1. Introduction
Pu-erh tea (PET) is a well-known fermented dark tea exclusively produced by the spontaneous fermentation of
Camellia assamica tea leaves [
1]. During the fermentation process, a series of chemical reactions are involved, such as decomposition, oxydoreduction, polymerzation, structural modification, methylation and glycosylation. The sensory quality and multiple health benefits of PET, including anti-diabetic [
2], anti-oxidative [
3], anti-cancerogenic [
4], anti-bacterial [
5], anti-inflammatory [
6] and free radical scavenging effects, are largely determined by microbial fermentation. Previous studies have reported that the chemicals of PET are hydrolyzed and conversed by microbial metabolites in hot and humid conditions [
7]. For instance, the content of catechin derivatives, flavonoids and their glycosides, phenolic acids, alkaloids and terpenoids is significantly changed during PET fermentation [
8]. The critical element involved in generating the bioactivity, quality and sensory characteristics of Pu-erh tea are interrelated with complex biochemical changes, which result from the microorganisms present and the metabolites during the fermentation process.
The natural solid-state fermentation (SSF) process is usually used for the manufacture of Pu-erh tea. In this process, sun-dried green tea leaves are moistened with water and fermented for a few weeks [
9]. Recently, both researchers and the agricultural industry have utilized microbial fermentation to increase the nutritional value, flavor and aroma of food. These products have been applied as feed additives to promote the performance of livestock [
10]. Traditional Pu-erh tea is fermented spontaneously using raw materials in hot and humid conditions without a seeding starter strain. The spontaneous fermentation process changes the microbial diversity and results in a rise in the abundance of certain microorganisms, such as
Aspergillus niger,
Rasamsonia emersonii and
Thermomyces lanuginosus [
11].
L. plantarum are generally recognized as safe and effective microorganisms that have been utilized in the processing of fermenting food for centuries. Previous studies showed that
L. plantarum strains isolated and identified from tea leaves could inhibit the growth of
Salmonella typhi,
Escherichia coli,
Staphylococcus aureus,
Enterococcus faecalis and
Citrobacter sp. [
12]. Furthermore,
L. plantarum produces extracellular tannase, which is beneficial for health. Both culture and culture-independent studies showed that
L. plantarum changes the tea leaf components, such as organic acids, free amino acids and catechins, during fermentation [
13].
S. cerevisiae, a unicellular fungus, is of great importance for various biotechnological applications relating to its fermentation ability, accompanied by the production of CO
2 and alcohol, and its tolerance to unfavorable conditions of osmolarity and low pH. The most prominent application involving the use of
S. cerevisiae is in food fermentation.
S. cerevisiae is a valuable tool for the fermentation process due to its “make–accumulate–consume” lifestyle [
14]. This results from the Crabtree effect, which consists of the fact that
S. cerevisiae does not use respiratory machinery to metabolize saccharides and instead produces ethanol and CO
2, even under aerobic conditions.
S. cerevisiae is the dominant sugar fermenter due to its remarkable resistance to high sugar levels and its production of different aromatic, volatile compounds. The function of
S. cerevisiae in beverage [
15], bread [
15,
16] and biofuel production [
17] has been characterized and explored. However, the roles of
S. cerevisiae in tea fermentation have not been investigated.
Both L. plantarum and S. cerevisiae are applied widely in food fermentation and the large-scope production of organic acids, enzymes and bioactive compounds. They produce many enzymes efficiently and these enzymes have been used commercially. Well-combined microorganism diversity during the fermentation process has the ability to control the metabolic outcome and therefore product quality. As such, it has been considered to combine L. plantarum and S. cerevisiae as a tea fermentation starter strain to give rise to the growth of diverse microorganisms and convert the tea compounds involved in the formation of the characteristic bioactive function. However, the relationships between the metabolites that characterize Pu-erh tea and a fermentation microbiome mainly composed of L. plantarum and S. cerevisiae have not been clarified. It is necessary to understand these relationships for the purpose of enhancing the quality of Pu-erh tea.
In this study, raw material tea, spontaneous fermentation tea and microbial fermentation tea were analyzed to investigate microbiome composition changes and the correlation between the microbiome and metabolites. The hundreds of endogenous metabolites from the Pu-erh tea fermentation process were characterized by the powerful metabolomics method combined with biochemical measurements. Multivariate analysis combined with microbiomics (high-throughput sequencing combined with qPCR) was applied to explore the metabolic potential in the microbial community. Moreover, the changes in functional compounds in Pu-erh tea during the manufacturing process were discussed. Furthermore, the intricate relationship between the effective microorganisms, metabolic pathways and dominant functional compounds in Pu-erh tea was evaluated. L. plantarum in conjunction with S. cerevisiae as the starter strain applied in Pu-erh tea fermentation exhibited the potential to improve the tea quality.
2. Materials and Methods
2.1. Preparation of Tea Leaves
Pu-erh tea raw materials (sun-dried green tea leaves) were obtained from the Yunnan LongRun Tea Industry Group (Yunnan, Kunming, China). A haploid control S. cerevisiae strain (BY4741) was obtained from Open Biosystems. Synthetic complete (SC) medium with 1.5% agar was added to culture S. cerevisiae in plates at 30 °C. L. plantarum (1.16089) were donated by the Institute of Microbiology, Chinese Academy of Sciences.
The Pu-erh tea raw materials (100 g) were mixed with distilled water (30 mL) to produce spontaneous fermentation tea (SFT). Li-mupirocin-modified MRS medium containing L. plantarum and S. cerevisiae (10 mL, 1 × 108 Cells/mL) was sprayed on sun-dried tea leaves (100 g) and fermented at 37 °C for 3 weeks; these samples were named microbial fermentation tea (MFT).
2.2. Chemicals
The standard reagents involved in tea metabolites were purchased from the Yuanye Biotechnology Company (Shanghai, China). Acetonitrile, formic acid and methanol of LC–MS grade were obtained from Sigma Aldrich (St. Louis, MO, USA). Other reagents (≥98%) were obtained from the China National Medicines Corporation Ltd. (Beijing, China).
2.3. Gene Sequencing and Quantitative PCR (qPCR) Detection
The FastDNA
® Spin Kit (MP Biomedicals, Norcross, GA, USA) was used to extract microbial DNA from tea samples according to the instructions. The extracted 16S rRNA gene in the V3-V4 region was amplified using forward primer 338F (5′-ACTCCTACGGGAGGCAGCA-3′) [
18] and reverse primer 806R (5′-GGACTACHVGGGT WTCTAAT-3′) [
19]. Before amplifying the DNA in triplicate, the sample was subjected to electrophoresis on 2% agarose gels. A total volume of 20 μL PCR products were purified with the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA). The QuantiFluotTM DNA Assay Kit (Pro-Mega, Madison, WI, USA) was applied for the quantification of the purified PCR products. Following quantification, the Illumina MiSeq system from Majorbio Bio-Pharm Technology Co. (Shanghai, China) was employed to identify the mixed sample amplicon sequencing.
The raw sequencing data were demultiplexed using Trimmomatic (v1.7.0) for denoising and trimming [
20], FLASH for quality filtering [
21], UPARSE for pairing [
22] and UCHIME for alignment [
23]. The UPARSE software (version 7.1
http://drive5.com/uparse/ accessed on 27 March 2022) clustered the resulting sequences into operational taxonomic units (OTUs) with a 97% similarity threshold. The RDP classifier (
http://rdp.cme.msu.edu/, accessed on 27 March 2022) assigned the OTUs from representative sequences in taxonomic information with a 70% threshold by comparison with the Silva database (SSU128). The Mothur software (version.1.47.0) developed by Dr. Patrick Schloss group (Michigan State University, Microbiological Sciences and Immunology, East Lansing, MI, USA), which was utilized to estimate the community richness index (CHAO index), community diversity (Shannon index) and the Good’s coverage of sequencing.
The ITS gene of
S. cerevisiae in a 188 bp fragment (091-279) was cloned using PCR. In the p416-TEF vector, specific primers (F: 5′-CGCGGATCCCCAGCCG GGCCTGCGCTTAAG, R: 5′- CCGCTCGAGCCTCTGGGCCCCGA TTGCTCG) were inserted into the BamHI and XhoI sites to produce the p416-TEF-ITS (188) plasmid for fungal standard curve generation. Meanwhile, the specific primers (F: 5′-CGCGGAT CCCGGCAGGCCTAACACATGCAAG, R: 5′-CCGCTCGAGGCATTTCACCG CTACACCTG) from a 659 bp fragment (031-690) of the E. coli 16s rDNA was inserted into the BamHI and XhoI sites in the p416-TEF vector. After confirming the gene sequences of all plasmids, the diluted plasmid DNA was used to generate standard curves for quantitative PCR (qTOWER 3.0G) [
24]. The molecular biology techniques followed previously established methods [
25]. Following quantitative PCR, TIANamp Soil DNA kit (Tiangen Biotech, Beijing, China) was used to extract DNA from the microorganisms for genomic analysis. The method of extracting microorganisms from different Pu-erh tea samples was based on our previous study [
11].
2.4. Metabolomics Analysis
First, 0.1 g tea powder was extracted using 3 mL 70% methanol for 30 min in an ultrasonic bath at 60 °C. The filtered extract liquid was subjected to metabolomics analysis using ultra-performance liquid chromatography coupled with electrospray time-of-flight mass spectrometry (UPLC-Q-TOF/MS) (waters, Milford, CT, USA). The chromatography separations were performed on a BEH C18 column (100 mm × 2.1 mm, 1.7 μm; Waters corporation, Milford, MA, USA). The column was eluted with water as mobile phase A (0.1% formic acid) and acetonitrile as mobile phase B (0.1% formic acid). The gradient sequence was as follows: 0–3 min, 5% B; 3–10 min, 20% B; 10–15 min, 100% B. The flow rate was 0.4 mL/min, column temperature 45 °C, injection volume 5 μL. The Q-TOF mass spectrometer was equipped with an electrospray ionization (ESI) source operating in negative ion mode, and the data were collected from 50 to 1000 m/s. The de-solvation temperature was 450 °C, and the source temperature was 115 °C, with a cone gas flow of 15 L/min. The capillary voltage, sampling cone voltage and collision energy were 2000 V, 40 V and 6 eV, respectively. Three independent extractions and analyses were performed.
The tea polyphenols, total amino acids, tea proteins and water-soluble sugars were extracted and quantitated using the method of Li et al. [
11]. The raw data obtained from LC–MS were initially processed using the MetaboAnalyst software (
https://www.metaboanalyst.ca/, accessed on 28 March 2022). The SIMCA-P software (version 14.1, Umetrics AB, Umea, Sweden) was applied to process the acquired data and evaluate the metabolite changes in different tea samples.
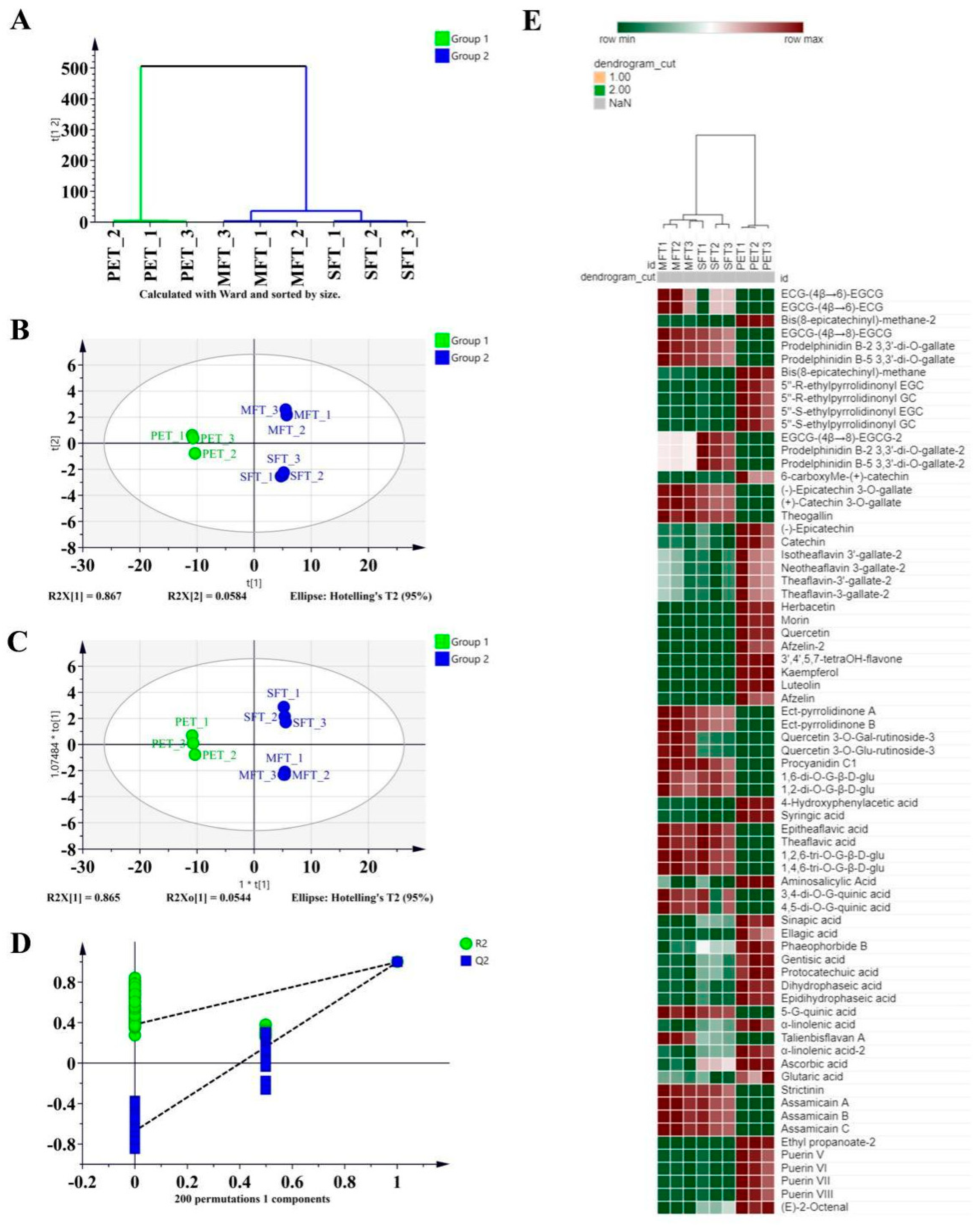
Hierarchical cluster analysis (HCA) was used to plot the respective dendrograms and their relationships. Unsupervised principal component analysis (PCA) distinguished the differences among the samples through the intrinsic variation from the collected data matrix. Meanwhile, supervised orthonormal partial least-squares discriminant analysis (OPLS-DA) was applied to classify samples of solely Y variables, and it was combined with a threshold of variable importance projection (VIP) > 1.16 to identify the critical metabolites that caused the metabolomic variations during the manufacturing process. The data were analyzed and a heat map was drawn through the website (
https://software.broadinstitute.org/morpheus/, accessed on 28 March 2022).
2.5. Statistical Analysis
All experiments were carried out in triplicate and the results were expressed as mean values followed by the standard deviation (n = 3). Differences in the relative abundance of operational taxonomic units (OTUs) were assessed using the Bray–Curtis distance, which was calculated using ANOSIM/Adonis dissimilarity analyses. The significance level of Pu-erh tea metabolites between different groups was calculated by one-way analysis of variance (ANOVA) with Dunnett’s multiple comparisons test using the GraphPad Prism 7.00 software (GraphPad Software Inc., San Diego, CA, USA). To estimate the community richness index (CHAO index) and community diversity (Shannon index), the statistical T-test method was used to determine the significant differences between samples. The linear correlation between the effects of microbes and the chemical composition of the tea samples was analyzed using the SMICA-P software (version 14.1, Umetrics AB, Umea, Sweden) and the probability values. p-values below 0.05 were considered significant.
4. Discussion
L. plantarum and
S. cerevisiae are critical microorganisms in the PET fermentation process, as they are the main contributors to enzyme secretion to change tea’s functional components [
12,
13,
14]. In our study, we compared the community modifications and metabolite changes in different manufactured PET samples by high-throughput Illumina MiSeq sequencing coupled with qPCR, HPLC–MS analysis, biochemical measurements and multivariate analysis. Fungal community analysis showed that
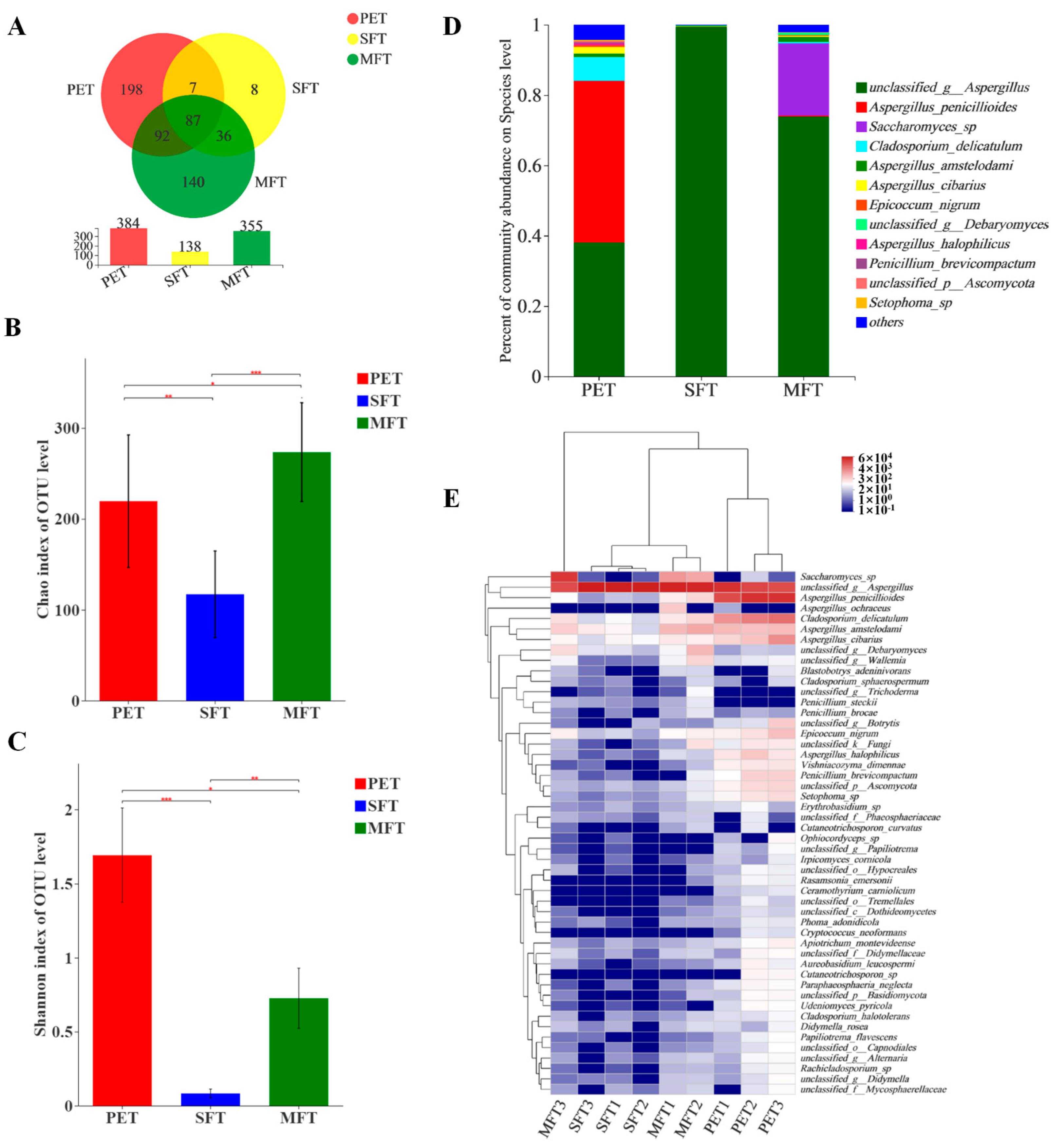
Aspergillus was dominant in the three samples.
Aspergillus was the predominant genus in the SFT sample, accounting for 99% of the total effective sequences. The
Aspergillus in different species are responsible for producing amylase, acid protease, cellulase, pectinase and glucose oxidase, which contribute to degrading macromolecular organic matter and transforming substances [
28,
29,
30].
A. penicillioid was the dominant species in the PET raw material, but was replaced by unclassified
Aspergillus after fermentation processing. Based on the culture experiment and observation, we conjectured that the increase in unclassified
Aspergillus was caused by
Eurotium cristatum. In addition, the
S. cerevisiae that were inoculated onto the tea leaves reproduced quickly in the MFT sample and competed with
A. penicillioides for nutrients, resulting in suppressed unclassified
Aspergillus growth during the fermentation.
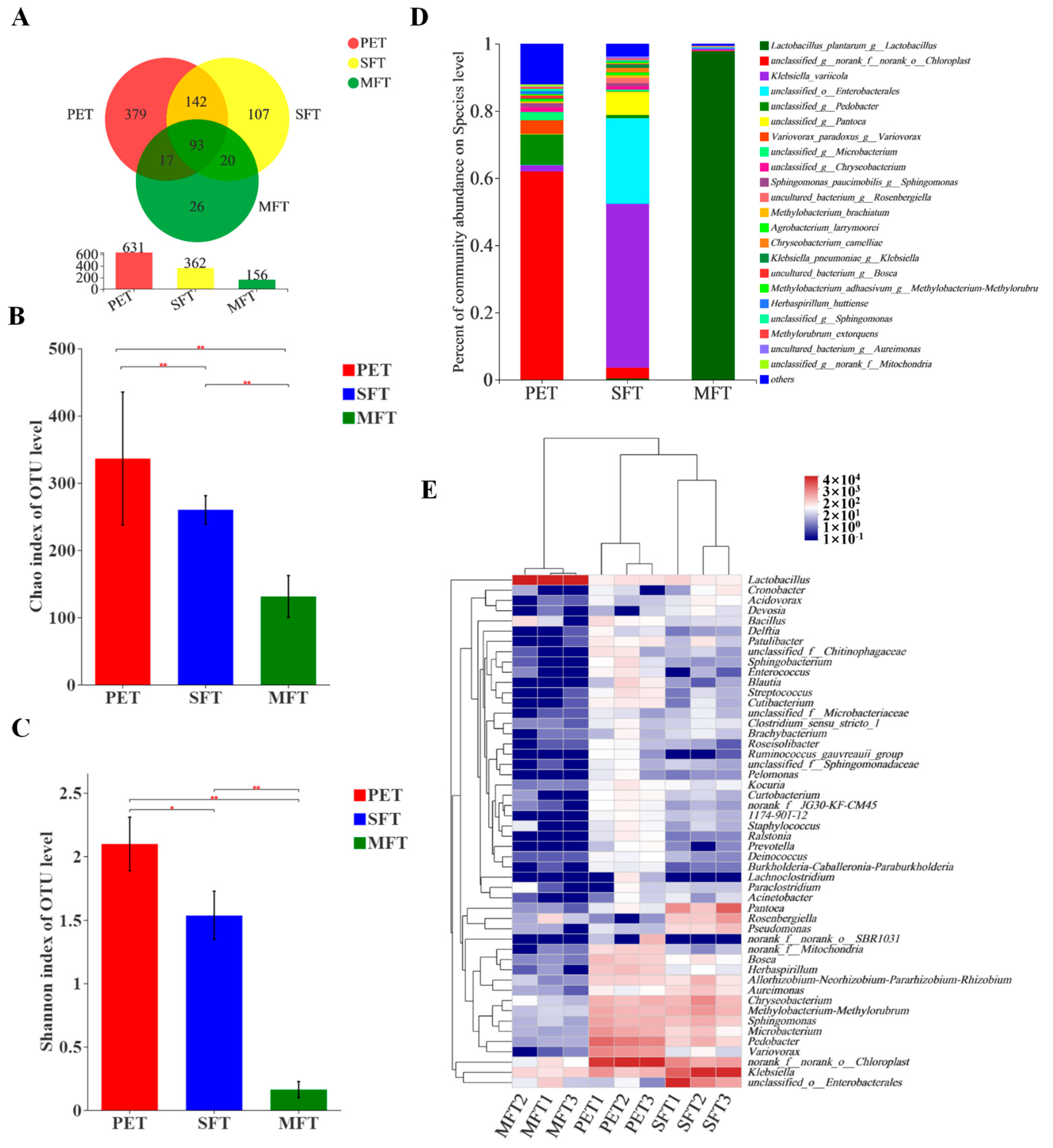
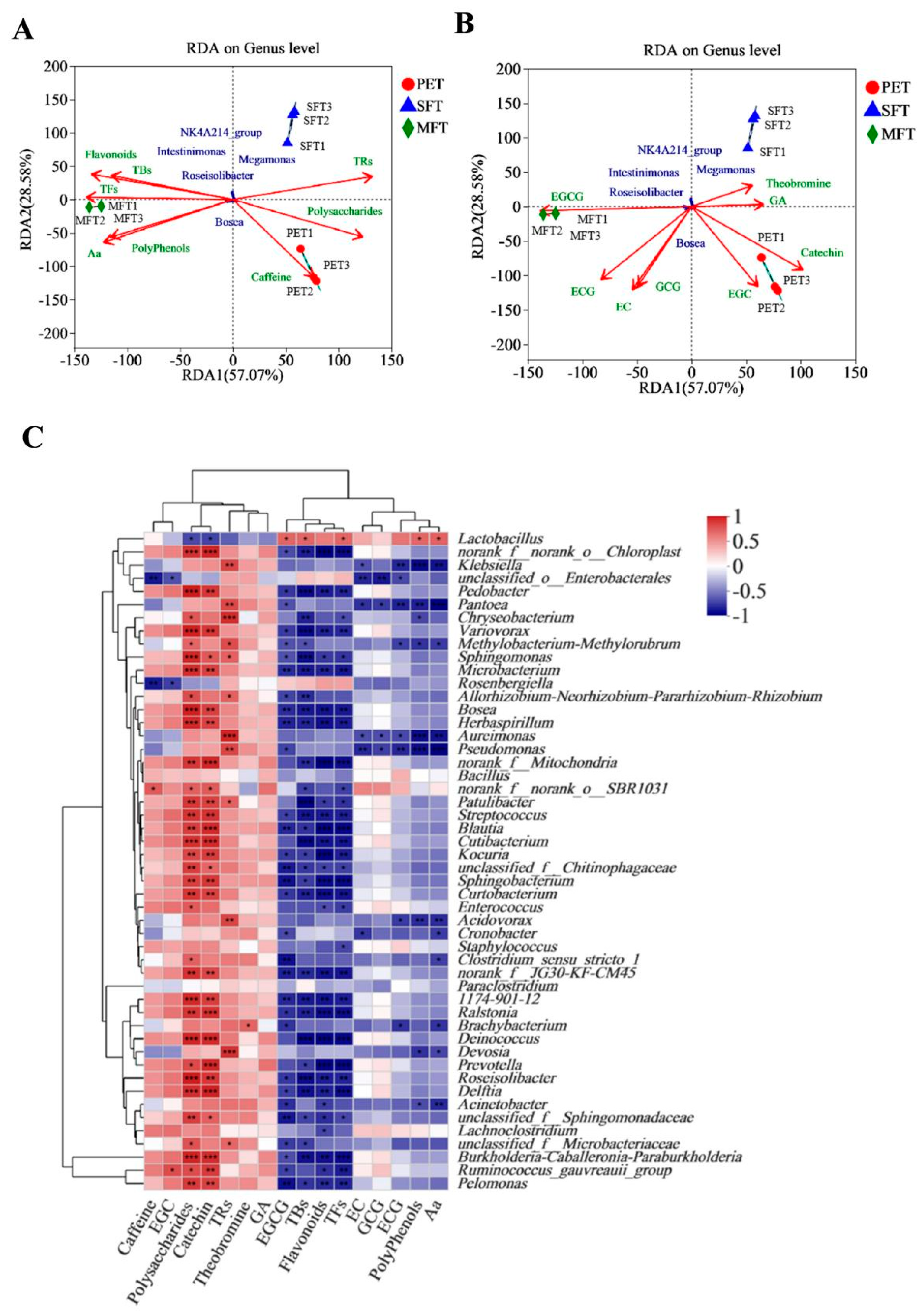
At the genus level, Chloroplast was the most dominant bacteria in the PET raw material. The richness of Klebsiella variicola, unclassified Enterobacterales and unclassified Pantoea was increased during spontaneous fermentation processing. The richness of L. plantarum was the highest among the bacterial genera in the MFT sample. In addition, the diversity of bacteria was decreased, as the inoculated L. plantarum restrained other bacteria’s growth.
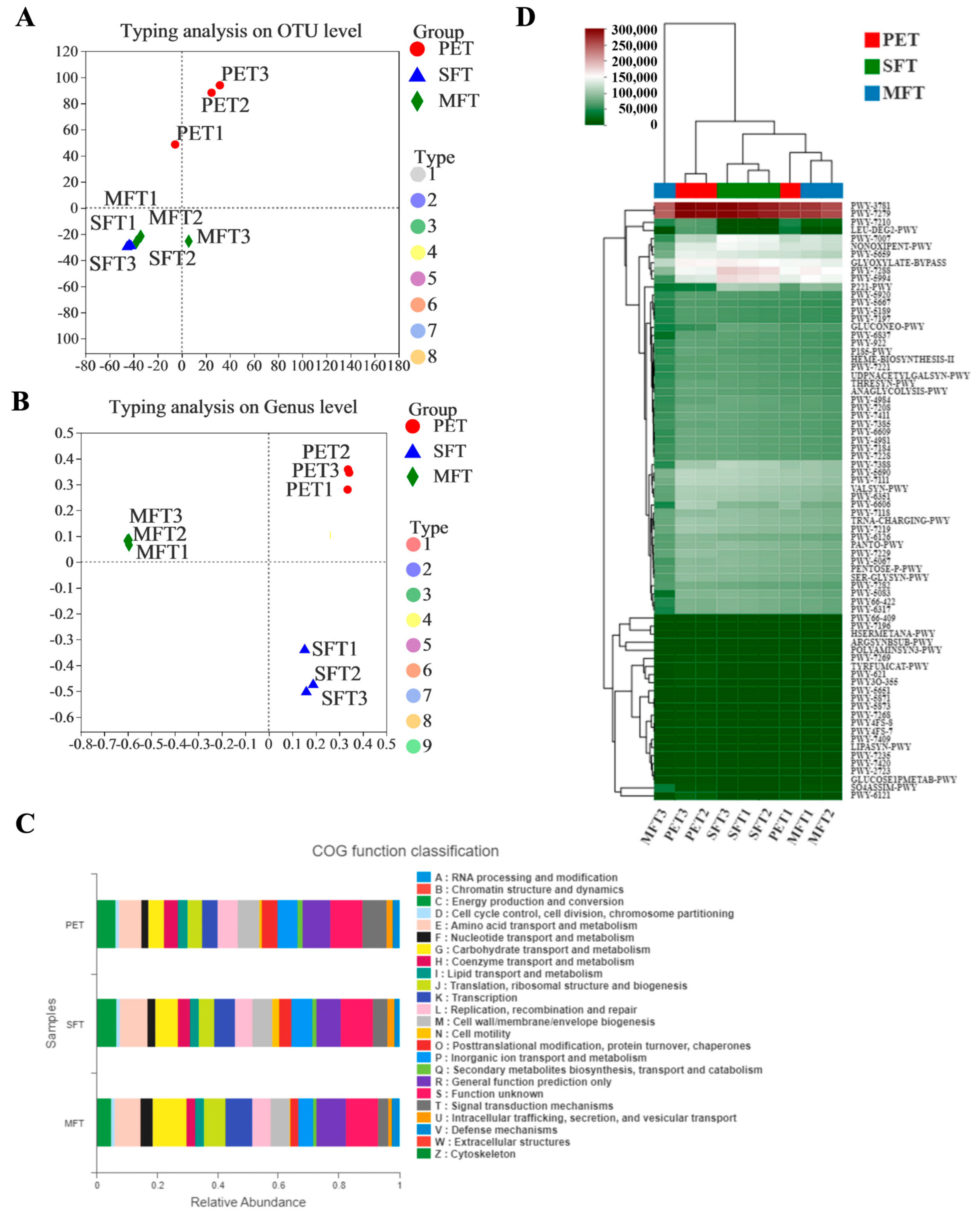
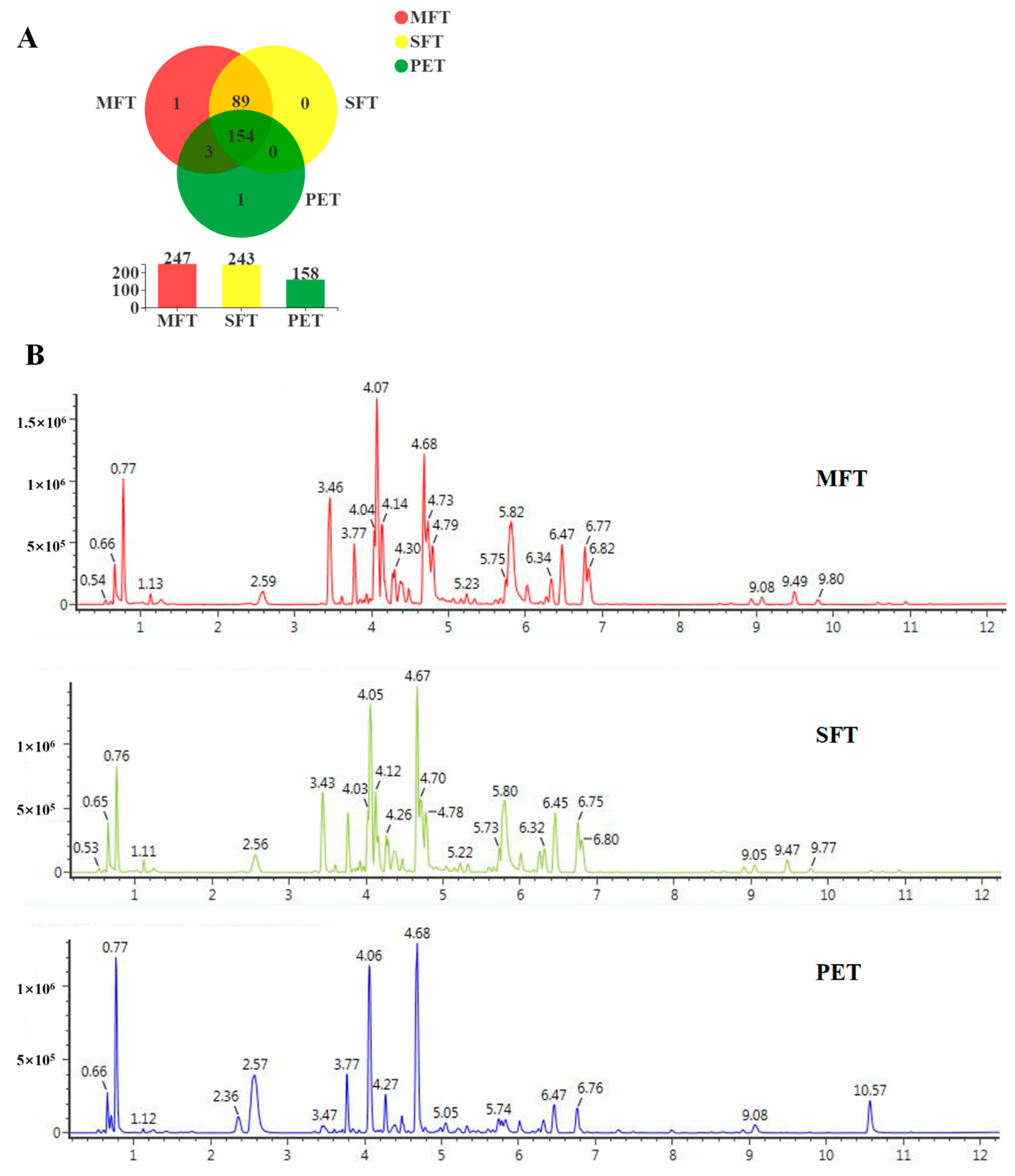
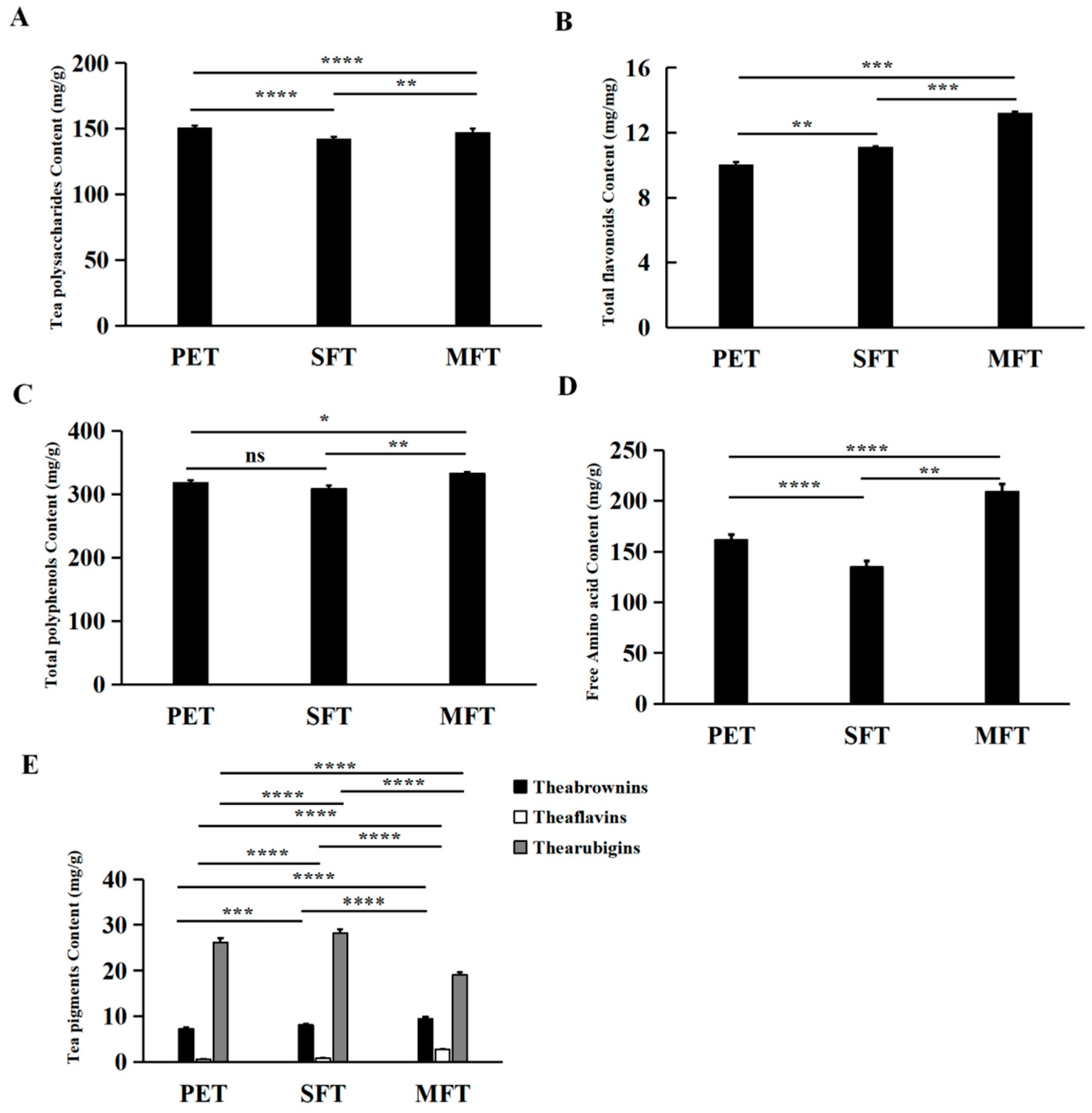
In exploring the relationships between metabolism and microorganisms, a total of 248 metabolites were identified in the tea samples. Among them, 93 particular metabolites were from the MFT sample. These results indicated that microbial fermentation processing caused the microorganisms to undergo community structure changes and metabolic profile transformations. We predicted and analyzed the metabolic pathways of the microorganisms, and the relative abundance of pathways associated with carbohydrate transport and metabolism increased in MFT. This indicated that fungal metabolism was mainly involved in the hydrolysis of macromolecular compounds, while bacterial metabolism transferred small molecular compounds in the fermentation process. A total of 16 critical metabolites were screened for the metabolic variation study by LC–MS metabolomics and multivariate analysis. The results revealed that the critical metabolites were significantly different between the raw tea materials and fermented tea samples. The content of quinic acid, strictinin, assamicain and theogallin increased during spontaneous fermentation, but it was further increased in tea samples that underwent microbial fermentation, indicating that the biochemical profiles of SFT and MFT were influenced uniquely by the fermentation processes. Furthermore, the effect of bacteria on metabolites was stronger than that of fungi, which may have played an auxiliary role in substance transformation, which accelerated the transformation reaction and reduced the fermentation cycle.
The critical functional components and microorganisms involved in fermentation processes were studied to improve the flavor and quality of Pu-erh tea. From the microbiome results,
L. plantarum and
S. cerevisiae had varied effects on tea metabolites. Moreover, the metabolic analysis showed that polysaccharides, flavonoids and free amino acids were increased in the MFT sample. Although
S. cerevisiae cannot secrete enzymes to break down polysaccharides, proteins and lipids during the fermentation process, we found that the metabolic activity of
S. cerevisiae increased in the MFT. These changes probably resulted from
L. plantarum secreting a variety of enzymes to promote
S. cerevisiae growth, such as proteolytic and peptidelytic enzymes [
31,
32]. Meanwhile, the origin of enzymatic activity could be attributed to dead microbial cells or the intracellular fraction of plant cells, and some of the metabolites could also be products of spontaneous reactions. The polyphenols increased after manufacturing; this result was consistent with a report that galloylated catechins were hydrolyzed during microbial fermentation, concurrent with an increase in their hydrolytic products [
33], which might be helpful in the bioactivity of MFT. The polysaccharides remained stable after fermentation, which was likely because the water-soluble tea polysaccharides in PET are digested by hydrolytic enzymes from fungi and utilized as carbon sources for microorganisms in fermentation, and other polysacchrides composed of complexes with water-insoluble protein and lipid are released from insoluble complexes. Amino acids are important ingredients contributing to the taste of tea infusions. Previous research found that theanine and aspartic acid were positively correlated with the umami taste of a tea infusion [
27]. The hydrolysis of extracellular enzymes from the secretion of microorganisms and consumption of microbes caused an increase in free amino acids. Protein was degraded into amino acids as a result of the hot/humid conditions and microbial catalysis. The changes in tea pigments (theabrownin, theaflavin and thearubigin) and their derivatives, flavonoids and flavonoid glycosides and simple phenols were consistent with the different dominant species in different tea samples [
34]. Catechins are either oxidized to characteristic pigments or degraded into phenolic acids during fermentation processing. For example, catechin/EC/GC/EGC can be metabolized into diOH-phenylacetic acid via C-ring fission [
33]. The growth of
L. plantarum in MFT was closely related to the increase in EGCG, TBs, TFs, polyphenols and Aa in the fermentation process. In summary, the 16 critical metabolites had a strong correlation with bacterial genera, and the change in
L. plantarum in the bacterial microbiome largely affected the metabolite transformation during fermentation.
This study reported the changes and correlations between the microbiome and metabolomics during the different manufacturing of Pu-erh tea samples. With a better understanding of the relationships between microorganisms and metabolites, it is possible to improve the compositions of bioactive compounds in PET through the exogenous inoculation of L. plantarum and S. cerevisiae to produce specific flavored PET and improve the quality of PET. Moreover, it is necessary to inoculate specific microorganisms and characterize the functional components from PET to further enhance the value of PET.
5. Conclusions
Investigating microbial fermentation in terms of producing beneficial bioactive compounds is a critical factor in Pu-erh tea manufacturing. To study the effects of the microbiome on the metabolite profile of fermented tea, L. plantarum and S. cerevisiae, two commonly studied microorganisms involved in fermentation, were inoculated onto tea leaves to increase the functional metabolites. Spontaneously fermented Pu-erh tea (no microbe pre-treatment) and raw material (non-fermented control leaves) were also studied as controls. To compare the three Pu-erh tea samples, the microbial community structure, correlation between microbiomes and metabolites and critical metabolite changes were characterized using metabolomics and microbiomics analyses. The spontaneously fermented tea displayed the lowest fungal diversity. More polyphenols, flavonoids and amino acids were identified in the L. plantarum- and S. cerevisiae-inoculated fermented tea (MFT). Compared to fungi, both the diversity and richness of bacteria were significantly decreased in the MFT sample. The conversion of metabolites in Pu-erh tea was mainly catalyzed by microbial enzymes secreted from microorganisms during the manufacturing process. In total, 71 critical metabolites were mostly responsible for the metabolic changes caused by the manufacturing process. Theabrownin, some novel phenolic acids and catechin derivatives formed, while the content of polysaccharides and tea pigments remained stable in MFT. These components are known to be responsible for the astringent and mellow tastes, as well as the brownish color and health benefits of Pu-erh tea.
In summary, our results advance the knowledge of the functions of L. plantarum and S. cerevisiae in the formation of the unique sensory characteristics of Pu-erh tea and reveal that the microbial composition is a critical factor in changing the tea’s metabolic profile. These findings provide a new method to improve the quality and safety of Pu-erh tea.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}