
A High-Throughput Absolute Abundance Quantification Method for the Characterisation of Daqu Core Fungal Communities


Abstract
:1. Introduction
2. Materials and Methods
2.1. Design and Verification of ISF and ISP
2.2. Screening and Fungi
2.3. Sample Collection
2.4. Construction of the HAQ Method
2.4.1. Selection of ISPs Addition Concentration
2.4.2. Establishment of the Core Fungal Standard Curve
2.5. Application of HAQ Method
2.6. Quantitative PCR
2.7. Amplification and Sequencing
2.8. Statistical Analysis
2.9. Data Availability
3. Results
3.1. Sequence Distribution of Fungal ITS2 and Construction of ISP
3.2. Selection of ISP Concentrations and Application Verification
3.3. Construction of the HAQ Method
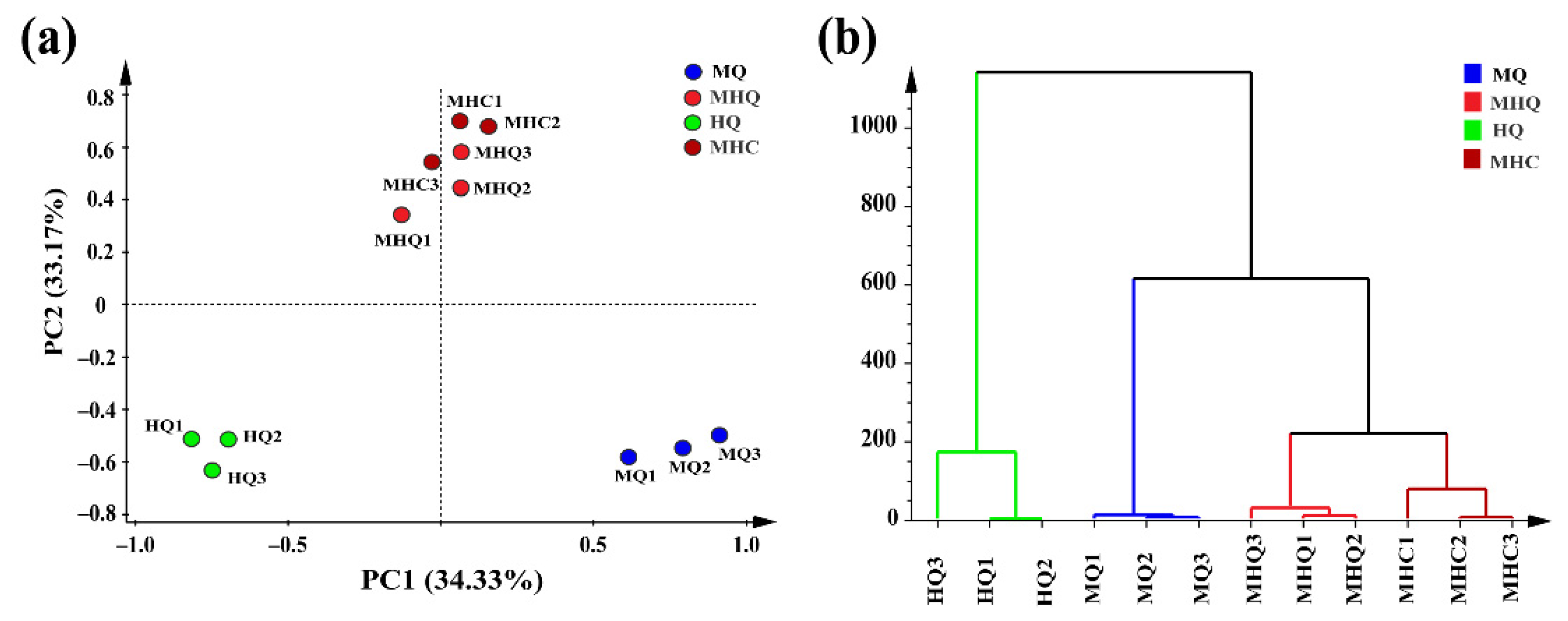
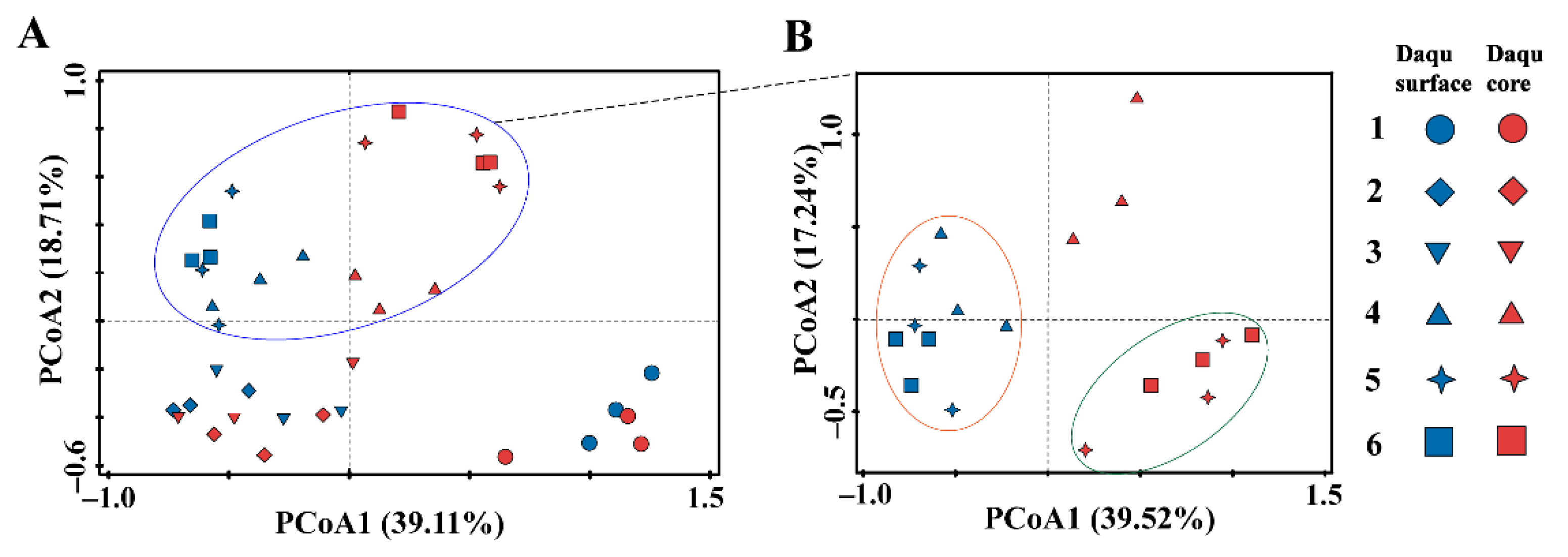
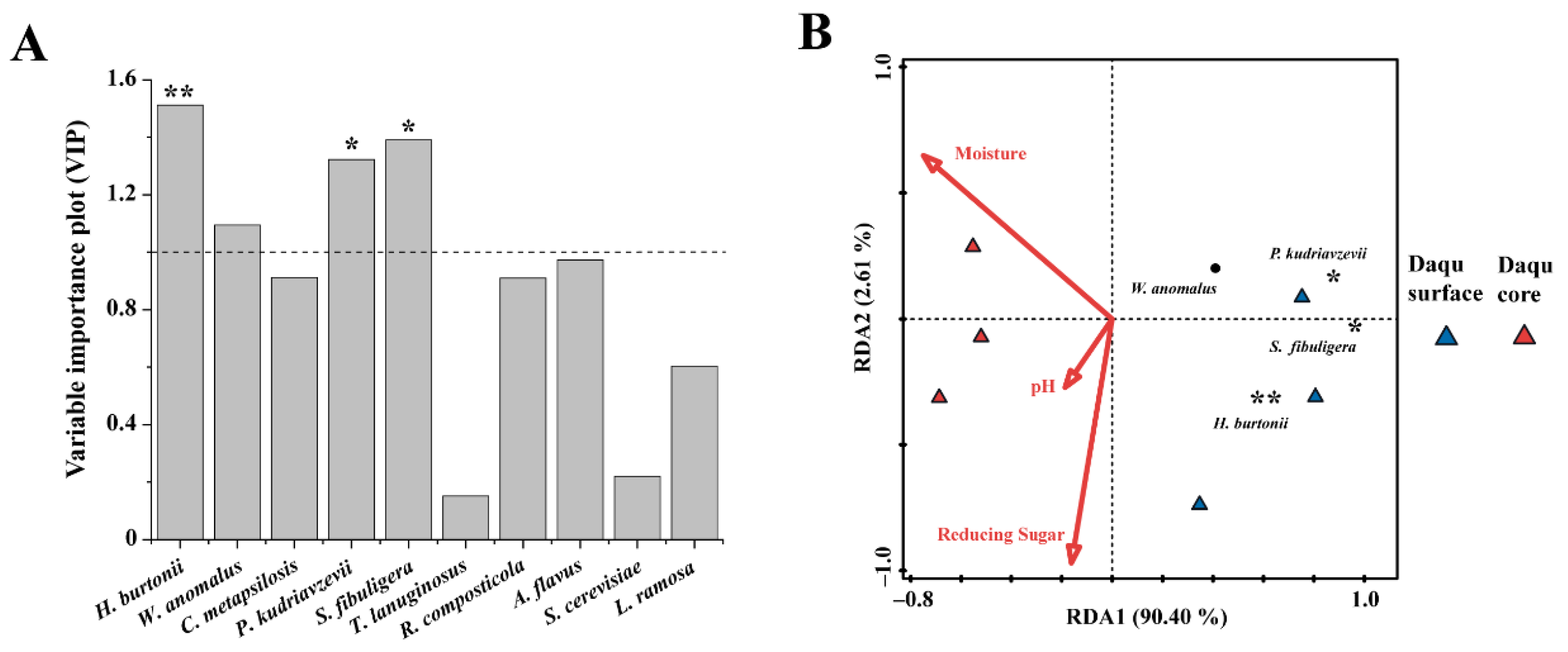
3.4. Application I: Case Studies of Different Mature Daqu
3.5. Application II: Case Studies of Medium-High Temperature Daqu during Fermentation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jin, G.; Zhu, Y.; Xu, Y. Mystery behind Chinese liquor fermentation. Trends Food Sci. Technol. 2017, 63, 18–28. [Google Scholar] [CrossRef]
- Xing, G.; Zonghua, A.O.; Wang, S.; Deng, B.; Wang, X.; Dong, Z. Analysis of the Change in Physiochemical Indexes during the Production Process of Daqu of Different Temperature. Liquor-Mak. Sci. Technol. 2014, 11, 592421. [Google Scholar]
- Zou, W.; Zhao, C.Q.; Luo, H.B. Diversity and Function of Microbial Community in Chinese Strong-Flavor Baijiu Ecosystem: A Review. Front. Microbiol. 2018, 9, 671. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Wang, X.S.; Zhang, Y.H.; Xu, Y. Exploring the impacts of raw materials and environments on the microbiota in Chinese Daqu starter. Int. J. Food Microbiol. 2019, 297, 32–40. [Google Scholar] [CrossRef]
- Fan, G.S.; Fu, Z.L.; Teng, C.; Liu, P.X.; Wu, Q.H.; Rahman, M.K.R.; Li, X.T. Effects of aging on the quality of roasted sesame-like flavor Daqu. Bmc Microbiol. 2020, 20, 67. [Google Scholar] [CrossRef]
- Pan, L.; Lin, W.; Xiong, L.; Wang, X.; Luo, L. Environmental Factors Affecting Microbiota Dynamics during Traditional Solid-state Fermentation of Chinese Daqu Starter. Front. Microbiol. 2016, 7, 1237. [Google Scholar]
- Li, P.; Sha, L.; Cheng, L.; Luo, L. Analyzing the relation between the microbial diversity of DaQu and the turbidity spoilage of traditional Chinese vinegar. Appl. Microbiol. Biotechnol. 2014, 98, 6073. [Google Scholar] [CrossRef]
- Pang, X.N.; Han, B.Z.; Huang, X.N.; Zhang, X.; Hou, L.F.; Cao, M.; Gao, L.J.; Hu, G.H.; Chen, J.Y. Effect of the environment microbiota on the flavour of light-flavour Baijiu during spontaneous fermentation. Sci. Rep. 2018, 8, 3396. [Google Scholar] [CrossRef]
- Li, P.; Lin, W.F.; Liu, X.; Wang, X.W.; Gan, X.; Luo, L.X.; Lin, W.T. Effect of bioaugmented inoculation on microbiota dynamics during solid-state fermentation of Daqu starter using autochthonous of Bacillus, Pediococcus, Wickerhamomyces and Saccharomycopsis. Food Microbiol. 2017, 61, 83–92. [Google Scholar] [CrossRef]
- Wang, X.S.; Du, H.; Zhang, Y.; Xu, Y. Environmental Microbiota Drives Microbial Succession and Metabolic Profiles during Chinese Liquor Fermentation. Appl. Environ. Microbiol. 2018, 84, e02369-17. [Google Scholar] [CrossRef]
- Zheng, X.W.; Yan, Z.; Nout, M.J.R.; Smid, E.J.; Zwietering, M.H.; Boekhout, T.; Han, J.S.; Han, B.Z. Microbiota dynamics related to environmental conditions during the fermentative production of Fen-Daqu, a Chinese industrial fermentation starter. Int. J. Food Microbiol. 2014, 182, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.S.; Fu, Z.L.; Teng, C.; Wu, Q.H.; Liu, P.X.; Yang, R.; Minhazul, K.; Li, X.T. Comprehensive analysis of different grades of roasted-sesame-like flavored Daqu. Int. J. Food Prop. 2019, 22, 1205–1222. [Google Scholar] [CrossRef]
- Tkacz, A.; Hortala, M.; Poole, P.S. Absolute quantitation of microbiota abundance in environmental samples. Microbiome 2018, 6, 110. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.J.; Gifford, S.; Ducklow, H.; Schofield, O.; Cassar, N. Towards Quantitative Microbiome Community Profiling Using Internal Standards. Appl. Environ. Microbiol. 2019, 85, e02634-18. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lou, J.; Wang, H.Z.; Wu, L.S.; Xu, J.M. Use of an improved high-throughput absolute abundance quantification method to characterize soil bacterial community and dynamics. Sci. Total Environ. 2018, 633, 360–371. [Google Scholar] [CrossRef]
- Dannemiller, K.C.; Lang-Yona, N.; Yamamoto, N.; Rudich, Y.; Peccia, J. Combining real-time PCR and next-generation DNA sequencing to provide quantitative comparisons of fungal aerosol populations. Atmos. Environ. 2014, 84, 113–121. [Google Scholar] [CrossRef]
- Nishino, S.; Okahashi, N.; Matsuda, F.; Shimizu, H. Absolute quantitation of glycolytic intermediates reveals thermodynamic shifts in Saccharomyces cerevisiae strains lacking PFK1 or ZWF1 genes. J. Biosci. Bioeng. 2015, 120, 280–286. [Google Scholar] [CrossRef]
- Nayfach, S.; Pollard, K.S. Toward Accurate and Quantitative Comparative Metagenomics. Cell 2016, 166, 1103–1116. [Google Scholar] [CrossRef]
- Props, R.; Kerckhof, F.M.; Rubbens, P.; De Vrieze, J.; Sanabria, E.H.; Waegeman, W.; Monsieurs, P.; Hammes, F.; Boon, N. Absolute quantification of microbial taxon abundances. ISME J. 2017, 11, 584–587. [Google Scholar] [CrossRef]
- Liu, X.R.; Li, J.; Yu, L.; Pan, H.; Liu, H.Y.; Liu, Y.M.; Di, H.J.; Li, Y.; Xu, J.M. Simultaneous measurement of bacterial abundance and composition in response to biochar in soybean field soil using 16S rRNA gene sequencing. Land Degrad. Dev. 2018, 29, 2172–2182. [Google Scholar] [CrossRef]
- Jiang, S.Q.; Yu, Y.N.; Gao, R.W.; Wang, H.; Zhang, J.; Li, R.; Long, X.H.; Shen, Q.R.; Chen, W.; Cai, F. High-throughput absolute quantification sequencing reveals the effect of different fertilizer applications on bacterial community in a tomato cultivated coastal saline soil. Sci. Total Environ. 2019, 687, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Du, R.B.; Wu, Q.; Xu, Y. Chinese Liquor Fermentation: Identification of Key Flavor-Producing Lactobacillus spp. by Quantitative Profiling with Indigenous Internal Standards. Appl. Environ. Microbiol. 2020, 86, e00456-20. [Google Scholar] [CrossRef] [PubMed]
- Vandeputte, D.; Kathagen, G.; D’Hoe, K.; Vieira-Silva, S.; Valles-Colomer, M.; Sabino, J.; Wang, J.; Tito, R.Y.; De Commer, L.; Darzi, Y.; et al. Quantitative microbiome profiling links gut community variation to microbial load. Nature 2017, 551, 507–511. [Google Scholar] [CrossRef]
- Lou, J.; Yang, L.; Wang, H.Z.; Wu, L.S.; Xu, J.M. Assessing soil bacterial community and dynamics by integrated high-throughput absolute abundance quantification. PeerJ 2018, 6, e4514. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.J.; Qu, Y.Y.; Li, S.Z.; Feng, K.; Wang, S.; Cai, W.W.; Liang, Y.T.; Li, H.; Xu, M.Y.; Yin, H.Q.; et al. Soil bacterial quantification approaches coupling with relative abundances reflecting the changes of taxa. Sci. Rep. 2017, 7, 4837. [Google Scholar] [CrossRef]
- Smets, W.; Leff, J.W.; Bradford, M.A.; McCulley, R.L.; Lebeer, S.; Fierer, N. A method for simultaneous measurement of soil bacterial abundances and community composition via 16S rRNA gene sequencing. Soil Biol. Biochem. 2016, 96, 145–151. [Google Scholar] [CrossRef]
- Tourlousse, D.M.; Yoshiike, S.; Ohashi, A.; Matsukura, S.; Noda, N.; Sekiguchi, Y. Synthetic spike-in standards for high-throughput 16S rRNA gene amplicon sequencing. Nucleic Acids Res. 2017, 45, e23. [Google Scholar] [CrossRef]
- Nilsson, R.H.; Anslan, S.; Bahram, M.; Wurzbacher, C.; Baldrian, P.; Tedersoo, L. Mycobiome diversity: High-throughput sequencing and identification of fungi. Nat. Rev. Microbiol. 2019, 17, 95–109. [Google Scholar] [CrossRef]
- Ban, S.B.; Chen, L.N.; Fu, S.X.; Wu, Q.; Xu, Y. Modelling and predicting population of core fungi through processing parameters in spontaneous starter (Daqu) fermentation. Int. J. Food Microbiol. 2022, 363, 109493. [Google Scholar] [CrossRef]
- Rama, T.; Davey, M.; Norden, J.; Halvorsen, R.; Blaalid, R.; Mathiassen, G.; Alsos, I.; Kauserud, H. Fungi Sailing the Arctic Ocean: Speciose Communities in North Atlantic Driftwood as Revealed by High-Throughput Amplicon Sequencing. Microb. Ecol. 2016, 72, 295–304. [Google Scholar] [CrossRef]
- Zhang, L.J.; Cao, Y.L.; Tong, J.N.; Xu, Y. An Alkylpyrazine Synthesis Mechanism Involving L-Threonine-3-Dehydrogenase Describes the Production of 2,5-Dimethylpyrazine and 2,3,5-Trimethylpyrazine by Bacillus subtilis. Appl. Environ. Microbiol. 2019, 85, e01807-19. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, S.; Doherty, T.M.; Kenneth, J.; Grp, T.B.T.S. Comparison of different standards for real-time PCR-based absolute quantification. J. Immunol. Methods 2010, 354, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.W.; Du, H.; Zhang, Y.; Xu, Y. Unraveling Core Functional Microbiota in Traditional Solid-State Fermentation by High-Throughput Amplicons and Metatranscriptomics Sequencing. Front. Microbiol. 2017, 8, 1294. [Google Scholar] [CrossRef]
- Xu, W.; Huang, Z.Y.; Zhang, X.J.; Li, Q.; Lu, Z.M.; Shi, J.S.; Xu, Z.H.; Ma, Y.H. Monitoring the microbial community during solid-state acetic acid fermentation of Zhenjiang aromatic vinegar. Food Microbiol. 2011, 28, 1175–1181. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef]
- Koljalg, U.; Larsson, K.H.; Abarenkov, K.; Nilsson, R.H.; Alexander, I.J.; Eberhardt, U.; Erland, S.; Hoiland, K.; Kjoller, R.; Larsson, E.; et al. UNITE: A database providing web-based methods for the molecular identification of ectomycorrhizal fungi. New Phytol. 2005, 166, 1063–1068. [Google Scholar] [CrossRef]
- French, K.E.; Tkacz, A.; Turnbull, L.A. Conversion of grassland to arable decreases microbial diversity and alters community composition. Appl. Soil Ecol. 2017, 110, 43–52. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef]
- Rodriguez, A.; Luque, M.I.; Andrade, M.J.; Rodriguez, M.; Asensio, M.A.; Cordoba, J.J. Development of real-time PCR methods to quantify patulin-producing molds in food products. Food Microbiol. 2011, 28, 1190–1199. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Blanch, J.F.; Sanchez, G.; Garay, E.; Aznar, R. Development of a real-time PCR assay for detection and quantification of enterotoxigenic members of Bacillus cereus group in food samples. Int. J. Food Microbiol. 2009, 135, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Wu, Q.; Xu, Y. Filamentous fungal diversity and community structure associated with the solid state fermentation of Chinese Maotai-flavor liquor. Int. J. Food Microbiol. 2014, 179, 80–84. [Google Scholar] [CrossRef]
- Li, H.; Huang, J.; Liu, X.P.; Zhou, R.Q.; Ding, X.F.; Xiang, Q.Y.; Zhang, L.Q.; Wu, C.D. Characterization of Interphase Microbial Community in Luzhou-Flavored Liquor Manufacturing Pits of Various Ages by Polyphasic Detection Methods. J. Microbiol. Biotechnol. 2017, 27, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Li, X.R.; Ma, E.B.; Yan, L.Z.; Meng, H.; Du, X.W.; Zhang, S.W.; Quan, Z.X. Bacterial and fungal diversity in the traditional Chinese liquor fermentation process. Int. J. Food Microbiol. 2011, 146, 31–37. [Google Scholar] [CrossRef]
- Benito, A.; Jeffares, D.; Palomero, F.; Calderon, F.; Bai, F.Y.; Bahler, J.; Benito, S. Selected Schizosaccharomyces pombe Strains Have Characteristics That Are Beneficial for Winemaking. PLoS ONE 2016, 11, e0151102. [Google Scholar] [CrossRef]
- Yuangsaard, N.; Yongmanitchai, W.; Yamada, M.; Limtong, S. Selection and characterization of a newly isolated thermotolerant Pichia kudriavzevii strain for ethanol production at high temperature from cassava starch hydrolysate. Antonie Van Leeuwenhoek Int. J. Gen. Mol. Microbiol. 2013, 103, 577–588. [Google Scholar] [CrossRef]
- Su, C.; Zhang, K.Z.; Cao, X.Z.; Yang, J.G. Effects of Saccharomycopsis fibuligera and Saccharomyces cerevisiae inoculation on small fermentation starters in Sichuan-style Xiaoqu liquor. Food Res. Int. 2020, 137, 109425. [Google Scholar] [CrossRef]
- Yang, Y.R.; Zhong, H.Y.; Yang, T.; Lan, C.H.; Zhu, H. Characterization of the key aroma compounds of a sweet rice alcoholic beverage fermented withSaccharomycopsis fibuligera. J. Food Sci. Technol.-Mysore 2021, 58, 3752–3764. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | ITS2 Length | ITS2 GC Content | ISF Length | ISF GC Content | Core Fungi |
|---|---|---|---|---|---|
| 1 | 231~300 bp | 39~42% GC | 272 bp | 40% GC | Kodamaea ohmeri, Hyphopichia burtonii, Wickerhamomyces anomalus |
| 2 | 231~300 bp | 45~52% GC | 272 bp | 50% GC | Candida versatilis, Candida metapsilosis, Kazachstania humilis, Pichia kudriavzevii. |
| 3 | 300~350 bp | 36~50% GC | 325 bp | 40% GC | Saccharomyces sp., Rhizopus arrhizus, Saccharomycopsis fibuligera, Rhizopus microsporus, Rhodotorula mucilaginosa, Rhizomucor pusillus. |
| 4 | 300~350 bp | 56~63% GC | 325 bp | 60% GC | Candida athensensis, Pichia sporocuriosa, Paecilomyces verrucosus, Aspergillus amstelodami, Aspergillus sp., Thermoascus crustaceus, Thermomyces lanuginosus, Rasamsonia composticola, Monascus purpureus, Thermoascus aurantiacus, Aspergillus flavus, Leiotheciume llipsoideum, Aspergillus costiformis. |
| 5 | 350~413 bp | 31~44% GC | 387 bp | 40% GC | Saccharomyces cerevisiae, Kazachstania bulderi, Lichtheimiaceaeramosa, Schizosaccharomyces pombe. |
| ISP a | Core Fungi | Source b |
|---|---|---|
| I | Hyphopichia burtonii | LBMAE |
| Wickerhamomyces anomalus | LBMAE | |
| II | Candida metapsilosis | LBMAE |
| Pichia kudriavzevii | LBMAE | |
| III | Saccharomycopsis fibuligera | LBMAE |
| Rhizopus microsporus | LBMAE | |
| Rhodotorula mucilaginosa | LBMAE | |
| IV | Paecilomyces verrucosus | LBMAE |
| Thermomyces lanuginosus | LBMAE | |
| Rasamsonia composticola | LBMAE | |
| Monascus purpureus | LBMAE | |
| Aspergillus flavus | LBMAE | |
| V | Saccharomyces cerevisiae | LBMAE |
| Kazachstania bulderi | LBMAE | |
| Lichtheimia ramosa | LBMAE | |
| Schizosaccharomyces pombe | LBMAE |
| Relative Abundance (%) | Absolute Abundance (Cells/g) | |||||
|---|---|---|---|---|---|---|
| Fungi | Medium-Temperature Daqu | Medium-High-Temperature Daqu | High-Temperature Daqu | Medium-Temperature Daqu | Medium-High-Temperature Daqu | High-Temperature Daqu |
| Hyphopichia burtonii | 2.02 ± 0.21 | 8.15 ± 0.10 | 17.31 ± 0.52 | 4.30 × 105 ± 1.16 × 105 | 2.66 × 105 ± 2.47 × 104 | 3.62 × 104 ± 7.14 × 103 |
| Wickerhamomyces anomalus | 0.26 ± 0.02 | 3.33 ± 0.53 | 3.13 ± 0.28 | 2.57 × 105 ± 7.80 × 104 | 1.05 × 106 ± 2.51 × 105 | 2.65 × 104 ± 1.75 × 103 |
| Candida membranifaciens | 0.23 ± 0.02 | 0.12 ± 0.01 | 0.12 ± 0.01 | 2.23 × 104 ± 3.60 × 103 | 1.12 × 104 ± 6.58 × 103 | 3.86 × 102 ± 3.66 × 101 |
| Pichia kudriavzevii | 6.13 ± 1.09 | 16.07 ± 0.71 | 5.91 ± 0.53 | 8.11 × 104 ± 4.01 × 105 | 7.28 × 104 ± 3.13 × 104 | 1.94 × 103 ± 2.41 × 102 |
| Saccharomyces fibuligera | 82.33 ± 2.75 | 41.79 ± 4.74 | 33.41 ± 1.59 | 1.74 × 106 ± 2.25 × 103 | 1.62 × 106 ± 2.57 × 105 | 3.35 × 104 ± 1.38 × 103 |
| Rhizopus microsporus | 0.03 ± 0.01 | 0.10 ± 0.03 | 0.12 ± 0.06 | 1.97 × 104 ± 7.20 × 103 | 9.68 × 104 ± 1.43 × 104 | 1.12 × 103 ± 6.01 × 102 |
| Rhodotorula mucilaginosa | 0.06 ± 0.01 | 0.08 ± 0.00 | 0.09 ± 0.01 | 1.81 × 104 ± 4.15 × 103 | 3.54 × 104 ± 4.19 × 103 | 4.69 × 102 ± 7.16 × 101 |
| Paecilomyces verrucosus | 0.03 ± 0.01 | 0.01 ± 0.01 | 1.90 ± 0.08 | 9.89 × 103 ± 2.87 × 104 | 5.68 × 103 ± 1.14 × 103 | 4.87 × 103 ± 1.01 × 103 |
| Thermomyces lanuginosus | 0.43 ± 0.06 | 8.61 ± 1.90 | 5.19 ± 0.93 | 4.56 × 105 ± 3.39 × 104 | 5.13 × 106 ± 7.67 × 103 | 6.72 × 104 ± 1.07 × 104 |
| Rasamsonia composticola | 0.03 ± 0.02 | 0.06 ± 0.03 | 1.3 ± 0.57 | 7.70 × 104 ± 5.51 × 104 | 1.44 × 105 ± 4.80 × 104 | 2.54 × 104 ± 1.93 × 104 |
| Monascus purpureus | 0.22 ± 0.03 | 0.19 ± 0.15 | 7.27 ± 0.67 | 2.48 × 105 ± 1.16 × 105 | 2.60 × 104 ± 4.62 × 104 | 7.04 × 104 ± 2.46 × 104 |
| Aspergillus flavus | 0.94 ± 0.61 | 0.52 ± 0.11 | 0.97 ± 0.03 | 7.96 × 104 ± 6.25 × 104 | 6.23 × 104 ± 4.58 × 104 | 2.43 × 103 ± 5.61 × 102 |
| Saccharomyces cerevisiae | 0.67 ± 0.09 | 1.05 ± 0.18 | 0.66 ± 0.10 | 2.18 × 105 ± 6.96 × 104 | 5.16 × 105 ± 9.12 × 104 | 7.14 × 103 ± 1.22 × 103 |
| Kazachstania bulderi | 0.11 ± 0.01 | 0.13 ± 0.01 | 0.17 ± 0.01 | 2.90 × 104 ± 8.87 × 103 | 5.66 × 104 ± 7.90 × 103 | 1.38 × 103 ± 6.60 × 101 |
| Lichtheimia ramosa | 0.04 ± 0.02 | 0.80 ± 0.13 | 0.04 ± 0.01 | 2.46 × 105 ± 2.28 × 105 | 7.37 × 106 ± 2.33 × 105 | 3.48 × 103 ± 5.67 × 102 |
| Schizosaccharomyces pombe | 0.01 ± 0.00 | 0.01 ± 0.00 | 0.01 ± 0.00 | 4.26 × 105 ± 5.21 × 104 | 8.72 × 105 ± 5.93 × 105 | 2.02 × 103 ± 1.45 × 103 |
| Total abundance | 93.52 ± 2.45 | 81.03 ± 1.80 | 77.59 ± 1.08 | 4.36 × 106 ± 6.12 × 105 | 1.76 × 107 ± 4.36 × 105 | 2.86 × 105 ± 5.09 × 104 |
| Name | Explains % | Contribution % | Psedo-F | p |
|---|---|---|---|---|
| Moisture | 51.8 | 55.5 | 4.3 | 0.136 |
| Reducing sugar | 33.5 | 35.9 | 6.8 | 0.042 |
| pH | 8.1 | 8.7 | 2.4 | 0.218 |
| Name | Explains % | Contribution % | Pseudo-F | p |
|---|---|---|---|---|
| Moisture | 19.1 | 43.9 | 1.7 | 0.194 |
| Reducing sugar | 15.4 | 35.5 | 1.4 | 0.268 |
| pH | 9 | 20.6 | 0.8 | 0.404 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, H.; Sun, J.; Zhou, T.; Xu, Y. A High-Throughput Absolute Abundance Quantification Method for the Characterisation of Daqu Core Fungal Communities. Fermentation 2022, 8, 345. https://doi.org/10.3390/fermentation8080345
Du H, Sun J, Zhou T, Xu Y. A High-Throughput Absolute Abundance Quantification Method for the Characterisation of Daqu Core Fungal Communities. Fermentation. 2022; 8(8):345. https://doi.org/10.3390/fermentation8080345
Chicago/Turabian StyleDu, Hai, Jia Sun, Tianci Zhou, and Yan Xu. 2022. "A High-Throughput Absolute Abundance Quantification Method for the Characterisation of Daqu Core Fungal Communities" Fermentation 8, no. 8: 345. https://doi.org/10.3390/fermentation8080345