
Genome-Wide Identification and Expression Analysis of the Mango (Mangifera indica L.) SWEET Gene Family

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Identification of Mango (Mangifera indica L.) SWEET Genes
2.2. Protein Physical and Chemical Properties Analysis
2.3. Phylogenetic Tree Construction
2.4. Gene Structure and Conserved Motif Analysis
2.5. Chromosome Localization and Covariance Analysis
2.6. Analysis of Promoter Cis-Elements
2.7. Specific Expression Analysis
2.8. Transcription Factor Prediction
3. Results
3.1. Identification of SWEET Family Members and Analysis of Physicochemical Properties of Mango
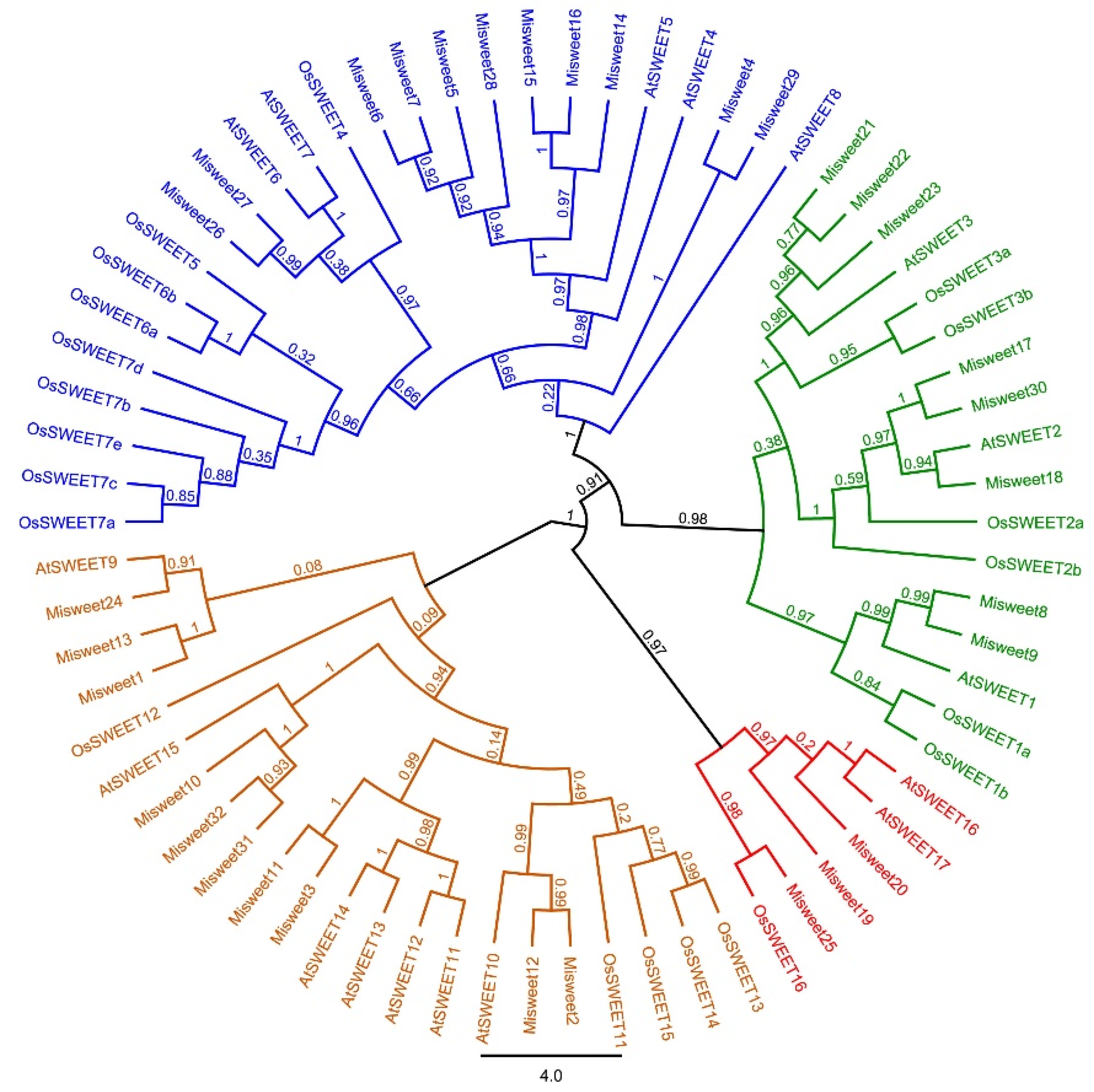
3.2. Phylogenetic Analysis of Mango SWEET Gene Family Members
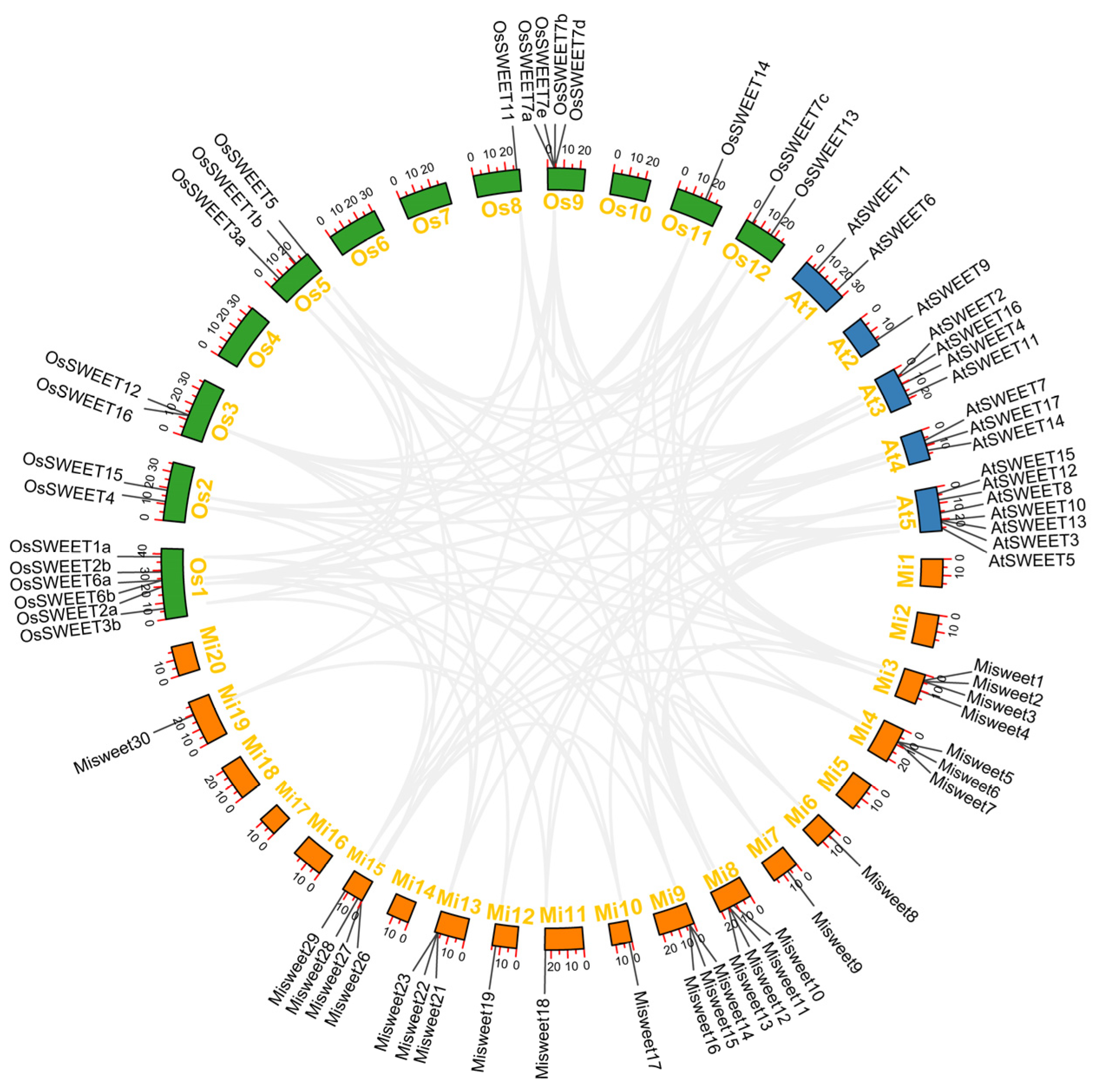
3.3. Chromosomal Localization and Collinearity Analysis of the Mango SWEET Gene
3.4. Motif, Conserved Domain, and Gene Structure Analysis of the Mango SWEET Family
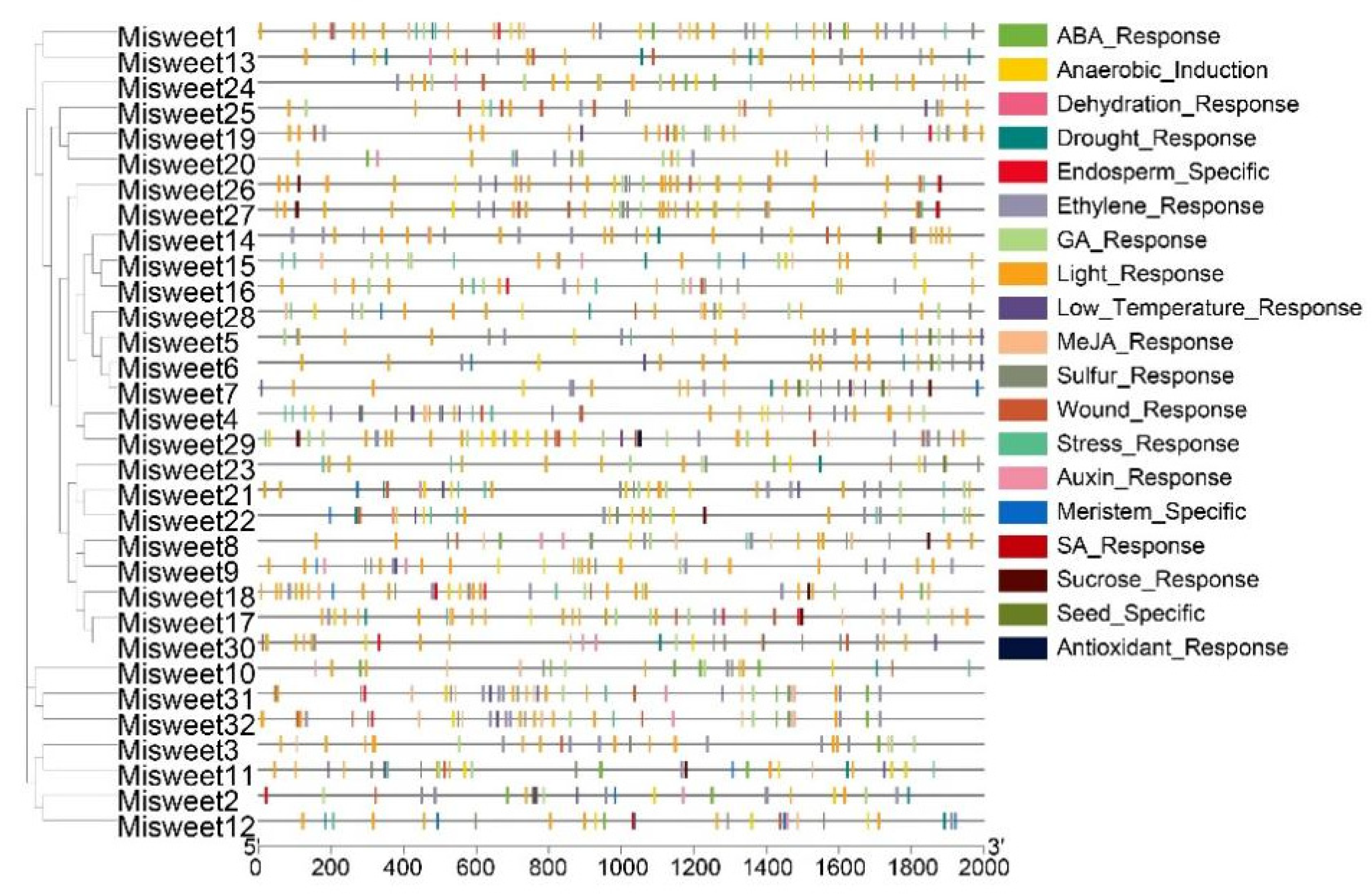
3.5. Prediction Analysis of Promoter Elements in the Mango SWEET Gene Family
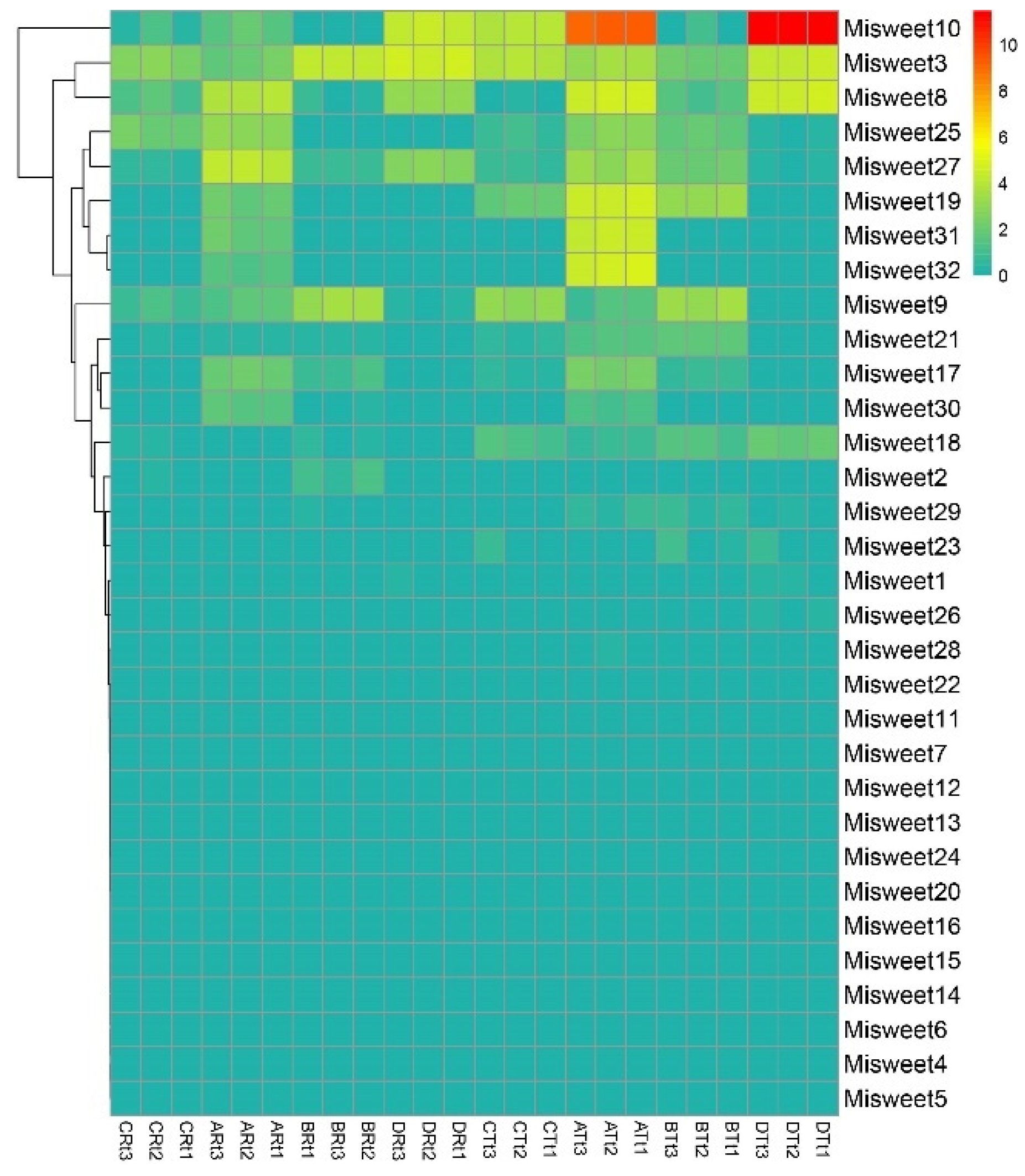
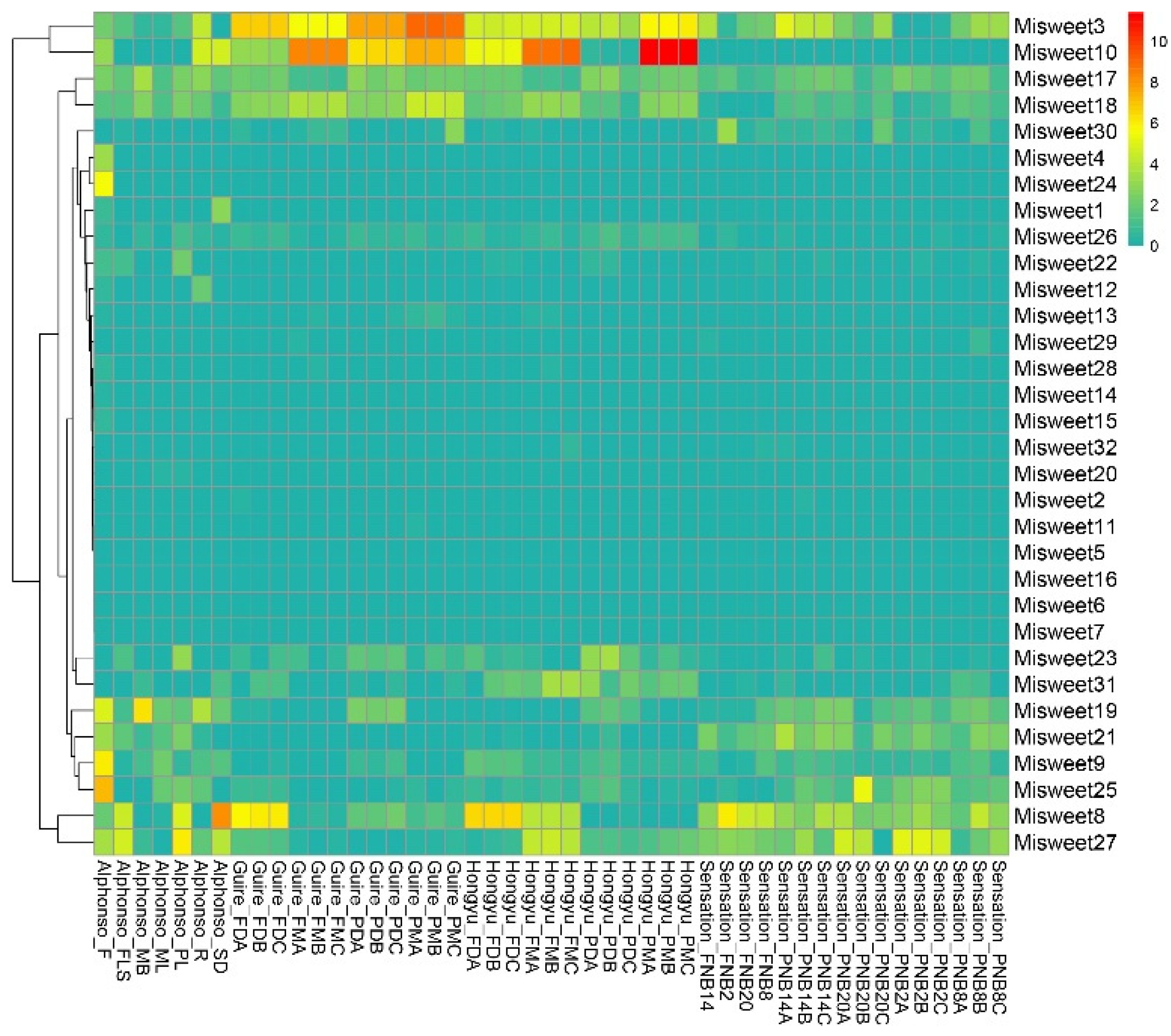
3.6. Expression Profile Analysis of the Mango SWEET Gene in Different Fruit Varieties and at Various Developmental Stages
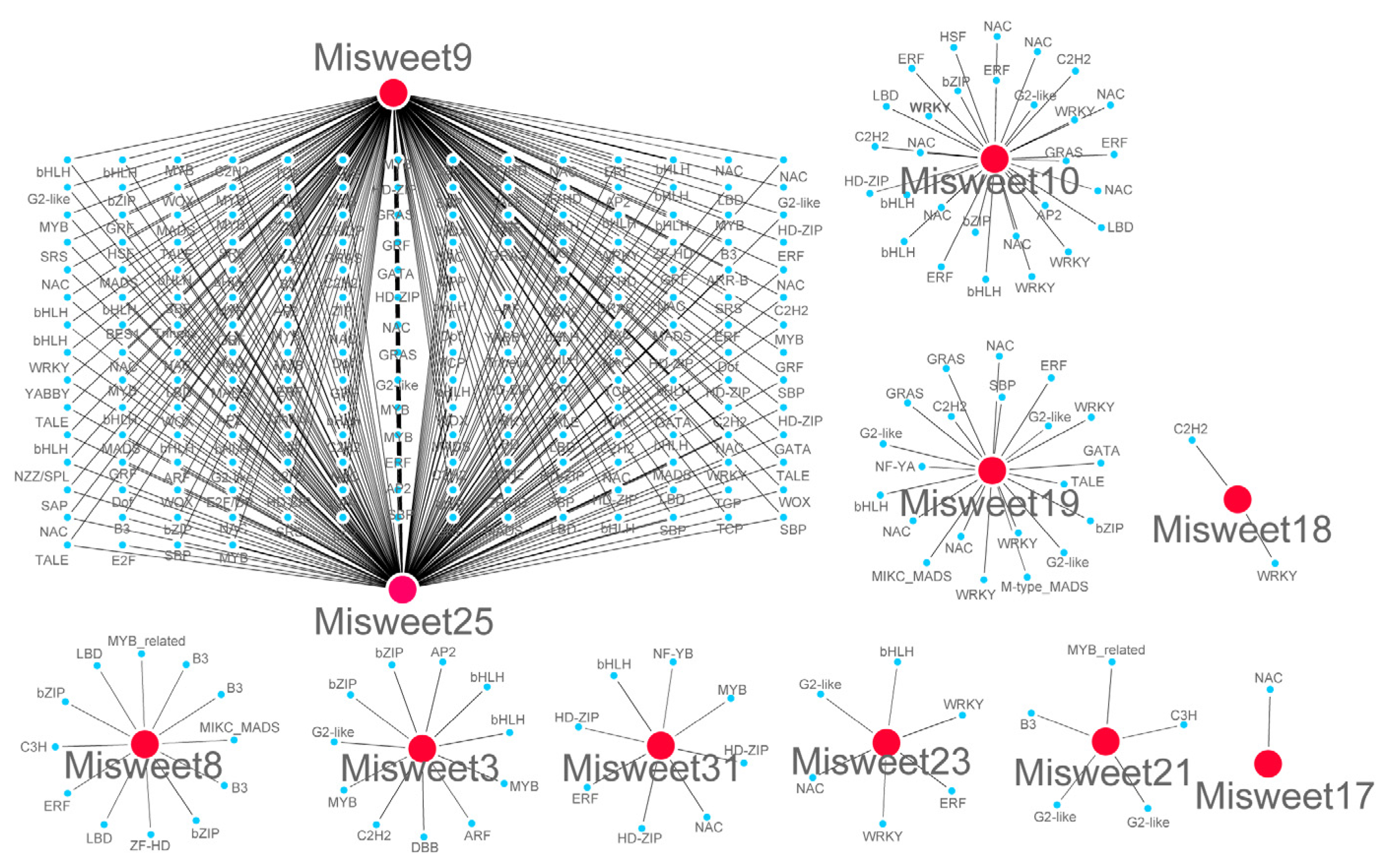
3.7. Analysis of the Mango SWEET Transcription Factor Regulatory Network
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sharan, M.B.; Mohan, S.; Ashverya, L. Role of Sugar and Auxin Crosstalk in Plant Growth and Development. Physiol. Plant. 2021, 174, e13546. [Google Scholar]
- Geigenberger, P. Regulation of starch biosynthesis in response to a fluctuating environment. Plant Physiol. 2011, 155, 1566–1577. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.J.; Foyer, C.H. Sink regulation of photosynthesis. J. Exp. Bot. 2001, 52, 1383–1400. [Google Scholar] [CrossRef]
- Ayre, B.G. Membrane-transport systems for sucrose in relation to whole-plant carbon partitioning. Mol. Plant 2011, 4, 377–394. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Gao, H.; Zhang, L.; Mao, D.; Li, Y.; Zhang, L.; Li, J.; Zhao, X.; Hou, H. Genome-wide identification and expression profiling of MST, SUT and SWEET transporters in Dendrobium catenatum. BMC Genomics 2024, 25, 1213. [Google Scholar] [CrossRef]
- Ji, J.; Yang, L.; Fang, Z.; Zhang, Y.; Zhuang, M.; Lv, H.; Wang, Y. Plant SWEET family of sugar transporters: Structure, evolution and biological functions. Biomolecules 2022, 12, 205. [Google Scholar] [CrossRef]
- Yuan, M.; Wang, S. Rice MtN3/saliva/SWEET family genes and their homologs in cellular organisms. Mol. Plant 2013, 6, 665–674. [Google Scholar] [CrossRef]
- Wu, Y.; Wu, P.; Xu, S.; Chen, Y.; Li, M.; Wu, G.; Jiang, H. Genome-wide identification, expression patterns and sugar transport of the physic nut SWEET gene family and a functional analysis of JcSWEET16 in Arabidopsis. Int. J. Mol. Sci. 2022, 23, 5391. [Google Scholar] [CrossRef]
- Patil, G.; Valliyodan, B.; Deshmukh, R.; Prince, S.; Nicander, B.; Zhao, M.; Sonah, H.; Song, L.; Lin, L.; Chaudhary, J. Soybean (Glycine max) SWEET gene family: Insights through comparative genomics, transcriptome profiling and whole genome re-sequence analysis. BMC Genomics 2015, 16, 520. [Google Scholar] [CrossRef]
- Li, M.; Xie, H.; He, M.; Su, W.; Yang, Y.; Wang, J.; Ye, G.; Zhou, Y. Genome-wide identification and expression analysis of the StSWEET family genes in potato (Solanum tuberosum L.). Genes Genomics 2020, 42, 135–153. [Google Scholar] [CrossRef]
- Geng, K.; Zhan, Z.; Xue, X.; Hou, C.; Li, D.; Wang, Z. Genome-wide identification of the SWEET gene family in grape (Vitis vinifera L.) and expression analysis of VvSWEET14a in response to water stress. Physiol. Mol. Biol. Plants 2024, 30, 1565–1579. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.-Y.; Han, J.-X.; Han, X.-X.; Jiang, J. Genome-wide identification, phylogeny, and expression analysis of the SWEET gene family in tomato. Gene 2015, 573, 261–272. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, S.; Ren, Y.; Gan, C.; Li, B.; Fan, Y.; Zhao, X.; Yuan, Z. Identification, analysis and gene cloning of the SWEET gene family provide insights into sugar transport in pomegranate (Punica granatum). Int. J. Mol. Sci. 2022, 23, 2471. [Google Scholar] [CrossRef] [PubMed]
- Fang, T.; Rao, Y.; Wang, M.; Li, Y.; Liu, Y.; Xiong, P.; Zeng, L. Characterization of the SWEET gene family in longan (Dimocarpus longan) and the role of DlSWEET1 in cold tolerance. Int. J. Mol. Sci. 2022, 23, 8914. [Google Scholar] [CrossRef]
- Xie, H.; Wang, D.; Qin, Y.; Ma, A.; Fu, J.; Qin, Y.; Hu, G.; Zhao, J. Genome-wide identification and expression analysis of SWEET gene family in Litchi chinensis reveal the involvement of LcSWEET2a/3b in early seed development. BMC Plant Biol. 2019, 19, 499. [Google Scholar] [CrossRef] [PubMed]
- Ahiakpa, J.K.; Karikari, B.; Magdy, M.; Munir, S.; Mumtaz, M.A.; Li, F.; Wang, Y.; Shang, L.; Zhang, Y. Regulation of invertase and sucrose for improving tomato fruit flavor: A review. Veg. Res. 2021, 1, 10. [Google Scholar] [CrossRef]
- Ren, Y.; Li, M.; Guo, S.; Sun, H.; Zhao, J.; Zhang, J.; Liu, G.; He, H.; Tian, S.; Yu, Y. Evolutionary gain of oligosaccharide hydrolysis and sugar transport enhanced carbohydrate partitioning in sweet watermelon fruits. Plant Cell 2021, 33, 1554–1573. [Google Scholar] [CrossRef]
- Li, J.; Qin, M.; Qiao, X.; Cheng, Y.; Li, X.; Zhang, H.; Wu, J. A new insight into the evolution and functional divergence of SWEET transporters in Chinese white pear (Pyrus bretschneideri). Plant Cell Physiol. 2017, 58, 839–850. [Google Scholar] [CrossRef]
- Zhang, X.; Feng, C.; Wang, M.; Li, T.; Liu, X.; Jiang, J. Plasma membrane-localized SlSWEET7a and SlSWEET14 regulate sugar transport and storage in tomato fruits. Hortic. Res. 2021, 8, 242. [Google Scholar] [CrossRef]
- Hu, L.-P.; Zhang, F.; Song, S.-H.; Tang, X.-W.; Hui, X.; Liu, G.-M.; Yaqin, W.; He, H.-J. Genome-wide identification, characterization, and expression analysis of the SWEET gene family in cucumber. J. Integr. Agric. 2017, 16, 1486–1501. [Google Scholar] [CrossRef]
- Guo, C.; Li, H.; Xia, X.; Liu, X.; Yang, L. Functional and evolution characterization of SWEET sugar transporters in Ananas comosus. Biochem. Biophys. Res. Commun. 2018, 496, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, S.; Lin, X.; Fang, H.; Shi, Y.; Grierson, D.; Chen, K. Transcription factor CitERF16 is involved in citrus fruit sucrose accumulation by activating CitSWEET11d. Front. Plant Sci. 2021, 12, 809619. [Google Scholar] [CrossRef]
- Zhu, Y.; Tian, Y.; Han, S.; Wang, J.; Liu, Y.; Yin, J. Structure, evolution, and roles of SWEET proteins in growth and stress responses in plants. Int. J. Biol. Macromol. 2024, 263, 130441. [Google Scholar] [CrossRef]
- Tharanathan, R.; Yashoda, H.; Prabha, T. Mango (Mangifera indica L.), “The king of fruits”—An overview. Food Rev. Int. 2006, 22, 95–123. [Google Scholar] [CrossRef]
- Wang, P.; Luo, Y.; Huang, J.; Gao, S.; Zhu, G.; Dang, Z.; Gai, J.; Yang, M.; Zhu, M.; Zhang, H. The genome evolution and domestication of tropical fruit mango. Genome Biol. 2020, 21, 60. [Google Scholar] [CrossRef]
- Ghassan, Z.; Yıldız, A.K.; Flavien, S.; Sara, I.; Azhar, N.M. Recent progress in omics and biotechnological approaches for improved mango cultivars in Pakistan. Genet. Resour. Crop Evol. 2022, 69, 2047–2065. [Google Scholar]
- Burton-Freeman, B.M.; Sandhu, A.K.; Edirisinghe, I. Mangos and their bioactive components: Adding variety to the fruit plate for health. Food Funct. 2017, 8, 3010–3032. [Google Scholar] [CrossRef] [PubMed]
- Hor, S.; Léchaudel, M.; Mith, H.; Bugaud, C. Fruit density: A reliable indicator of sensory quality for mango. Sci. Hortic. 2020, 272, 109548. [Google Scholar] [CrossRef]
- Datir, S.; Regan, S. Advances in physiological, transcriptomic, proteomic, metabolomic, and molecular genetic approaches for enhancing mango fruit quality. J. Agric. Food Chem. 2022, 71, 20–34. [Google Scholar] [CrossRef]
- Liang, Q.; Pan, H.; He, X.; Wang, S.; Hou, Y.; Xiao, H.; Xu, G.; Yi, R.; Lin, D.; Yang, Z. Population structure and genetic diversity of mango (Mangifera indica L.) germplasm resources as revealed by single-nucleotide polymorphism markers. Front. Plant Sci. 2024, 15, 1328126. [Google Scholar] [CrossRef]
- Ma, X.; Wu, H.; Liu, B.; Wang, S.; Zhang, Y.; Su, M.; Zheng, B.; Pan, H.; Du, B.; Wang, J. Genomic diversity, population structure, and genome-wide association reveal genetic differentiation and trait improvements in mango. Hortic. Res. 2024, 11, uhae153. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Wang, H.; Fan, Z.; Huang, H.; Ma, H. Advances in sequencing and key character analysis of mango (Mangifera indica L.). Hortic. Res. 2023, 10, uhac259. [Google Scholar] [CrossRef] [PubMed]
- Alghanem, S.M.; Alnusairi, G.S.; Alkhateeb, M.A.; Alwutayd, K.M.; Alaklabi, A.; Alharbi, B.M.; Albalawi, D.A.; Alshehri, D.; Al-Harbi, N.A.; Al-Qahtani, S.M. Genome-wide identification and characterization of the dof gene family in mango (Mangifera indica L.). Genet. Resour. Crop Evol. 2024, 71, 2749–2765. [Google Scholar] [CrossRef]
- Shi, B.; Wu, H.; Zhu, W.; Zheng, B.; Wang, S.; Zhou, K.; Qian, M. Genome-wide identification and expression analysis of WRKY genes during anthocyanin biosynthesis in the mango (Mangifera indica L.). Agriculture 2022, 12, 821. [Google Scholar] [CrossRef]
- Li, L.; Luo, C.; Huang, F.; Liu, Z.; An, Z.; Dong, L.; He, X. Identification and characterization of the mango eIF gene family reveals MieIF1A-a, which confers tolerance to salt stress in transgenic Arabidopsis. Sci. Hortic. 2019, 248, 274–281. [Google Scholar] [CrossRef]
- Lei, C.; Dang, Z.; Zhu, M.; Zhang, M.; Wang, H.; Chen, Y.; Zhang, H. Identification of the ERF gene family of Mangifera indica and the defense response of MiERF4 to Xanthomonas campestris pv. Mangiferaeindicae. Gene 2024, 912, 148382. [Google Scholar] [CrossRef]
- Peng, L.; Gao, W.; Song, M.; Li, M.; He, D.; Wang, Z. Integrated metabolome and transcriptome analysis of fruit flavor and carotenoids biosynthesis differences between mature-green and tree-ripe of cv. “Golden Phoenix” mangoes (Mangifera indica L.). Front. Plant Sci. 2022, 13, 816492. [Google Scholar] [CrossRef]
- Hu, W.-L.; Luo, C.; Xia, L.-M.; Liang, R.-Z.; Zhu, J.-W.; Li, Y.-Z.; Zhang, Y.-L.; Lan, M.-Y.; Liu, Y.; Nong, S.-F. Genome-wide identification of the mango auxin response factor family and the ectopic expression of two ARF (MiARF18A) genes confers early flowering and increases silique number in transgenic Arabidopsis. Sci. Hortic. 2025, 342, 114017. [Google Scholar] [CrossRef]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef]
- Duvaud, S.; Gabella, C.; Lisacek, F.; Stockinger, H.; Ioannidis, V.; Durinx, C. Expasy, the Swiss Bioinformatics Resource Portal, as designed by its users. Nucleic Acids Res. 2021, 49, W216–W227. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; Von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef]
- Yu, C.S.; Chen, Y.C.; Lu, C.H.; Hwang, J.K. Prediction of protein subcellular localization. Proteins Struct. Funct. Bioinform. 2006, 64, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef]
- Chen, C.; Wu, Y.; Li, J.; Wang, X.; Zeng, Z.; Xu, J.; Liu, Y.; Feng, J.; Chen, H.; He, Y. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Higo, K.; Ugawa, Y.; Iwamoto, M.; Korenaga, T. Plant cis-acting regulatory DNA elements (PLACE) database: 1999. Nucleic Acids Res. 1999, 27, 297–300. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing (Version 3.6.1) [Computer Software]; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Jin, J.; Tian, F.; Yang, D.-C.; Meng, Y.-Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2016, 45, gkw982. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.; Gai, X.T.; Pu, Z.J.; Gao, Y.; Xuan, Y.H. From functional characterization to the application of SWEET sugar transporters in plant resistance breeding. J. Agric. Food Chem. 2022, 70, 5273–5283. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Cai, M.; Liu, J.; Jiang, X.; Liu, J.; Zhenxing, W.; Wang, Y.; Li, Y. Genome-wide identification and expression analyses of SWEET gene family reveal potential roles in plant development, fruit ripening and abiotic stress responses in cranberry (Vaccinium macrocarpon Ait). PeerJ 2024, 12, e17974. [Google Scholar] [CrossRef]
- Liu, J.; Jiang, X.; Yang, L.; Zhao, D.; Wang, Y.; Zhang, Y.; Sun, H.; Chen, L.; Li, Y. Characterization of the SWEET Gene Family in Blueberry (Vaccinium corymbosum L.) and the Role of VcSWEET6 Related to Sugar Accumulation in Fruit Development. Int. J. Mol. Sci. 2025, 26, 1055. [Google Scholar] [CrossRef]
- Xuan, C.; Lan, G.; Si, F.; Zeng, Z.; Wang, C.; Yadav, V.; Wei, C.; Zhang, X. Systematic genome-wide study and expression analysis of SWEET gene family: Sugar transporter family contributes to biotic and abiotic stimuli in watermelon. Int. J. Mol. Sci. 2021, 22, 8407. [Google Scholar] [CrossRef]
- Conklin, P.A.; Strable, J.; Li, S.; Scanlon, M.J. On the mechanisms of development in monocot and eudicot leaves. New Phytol. 2019, 221, 706–724. [Google Scholar] [CrossRef] [PubMed]
- Scarpella, E.; Meijer, A.H. Pattern formation in the vascular system of monocot and dicot plant species. New Phytol. 2004, 164, 209–242. [Google Scholar] [CrossRef]
- Jiang, S.; Balan, B.; Assis, R.d.A.; Sagawa, C.H.; Wan, X.; Han, S.; Wang, L.; Zhang, L.; Zaini, P.A.; Walawage, S.L. Genome-wide profiling and phylogenetic analysis of the SWEET sugar transporter gene family in walnut and their lack of responsiveness to Xanthomonas arboricola pv. Juglandis infection. Int. J. Mol. Sci. 2020, 21, 1251. [Google Scholar] [CrossRef]
- Gautam, T.; Saripalli, G.; Gahlaut, V.; Kumar, A.; Sharma, P.; Balyan, H.; Gupta, P. Further studies on sugar transporter (SWEET) genes in wheat (Triticum aestivum L.). Mol. Biol. Rep. 2019, 46, 2327–2353. [Google Scholar] [CrossRef]
- Miao, H.; Sun, P.; Liu, Q.; Miao, Y.; Liu, J.; Zhang, K.; Hu, W.; Zhang, J.; Wang, J.; Wang, Z. Genome-wide analyses of SWEET family proteins reveal involvement in fruit development and abiotic/biotic stress responses in banana. Sci. Rep. 2017, 7, 3536. [Google Scholar] [CrossRef]
- Wei, Y.; Xiao, D.; Zhang, C.; Hou, X. The expanded SWEET gene family following whole genome triplication in Brassica rapa. Genes 2019, 10, 722. [Google Scholar] [CrossRef] [PubMed]
- Leung, J.; Giraudat, J. Abscisic acid signal transduction. Annu. Rev. Plant Biol. 1998, 49, 199–222. [Google Scholar] [CrossRef] [PubMed]
- Marand, A.P.; Eveland, A.L.; Kaufmann, K.; Springer, N.M. cis-Regulatory elements in plant development, adaptation, and evolution. Annu. Rev. Plant Biol. 2023, 74, 111–137. [Google Scholar] [CrossRef]
- Xiao, Y.; Yi, F.; Ling, J.; Yang, G.; Lu, N.; Jia, Z.; Wang, J.; Zhao, K.; Wang, J.; Ma, W. Genome-wide analysis of lncRNA and mRNA expression and endogenous hormone regulation during tension wood formation in Catalpa bungei. BMC Genomics 2020, 21, 609. [Google Scholar] [CrossRef]
- Ntsoane, M.L.; Zude-Sasse, M.; Mahajan, P.; Sivakumar, D. Quality assesment and postharvest technology of mango: A review of its current status and future perspectives. Sci. Hortic. 2019, 249, 77–85. [Google Scholar] [CrossRef]
- Hir, R.L.; Spinner, L.; Klemens, P.A.W.; Chakraborti, D.; Marco, F.d.; Vilaine, F.; Wolff, N.; Lemoine, R.; Porcheron, B.; Géry, C.; et al. Disruption of the Sugar Transporters AtSWEET11 and AtSWEET12 Affects Vascular Development and Freezing Tolerance in Arabidopsis. Mol. Plant 2015, 8, 1687–1690. [Google Scholar]
- Urooj, F.; Anjali, A.; Muthappa, S.-K. AtSWEET11 and AtSWEET12: The twin traders of sucrose. Trends Plant Sci. 2022, 27, 958–960. [Google Scholar]
- Jiang, W.; Xueyi, X.; Houqing, Z.; Jiankun, L.; LiQing, C. Sucrose rather than GA transported by AtSWEET13 and AtSWEET14 supports pollen fitness at late anther development stages. New Phytol. 2022, 236, 525–537. [Google Scholar]
- Yuri, K.; Takaya, O.; Yasutaka, C.; Yasuhiro, I.; Takafumi, S.; Naoto, S.; Tomokazu, K.; Yuji, K.; Minoru, U.; Mitsunori, S. AtSWEET13 and AtSWEET14 regulate gibberellin-mediated physiological processes. Nat. Commun. 2016, 7, 13245. [Google Scholar]
- Gao, Y.; Yao, Y.; Chen, X.; Wu, J.; Wu, Q.; Liu, S.; Guo, A.; Zhang, X. Metabolomic and transcriptomic analyses reveal the mechanism of sweet-acidic taste formation during pineapple fruit development. Front. Plant Sci. 2022, 13, 971506. [Google Scholar] [CrossRef]
- Liu, H.-T.; Lyu, W.-Y.; Tian, S.-H.; Zou, X.-H.; Zhang, L.-Q.; Gao, Q.-H.; Ni, D.-A.; Duan, K. The SWEET family genes in strawberry: Identification and expression profiling during fruit development. S. Afr. J. Bot. 2019, 125, 176–187. [Google Scholar] [CrossRef]
- Wu, Y.; Lee, S.-K.; Yoo, Y.; Wei, J.; Kwon, S.-Y.; Lee, S.-W.; Jeon, J.-S.; An, G. Rice transcription factor OsDOF11 modulates sugar transport by promoting expression of sucrose transporter and SWEET genes. Mol. Plant 2018, 11, 833–845. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Gao, Z.; Wang, J.; Huang, Y.; Chen, Y.; Li, J.; Lv, M.; Wang, J.; Luo, M.; Zuo, K. Cotton fiber elongation requires the transcription factor Gh MYB 212 to regulate sucrose transportation into expanding fibers. New Phytol. 2019, 222, 864–881. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | ORF Length (bp) | Protein Length (aa) | Moleculer Weight (kD) | PI | Subcellular Localization | TMHs | GRAVY |
|---|---|---|---|---|---|---|---|
| Misweet1 | 813 | 270 | 30.21 | 8.89 | PM | 7 | 0.513 |
| Misweet2 | 861 | 286 | 32.06 | 9.18 | PM | 7 | 0.888 |
| Misweet3 | 1044 | 347 | 38.94 | 6.71 | PM | 6 | 0.157 |
| Misweet4 | 720 | 239 | 26.99 | 9.03 | PM | 7 | 0.588 |
| Misweet5 | 522 | 173 | 19.52 | 6.55 | PM | 5 | 0.762 |
| Misweet6 | 714 | 237 | 26.74 | 8.68 | PM | 7 | 0.705 |
| Misweet7 | 546 | 181 | 20.62 | 5.72 | PM | 5 | 0.806 |
| Misweet8 | 735 | 244 | 26.89 | 8.93 | PM | 7 | 0.762 |
| Misweet9 | 723 | 240 | 26.21 | 8.83 | PM | 7 | 0.736 |
| Misweet10 | 903 | 300 | 33.71 | 8.21 | PM | 7 | 0.652 |
| Misweet11 | 792 | 263 | 29.45 | 9.73 | PM | 7 | 0.58 |
| Misweet12 | 978 | 325 | 36.73 | 9.01 | PM | 7 | 0.711 |
| Misweet13 | 786 | 261 | 29.45 | 8.87 | PM | 7 | 0.671 |
| Misweet14 | 549 | 182 | 20.87 | 8.46 | PM | 5 | 0.674 |
| Misweet15 | 525 | 174 | 19.83 | 8.39 | PM | 5 | 0.67 |
| Misweet16 | 525 | 174 | 19.89 | 8.72 | PM | 5 | 0.614 |
| Misweet17 | 690 | 229 | 25.81 | 9.32 | PM | 7 | 0.903 |
| Misweet18 | 876 | 291 | 32.06 | 8.67 | PM | 7 | 0.76 |
| Misweet19 | 741 | 246 | 27.43 | 7.71 | PM | 7 | 0.484 |
| Misweet20 | 711 | 236 | 25.68 | 7.71 | PM | 7 | 0.698 |
| Misweet21 | 750 | 249 | 27.76 | 9.21 | PM | 7 | 0.573 |
| Misweet22 | 744 | 247 | 27.54 | 9.03 | PM | 7 | 0.554 |
| Misweet23 | 309 | 102 | 11.36 | 9.30 | PM | 3 | 1.039 |
| Misweet24 | 711 | 236 | 26.07 | 9.04 | PM | 6 | 0.619 |
| Misweet25 | 903 | 300 | 33.14 | 9.26 | PM | 6 | 0.382 |
| Misweet26 | 1029 | 342 | 38.23 | 8.55 | PM | 6 | 0.543 |
| Misweet27 | 771 | 256 | 27.93 | 9.44 | PM | 6 | 0.923 |
| Misweet28 | 717 | 238 | 26.83 | 6.56 | PM | 7 | 0.695 |
| Misweet29 | 771 | 256 | 28.59 | 8.66 | PM | 6 | 0.664 |
| Misweet30 | 708 | 235 | 26.05 | 8.93 | PM | 7 | 0.804 |
| Misweet31 | 894 | 297 | 33.16 | 6.96 | PM | 7 | 0.578 |
| Misweet32 | 894 | 297 | 33.15 | 6.96 | PM | 7 | 0.581 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, L.; Liu, X.; Leng, X.; Zhang, M.; Yang, Z.; Xu, W.; Wang, S.; Wu, H.; Liang, Q. Genome-Wide Identification and Expression Analysis of the Mango (Mangifera indica L.) SWEET Gene Family. Horticulturae 2025, 11, 675. https://doi.org/10.3390/horticulturae11060675
Zhou L, Liu X, Leng X, Zhang M, Yang Z, Xu W, Wang S, Wu H, Liang Q. Genome-Wide Identification and Expression Analysis of the Mango (Mangifera indica L.) SWEET Gene Family. Horticulturae. 2025; 11(6):675. https://doi.org/10.3390/horticulturae11060675
Chicago/Turabian StyleZhou, Lirong, Xinyu Liu, Xiangchi Leng, Meng Zhang, Zhuanying Yang, Wentian Xu, Songbiao Wang, Hongxia Wu, and Qingzhi Liang. 2025. "Genome-Wide Identification and Expression Analysis of the Mango (Mangifera indica L.) SWEET Gene Family" Horticulturae 11, no. 6: 675. https://doi.org/10.3390/horticulturae11060675
APA StyleZhou, L., Liu, X., Leng, X., Zhang, M., Yang, Z., Xu, W., Wang, S., Wu, H., & Liang, Q. (2025). Genome-Wide Identification and Expression Analysis of the Mango (Mangifera indica L.) SWEET Gene Family. Horticulturae, 11(6), 675. https://doi.org/10.3390/horticulturae11060675