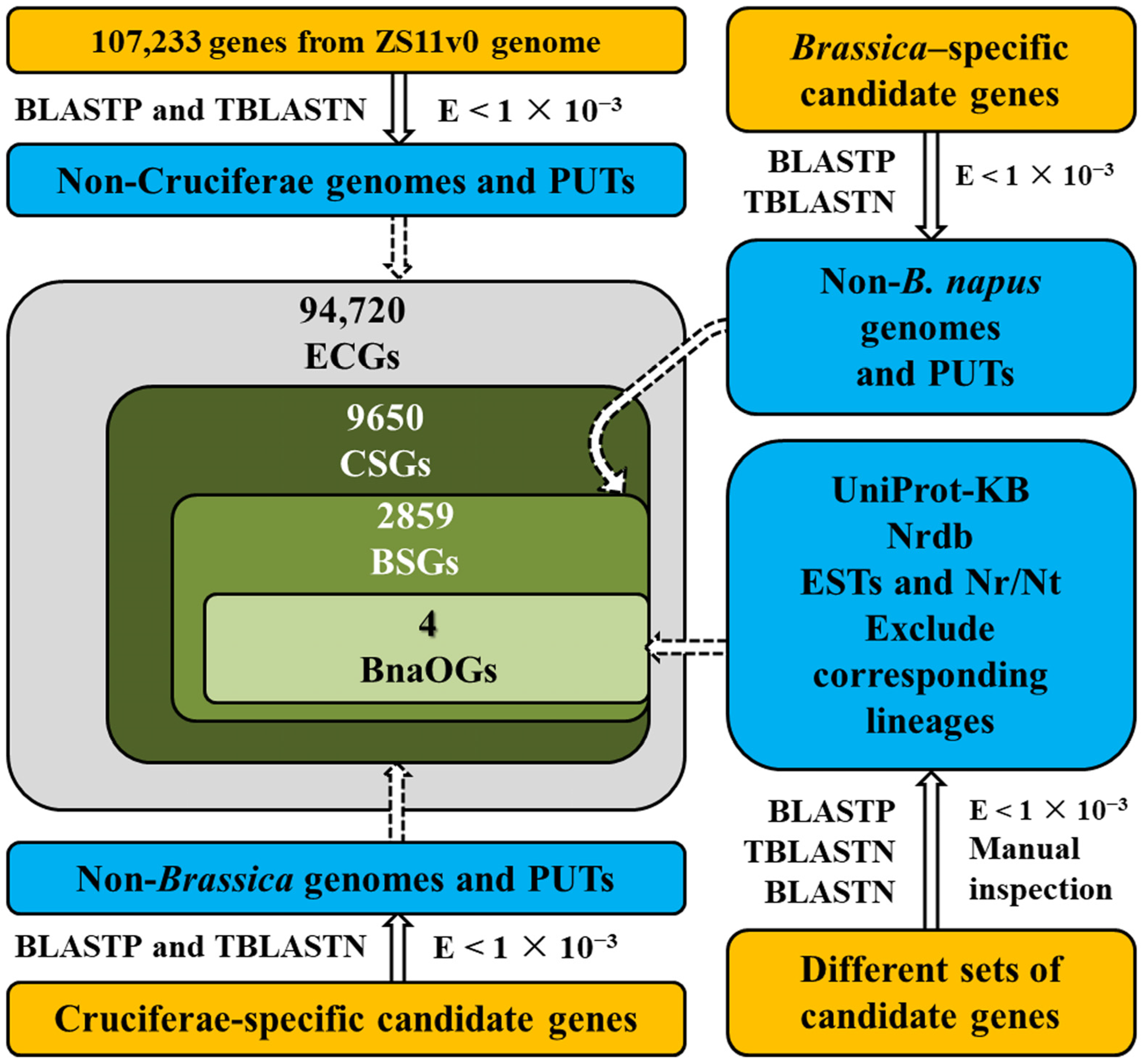
2.2. Homology Screening and Classification
2.3. Bioinformatic Characterization
2.4. Functional Annotation and Expression Profiling
2.5. Sampling and qRT-PCR Analysis
2.6. Statistical Analysis and Data Visualization
3. Results
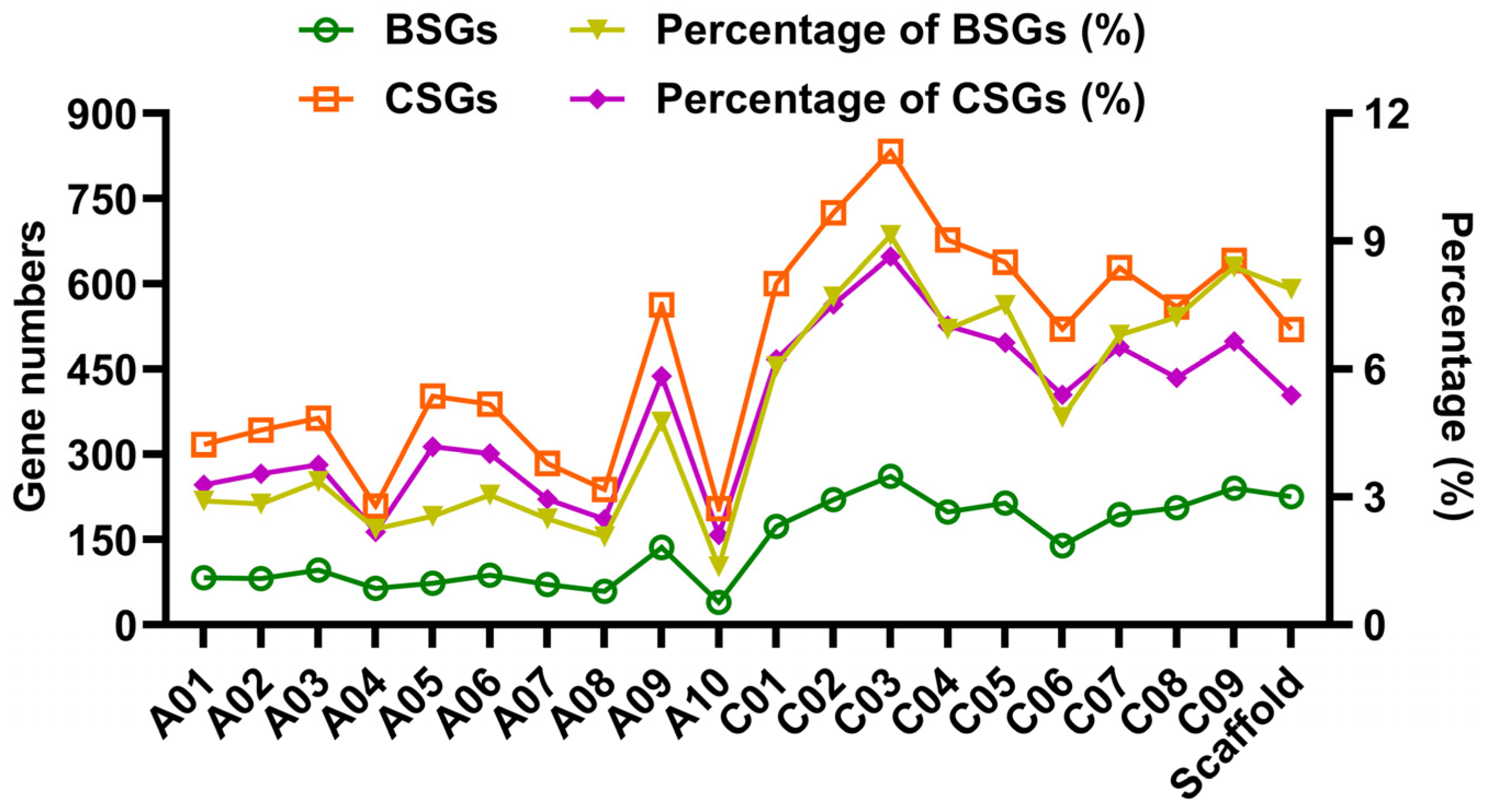
3.1. Identification and Classification of OGs in Rapeseed
3.2. Structural and Evolutionary Divergence of Different Gene Categories
3.3. Functional Annotation of Different Gene Categories
3.4. Dynamic Expression Patterns of BnaOGs During Different Tissues and Stress Adaptation
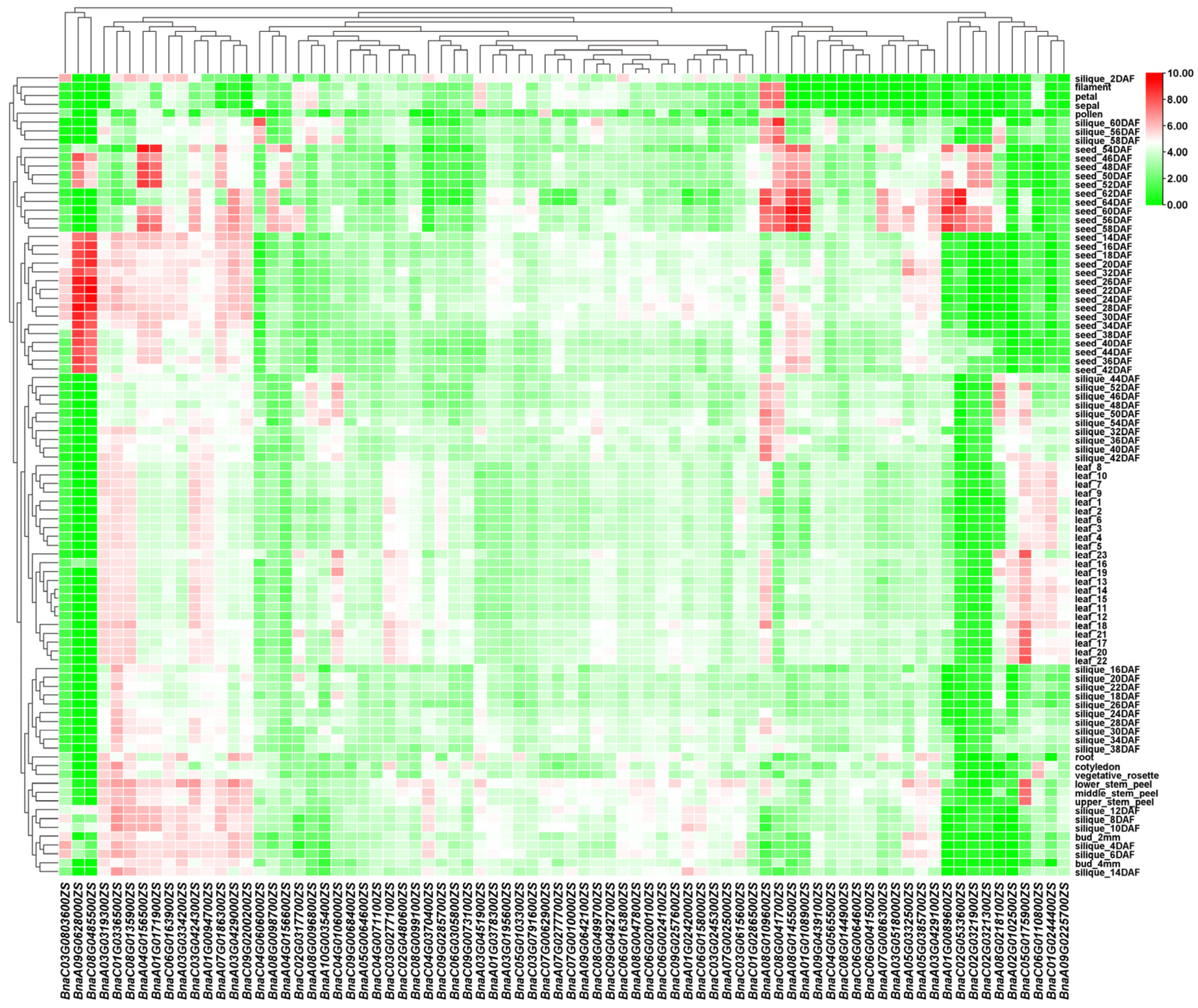
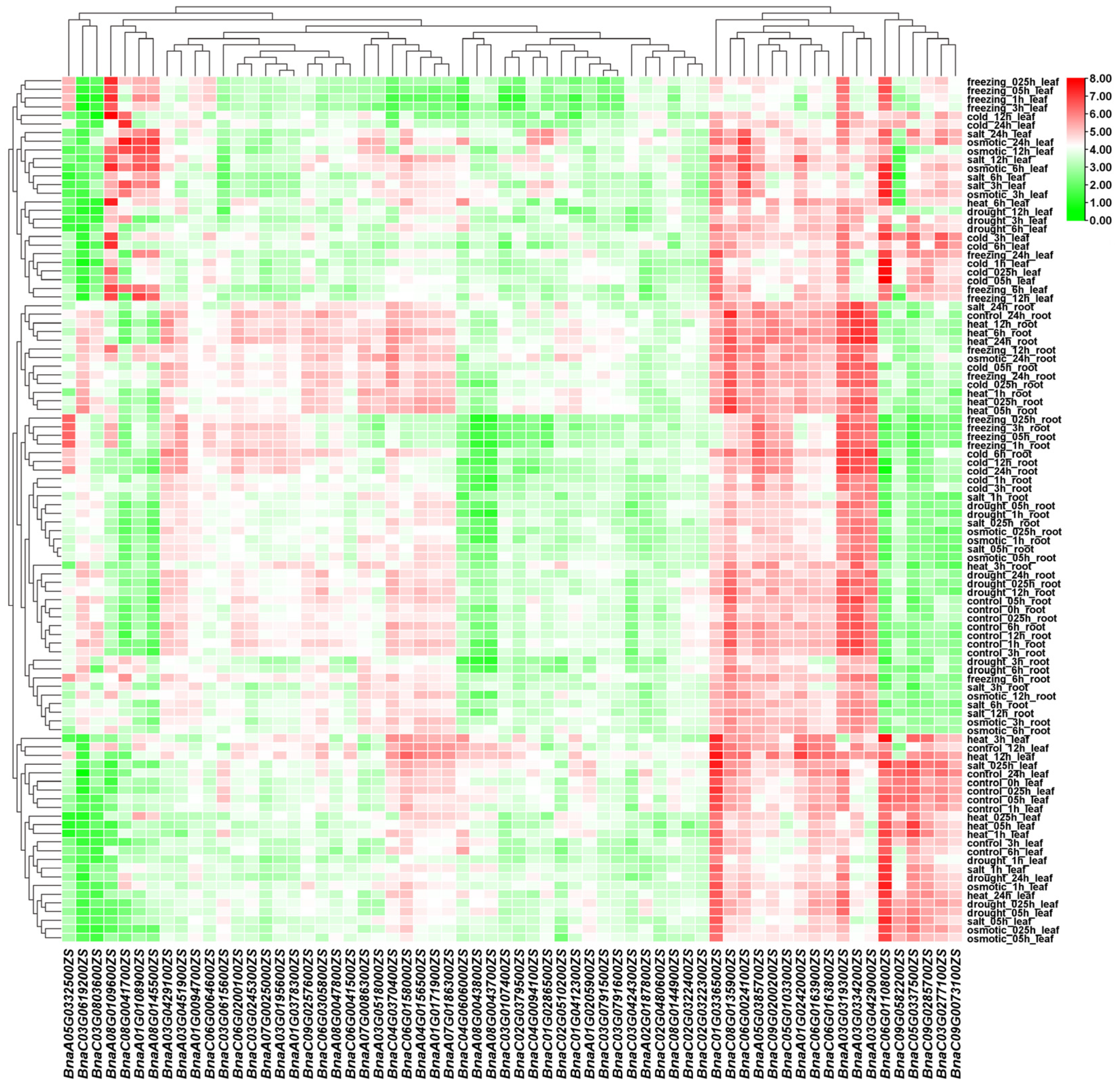
3.5. Tissue-Specific and Stress-Responsive Expression Profiles of BSGs
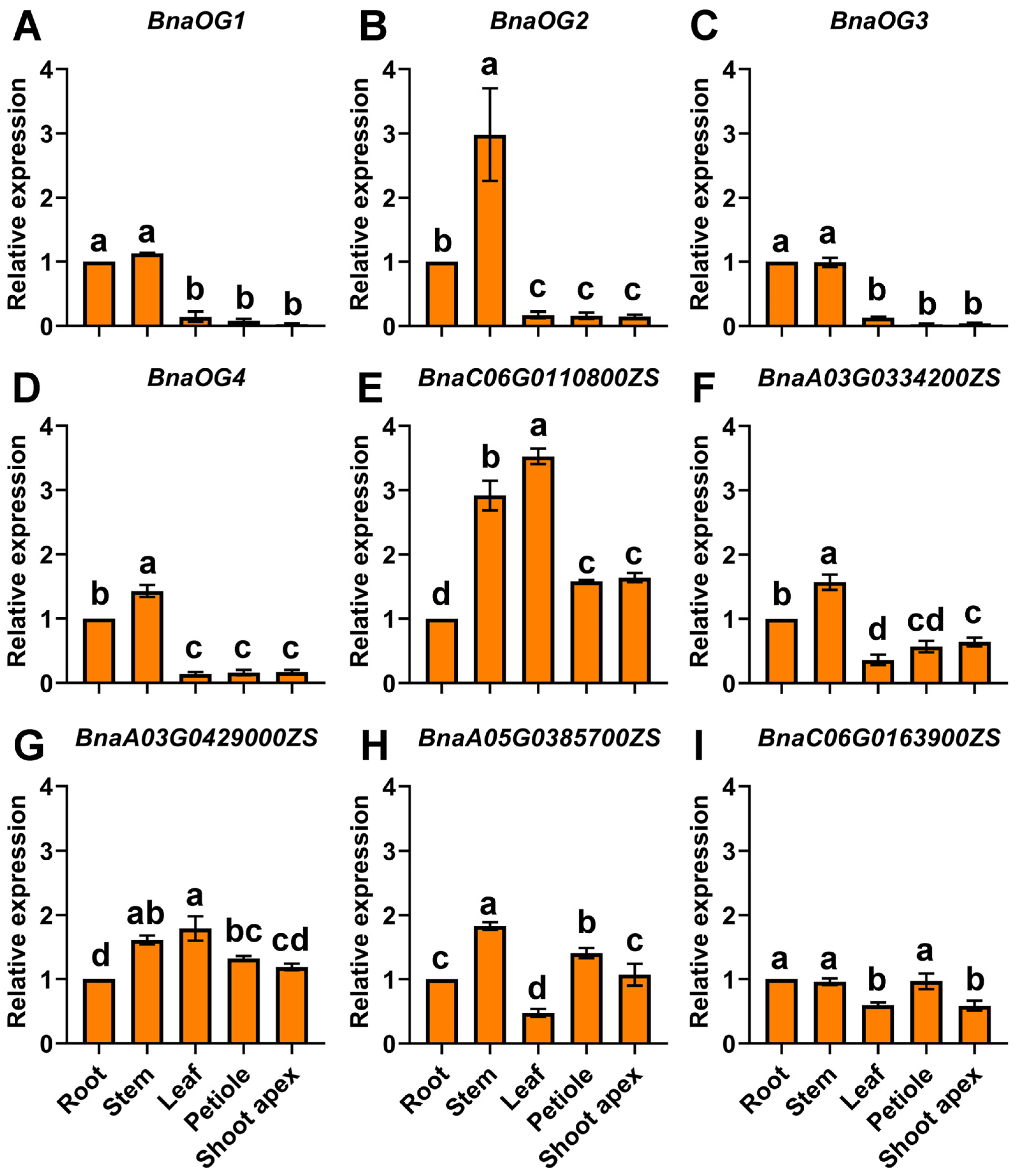
3.6. Tissue-Specific Expression of BnaOGs and Core BSGs
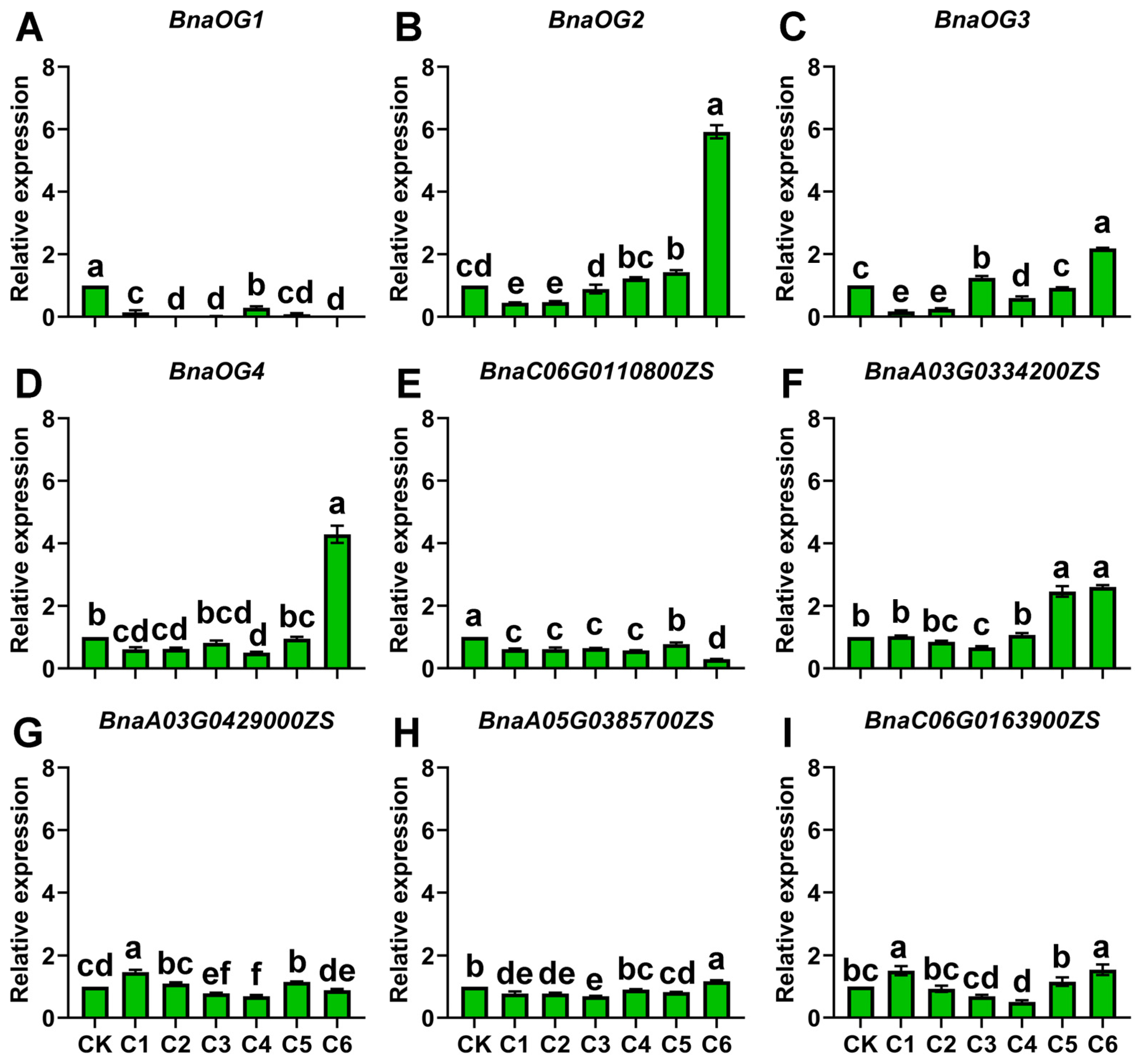
3.7. Cold-Responsive Expression of BnaOGs and Core BSGs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| ANOVA | One-way analysis of variance |
| BnaOGs | B. napus OGs |
| BSGs | Brassica-specific genes |
| CDS | Coding sequence |
| CSGs | Cruciferae-specific genes |
| ECGs | Evolutionarily conserved genes |
| EST | Expressed Sequence Tag |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| Nrdb | Non-redundant protein database |
| OGs | Orphan genes |
| PUTs | PlantGDB-generated unique transcripts |
| TFs | Transcription factors |
| UniProt-KB | UniProt Knowledgebase |
References
- Jiang, M.; Li, X.; Dong, X.; Zu, Y.; Zhan, Z.; Piao, Z.; Lang, H. Research Advances and Prospects of Orphan Genes in Plants. Front. Plant Sci. 2022, 13, 947129. [Google Scholar] [CrossRef]
- Jin, G.H.; Zhou, Y.L.; Yang, H.; Hu, Y.T.; Shi, Y.; Li, L.; Siddique, A.N.; Liu, C.N.; Zhu, A.D.; Zhang, C.J.; et al. Genetic innovations: Transposable element recruitment and de novo formation lead to the birth of orphan genes in the rice genome. J. Syst. Evol. 2019, 59, 341–351. [Google Scholar] [CrossRef]
- Fakhar, A.Z.; Liu, J.; Pajerowska-Mukhtar, K.M.; Mukhtar, M.S. The ORFans’ tale: New insights in plant biology. Trends Plant Sci. 2023, 28, 1379–1390. [Google Scholar] [CrossRef]
- Fakhar, A.Z.; Liu, J.; Pajerowska-Mukhtar, K.M.; Mukhtar, M.S. The Lost and Found: Unraveling the Functions of Orphan Genes. J. Dev. Biol. 2023, 11, 27. [Google Scholar] [CrossRef]
- Jiang, M.; Dong, X.; Lang, H.; Pang, W.; Zhan, Z.; Li, X.; Piao, Z. Mining of Brassica-Specific Genes (BSGs) and Their Induction in Different Developmental Stages and under Plasmodiophora brassicae Stress in Brassica rapa. Int. J. Mol. Sci. 2018, 19, 2064. [Google Scholar] [CrossRef]
- Xu, Y.; Wu, G.; Hao, B.; Chen, L.; Deng, X.; Xu, Q. Identification, characterization and expression analysis of lineage-specific genes within sweet orange (Citrus sinensis). BMC Genomics 2015, 16, 995. [Google Scholar] [CrossRef]
- Zhang, M.; Zhu, M.; Lang, H.; Wang, W.; Li, X.; Jiang, M. Genome-Wide Identification, Characterization, and Expression Analysis of Orphan Genes Within Coriander. Plants 2025, 14, 778. [Google Scholar] [CrossRef]
- Ma, Y.; Zhai, Q.; Liu, Z.; Liu, W. Genome-wide identification and characterization of alfalfa-specific genes in drought stress tolerance. Plant Physiol. Bioch. 2025, 220, 109474. [Google Scholar] [CrossRef]
- Ren, Q.; Lim, Y.-Y.; Teo, C.H. Genome-wide identification and expression analysis of orphan genes in twelve Musa (sub) species. 3 Biotech 2025, 15, 41. [Google Scholar] [CrossRef]
- Arendsee, Z.W.; Li, L.; Wurtele, E.S. Coming of age: Orphan genes in plants. Trends Plant Sci. 2014, 19, 698–708. [Google Scholar] [CrossRef]
- Feyissa, B.A.; de Becker, E.M.; Salesse-Smith, C.E.; Shu, M.; Zhang, J.; Yates, T.B.; Xie, M.; De, K.; Gotarkar, D.; Chen, M.S.S.; et al. An orphan gene BOOSTER enhances photosynthetic efficiency and plant productivity. Dev. Cell 2025, 60, 723–734.e727. [Google Scholar] [CrossRef]
- Zu, Y.; Jiang, M.; Zhan, Z.; Li, X.; Piao, Z. Orphan gene BR2 positively regulates bolting resistance through the vernalization pathway in Chinese cabbage. Hortic. Res. 2024, 11, uhae216. [Google Scholar] [CrossRef]
- Jiang, M.; Zhan, Z.; Li, X.; Piao, Z. Construction and evaluation of Brassica rapa orphan genes overexpression library. Front. Plant Sci. 2025, 16, 1532449. [Google Scholar] [CrossRef]
- Jiang, M.; Zhang, Y.; Yang, X.; Li, X.; Lang, H. Brassica rapa orphan gene BR1 delays flowering time in Arabidopsis. Front. Plant Sci. 2023, 14, 1135684. [Google Scholar] [CrossRef]
- Wang, D.-H.; Xu, Z.-H.; Bai, S.-N. OsFON879, an orphan gene, regulates floral organ homeostasis in rice. Plant Biotechnol. J. 2025, 23, 2888–2890. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiang, M.; Sun, S.; Zhan, Z.; Li, X.; Piao, Z. Chinese cabbage orphan gene BR3 confers bolting resistance to Arabidopsis through the gibberellin pathway. Front. Plant Sci. 2025, 15, 1518962. [Google Scholar] [CrossRef]
- Jiang, M.; Zhan, Z.; Li, H.; Dong, X.; Cheng, F.; Piao, Z. Brassica rapa orphan genes largely affect soluble sugar metabolism. Hortic. Res. 2020, 7, 181. [Google Scholar] [CrossRef]
- Li, L.; Wurtele, E.S. The QQS orphan gene of Arabidopsis modulates carbon and nitrogen allocation in soybean. Plant Biotechnol. J. 2015, 13, 177–187. [Google Scholar] [CrossRef]
- Li, L.; Zheng, W.; Zhu, Y.; Ye, H.; Tang, B.; Arendsee, Z.W.; Jones, D.; Li, R.; Ortiz, D.; Zhao, X.; et al. QQS orphan gene regulates carbon and nitrogen partitioning across species via NF-YC interactions. Proc. Natl. Acad. Sci. USA 2015, 112, 14734–14739. [Google Scholar] [CrossRef]
- Cui, X.; Lv, Y.; Chen, M.; Nikoloski, Z.; Twell, D.; Zhang, D. Young Genes out of the Male: An Insight from Evolutionary Age Analysis of the Pollen Transcriptome. Mol. Plant 2015, 8, 935–945. [Google Scholar] [CrossRef]
- Ni, F.; Qi, J.; Hao, Q.; Lyu, B.; Luo, M.C.; Wang, Y.; Chen, F.; Wang, S.; Zhang, C.; Epstein, L.; et al. Wheat Ms2 encodes for an orphan protein that confers male sterility in grass species. Nat. Commun. 2017, 8, 15121. [Google Scholar] [CrossRef]
- Dossa, K.; Zhou, R.; Li, D.; Liu, A.; Qin, L.; Mmadi, M.A.; Su, R.; Zhang, Y.; Wang, J.; Gao, Y.; et al. A novel motif in the 5′-UTR of an orphan gene ‘Big Root Biomass’ modulates root biomass in sesame. Plant Biotechnol. J. 2021, 19, 1065–1079. [Google Scholar] [CrossRef]
- Cardoso-Silva, C.B.; Aono, A.H.; Mancini, M.C.; Sforça, D.A.; da Silva, C.C.; Pinto, L.R.; Adams, K.L.; de Souza, A.P. Taxonomically Restricted Genes Are Associated With Responses to Biotic and Abiotic Stresses in Sugarcane (Saccharum spp.). Front. Plant Sci. 2022, 13, 923069. [Google Scholar] [CrossRef]
- Perochon, A.; Jianguang, J.; Kahla, A.; Arunachalam, C.; Scofield, S.R.; Bowden, S.; Wallington, E.; Doohan, F.M. TaFROG Encodes a Pooideae Orphan Protein That Interacts with SnRK1 and Enhances Resistance to the Mycotoxigenic Fungus Fusarium graminearum. Plant Physiol. 2015, 169, 2895–2906. [Google Scholar] [CrossRef]
- Jiang, C.; Hei, R.; Yang, Y.; Zhang, S.; Wang, Q.; Wang, W.; Zhang, Q.; Yan, M.; Zhu, G.; Huang, P. An orphan protein of Fusarium graminearum modulates host immunity by mediating proteasomal degradation of TaSnRK1α. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Fang, H.; Shen, S.; Wang, D.; Zhang, F.; Zhang, C.; Wang, Z.; Zhou, Q.; Wang, R.; Tao, H.; He, F.; et al. A monocot-specific hydroxycinnamoylputrescine gene cluster contributes to immunity and cell death in rice. Sci. Bull. 2021, 66, 2381–2393. [Google Scholar] [CrossRef]
- Brennan, C.J.; Zhou, B.; Benbow, H.R.; Ajaz, S.; Karki, S.J.; Hehir, J.G.; O’Driscoll, A.; Feechan, A.; Mullins, E.; Doohan, F.M. Taxonomically Restricted Wheat Genes Interact With Small Secreted Fungal Proteins and Enhance Resistance to Septoria tritici Blotch Disease. Front. Plant Sci. 2020, 11, 433. [Google Scholar] [CrossRef]
- Song, J.M.; Guan, Z.; Hu, J.; Guo, C.; Yang, Z.; Wang, S.; Liu, D.; Wang, B.; Lu, S.; Zhou, R.; et al. Eight high-quality genomes reveal pan-genome architecture and ecotype differentiation of Brassica napus. Nat. Plants 2020, 6, 34–45. [Google Scholar] [CrossRef]
- Lu, K.; Wei, L.; Li, X.; Wang, Y.; Wu, J.; Liu, M.; Zhang, C.; Chen, Z.; Xiao, Z.; Jian, H.; et al. Whole-genome resequencing reveals Brassica napus origin and genetic loci involved in its improvement. Nat. Commun. 2019, 10, 1154. [Google Scholar] [CrossRef]
- Tan, Z.; Han, X.; Dai, C.; Lu, S.; He, H.; Yao, X.; Chen, P.; Yang, C.; Zhao, L.; Yang, Q.Y.; et al. Functional genomics of Brassica napus: Progresses, challenges, and perspectives. J Integr. Plant Biol. 2024, 66, 484–509. [Google Scholar] [CrossRef]
- Wu, D.; Liang, Z.; Yan, T.; Xu, Y.; Xuan, L.; Tang, J.; Zhou, G.; Lohwasser, U.; Hua, S.; Wang, H.; et al. Whole-Genome Resequencing of a Worldwide Collection of Rapeseed Accessions Reveals the Genetic Basis of Ecotype Divergence. Mol. Plant 2019, 12, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Guan, Z.; Jiao, Y.; Liu, K.; Hong, D. The story of a decade: Genomics, functional genomics, and molecular breeding in Brassica napus. Plant Commun. 2024, 5, 100884. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xiao, H.; Lv, X.; Wang, D.; Chen, H.; Wei, F. Comprehensive review of composition distribution and advances in profiling of phenolic compounds in oilseeds. Front. Nutr. 2022, 9, 1044871. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Y.; Lu, H.Q.; Jiang, K.X.; Wang, Y.R.; Wang, Y.P.; Jiang, J.J. The Flavonoid Biosynthesis and Regulation in Brassica napus: A Review. Int. J. Mol. Sci. 2022, 24, 357. [Google Scholar] [CrossRef]
- Shen, J.; Liu, Y.; Wang, X.; Bai, J.; Lin, L.; Luo, F.; Zhong, H. A Comprehensive Review of Health-Benefiting Components in Rapeseed Oil. Nutrients 2023, 15, 999. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, S.; Wei, L.; Huang, Y.; Liu, D.; Jia, Y.; Luo, C.; Lin, Y.; Liang, C.; Hu, Y.; et al. BnIR: A multi-omics database with various tools for Brassica napus research and breeding. Mol. Plant 2023, 16, 775–789. [Google Scholar] [CrossRef]
- Zou, J.; Mao, L.; Qiu, J.; Wang, M.; Jia, L.; Wu, D.; He, Z.; Chen, M.; Shen, Y.; Shen, E.; et al. Genome-wide selection footprints and deleterious variations in young Asian allotetraploid rapeseed. Plant Biotechnol. J. 2019, 17, 1998–2010. [Google Scholar] [CrossRef]
- Sun, F.; Fan, G.; Hu, Q.; Zhou, Y.; Guan, M.; Tong, C.; Li, J.; Du, D.; Qi, C.; Jiang, L.; et al. The high-quality genome of Brassica napus cultivar ‘ZS11’ reveals the introgression history in semi-winter morphotype. Plant J. 2017, 92, 452–468. [Google Scholar] [CrossRef]
- Jiang, J.; Li, R.; Wang, K.; Xu, Y.; Lu, H.; Zhang, D. Combined Bulked Segregant Analysis-Sequencing and Transcriptome Analysis to Identify Candidate Genes Associated with Cold Stress in Brassica napus L. Int. J. Mol. Sci. 2025, 26, 1148. [Google Scholar] [CrossRef]
- Liu, X.; Wang, T.; Ruan, Y.; Xie, X.; Tan, C.; Guo, Y.; Li, B.; Qu, L.; Deng, L.; Li, M.; et al. Comparative Metabolome and Transcriptome Analysis of Rapeseed (Brassica napus L.) Cotyledons in Response to Cold Stress. Plants 2024, 13, 2212. [Google Scholar] [CrossRef]
- Waseem, M.; Peng, J.; Basharat, S.; Peng, Q.; Li, Y.; Yang, G.; Cheng, S.; Liu, P. A comprehensive analysis of transcriptomic data for comparison of cold tolerance in two Brassica napus genotypes. Physiol. Plant 2024, 176, e14213. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wu, Y.; Li, J.; Wang, X.; Zeng, Z.; Xu, J.; Liu, Y.; Feng, J.; Chen, H.; He, Y.; et al. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Raza, A.; Su, W.; Hussain, M.A.; Mehmood, S.S.; Zhang, X.; Cheng, Y.; Zou, X.; Lv, Y. Integrated Analysis of Metabolome and Transcriptome Reveals Insights for Cold Tolerance in Rapeseed (Brassica napus L.). Front. Plant Sci. 2021, 12, 721681. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Liu, J.; Huang, S.; Guo, T.; Deng, L.; Hua, W. Selection and evaluation of novel reference genes for quantitative reverse transcription PCR (qRT-PCR) based on genome and transcriptome data in Brassica napus L. Gene 2014, 538, 113–122. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Dong, X.-M.; Pu, X.-J.; Zhou, S.-Z.; Li, P.; Luo, T.; Chen, Z.-X.; Chen, S.-L.; Liu, L. Orphan gene PpARDT positively involved in drought tolerance potentially by enhancing ABA response in Physcomitrium (Physcomitrella) patens. Plant Sci. 2022, 319, 111222. [Google Scholar] [CrossRef]
- Li, G.; Wu, X.; Hu, Y.; Muñoz-Amatriaín, M.; Luo, J.; Zhou, W.; Wang, B.; Wang, Y.; Wu, X.; Huang, L.; et al. Orphan genes are involved in drought adaptations and ecoclimatic-oriented selections in domesticated cowpea. J. Exp. Bot. 2019, 70, 3101–3110. [Google Scholar] [CrossRef]
- Jin, G.-T.; Xu, Y.-C.; Hou, X.-H.; Jiang, J.; Li, X.-X.; Xiao, J.-H.; Bian, Y.-T.; Gong, Y.-B.; Wang, M.-Y.; Zhang, Z.-Q.; et al. A de novo Gene Promotes Seed Germination Under Drought Stress in Arabidopsis. Mol. Biol. Evol. 2025, 42, msae262. [Google Scholar] [CrossRef]
- Cheng, L.; Han, Q.; Hao, Y.; Qiao, Z.; Li, M.; Liu, D.; Yin, H.; Li, T.; Long, W.; Luo, S.; et al. Genome assembly of Stewartia sinensis reveals origin and evolution of orphan genes in Theaceae. Commun. Biol. 2025, 8, 354. [Google Scholar] [CrossRef]
- Cai, J.J.; Petrov, D.A. Relaxed purifying selection and possibly high rate of adaptation in primate lineage-specific genes. Genome Biol. Evol. 2010, 2, 393–409. [Google Scholar] [CrossRef]
- Lin, H.; Moghe, G.; Ouyang, S.; Iezzoni, A.; Shiu, S.H.; Gu, X.; Buell, C.R. Comparative analyses reveal distinct sets of lineage-specific genes within Arabidopsis thaliana. BMC Evol. Biol. 2010, 10, 41. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.; Jeong, A.R.; Kwon, O.-K.; Park, C.-J. Oryza-Specific Orphan Protein Triggers Enhanced Resistance to Xanthomonas oryzae pv. oryzae in Rice. Front. Plant Sci. 2022, 13, 859375. [Google Scholar] [CrossRef] [PubMed]
- Beike, A.K.; Lang, D.; Zimmer, A.D.; Wüst, F.; Trautmann, D.; Wiedemann, G.; Beyer, P.; Decker, E.L.; Reski, R. Insights from the cold transcriptome of Physcomitrella patens: Global specialization pattern of conserved transcriptional regulators and identification of orphan genes involved in cold acclimation. New Phytol. 2015, 205, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Kosová, K.; Vítámvás, P.; Urban, M.O.; Klíma, M.; Roy, A.; Prášil, I.T. Biological Networks Underlying Abiotic Stress Tolerance in Temperate Crops--A Proteomic Perspective. Int. J. Mol. Sci. 2015, 16, 20913–20942. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene IDs | Forward Primer | Reverse Primer |
|---|---|---|
| BnaOG1 | AGAGTGAGGGAGAGAGAGCG | TCAGGTCCTCTCCCCCCG |
| BnaOG2 | CCCCTTTCCCTTCGATTGAGAA | CAGTAGCATGGTCACCTTTCCT |
| BnaOG3 | ACCAAACATGGGTCATCTCCTC | AGGAGAGAGATGGCCCTTTACT |
| BnaOG4 | ACCACCAGAGCCTAAACTGATG | CTGGTTCTTGTTGCTCGAGTTG |
| BnaC06G0110800ZS | TGTGTTTCTTCCTCCCTCATCT | CCAGCAAGAAGACAGGTCAAGA |
| BnaA03G0334200ZS | AGGCGTTGGATTTCCATCTCTT | GAGCCAAACACCACTTGAAGAC |
| BnaA03G0429000ZS | AGAGACTAACCCTCCAGAGACC | CTCGTTTCTGCCTCTCTCCAAT |
| BnaA05G0385700ZS | TCATTGGAGTGAGGTCTTTCCG | AGAGCCACATCTTGATGACGTT |
| BnaC06G0163900ZS | GGCTCAAGGGAGGTGATAGATG | CCTTCAAACCGGCAAGAAAGTT |
| BnaPPR | TGGTGTGCGATAAGTGTGAGA | GGTGTCCATCTGTTCTTCTTGG |
| BnaGDI1 | GAGTCCCTTGCTCGTTTCC | TGGCAGTCTCTCCCTCAGAT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lang, H.; Zhang, Y.; Wang, B.; Li, K.; Jiang, M. Characterization and Expression Profiling of Orphan Genes in Rapeseed (Brassica napus) Provide Insights into Tissue Development and Cold Stress Adaptation. Horticulturae 2025, 11, 826. https://doi.org/10.3390/horticulturae11070826
Lang H, Zhang Y, Wang B, Li K, Jiang M. Characterization and Expression Profiling of Orphan Genes in Rapeseed (Brassica napus) Provide Insights into Tissue Development and Cold Stress Adaptation. Horticulturae. 2025; 11(7):826. https://doi.org/10.3390/horticulturae11070826
Chicago/Turabian StyleLang, Hong, Yuting Zhang, Baofeng Wang, Kexin Li, and Mingliang Jiang. 2025. "Characterization and Expression Profiling of Orphan Genes in Rapeseed (Brassica napus) Provide Insights into Tissue Development and Cold Stress Adaptation" Horticulturae 11, no. 7: 826. https://doi.org/10.3390/horticulturae11070826
APA StyleLang, H., Zhang, Y., Wang, B., Li, K., & Jiang, M. (2025). Characterization and Expression Profiling of Orphan Genes in Rapeseed (Brassica napus) Provide Insights into Tissue Development and Cold Stress Adaptation. Horticulturae, 11(7), 826. https://doi.org/10.3390/horticulturae11070826