The Use of CRISPR-Cas9 Genome Editing to Determine the Importance of Glycerol Uptake in Wine Yeast During Icewine Fermentation

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Yeast Strains and Media
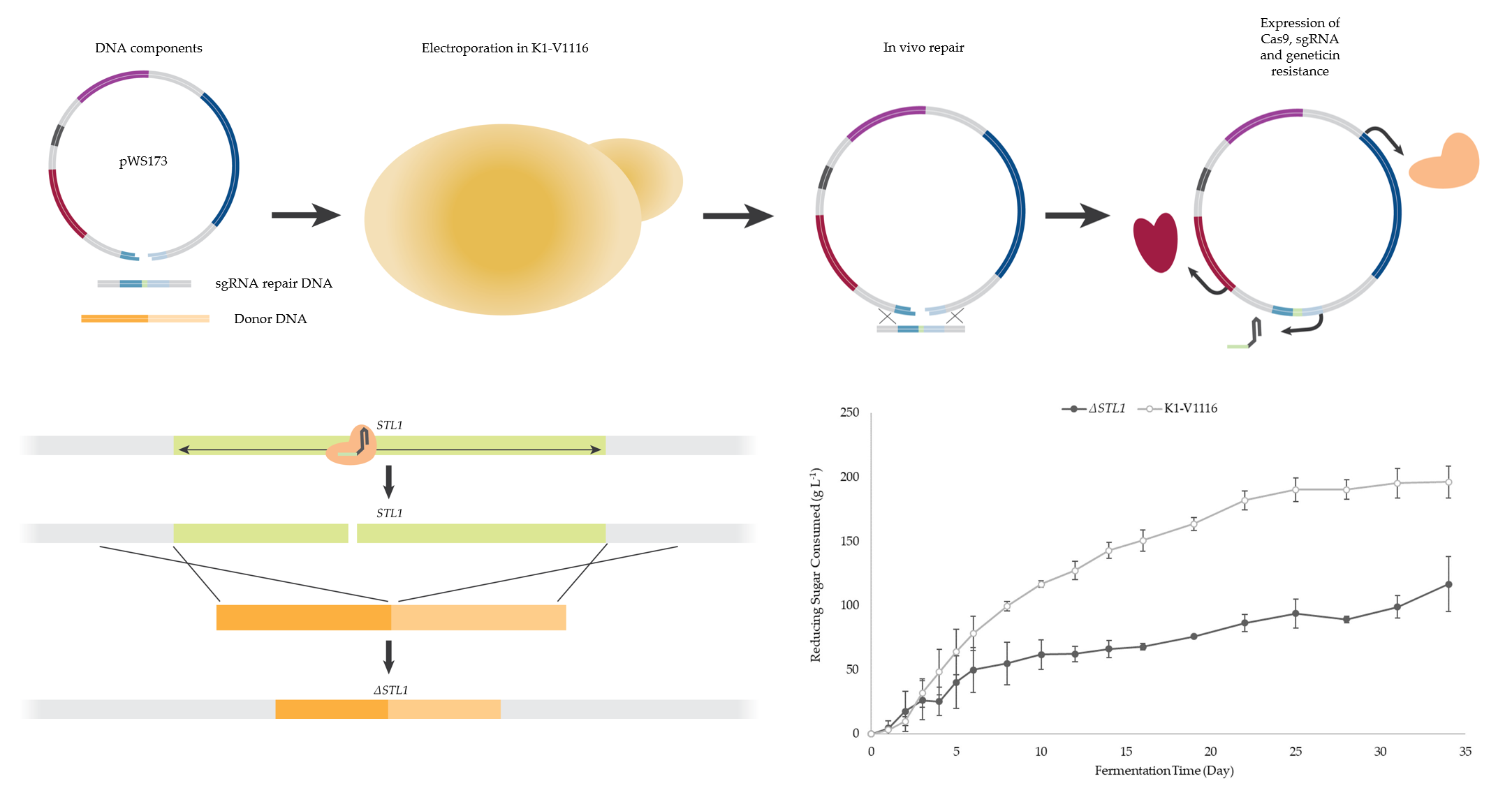
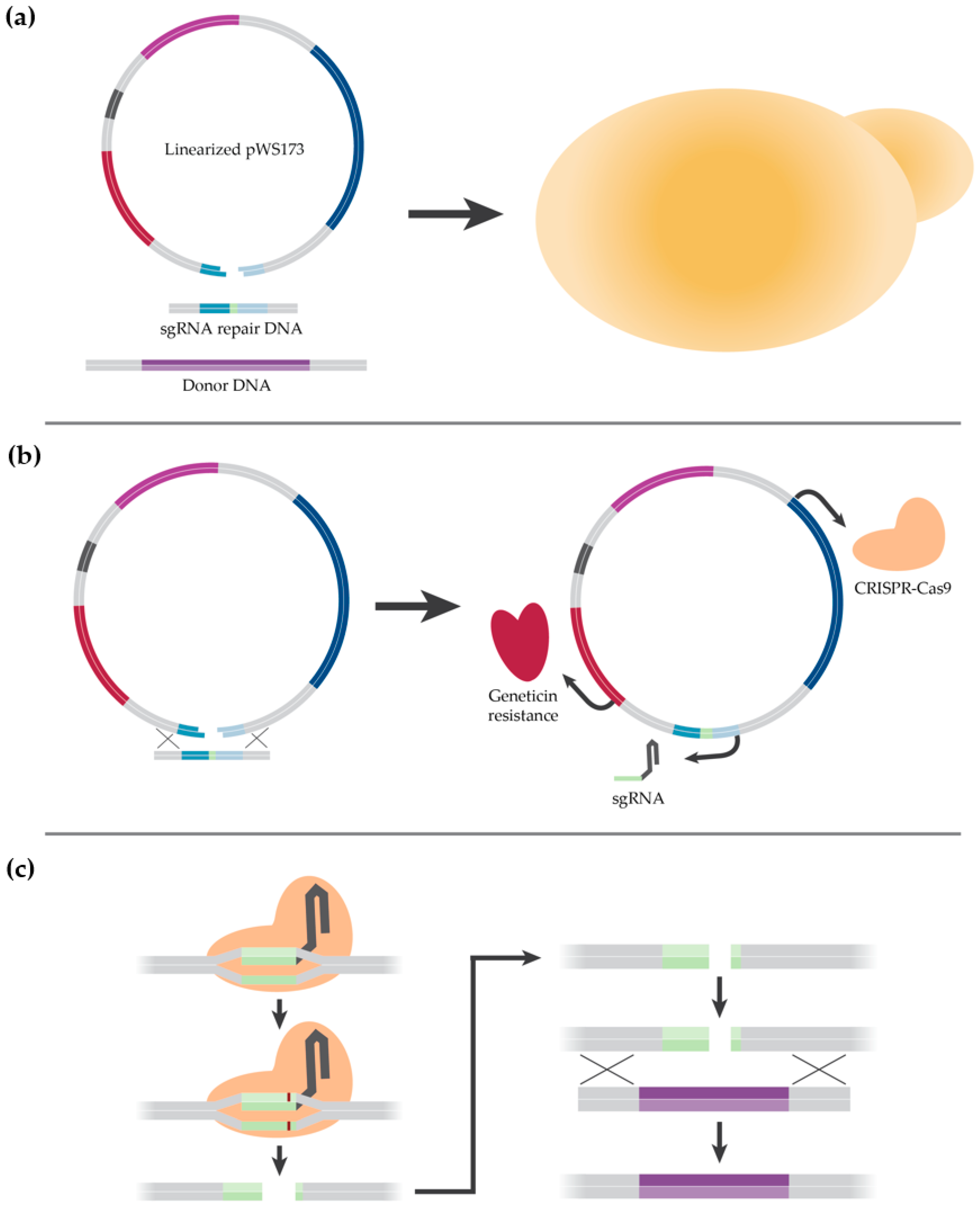
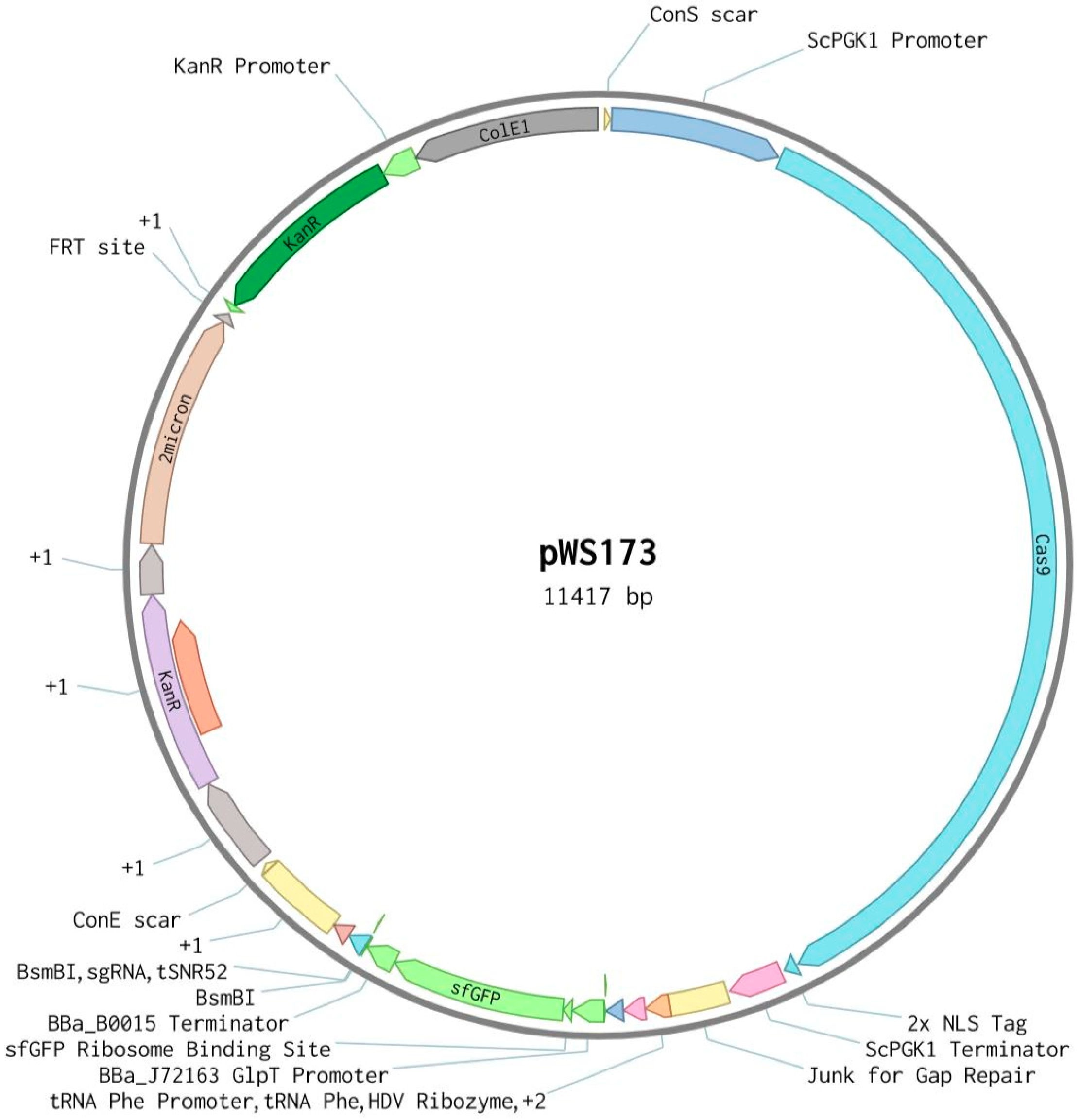
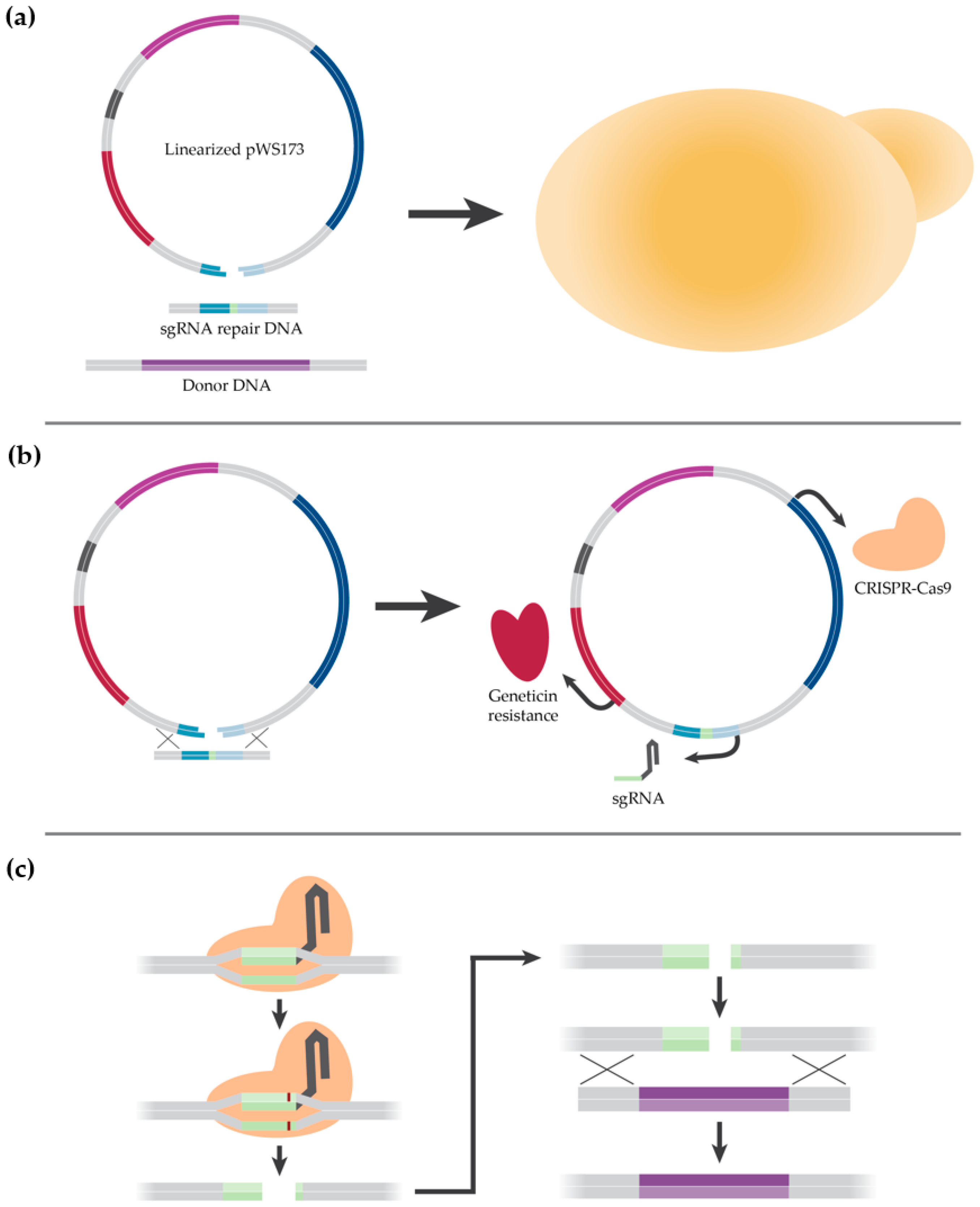
2.2. Description of CRISPR-Cas9 Method
2.3. Cloning
2.4. Strain Construction
2.5. Icewine Juice
2.6. Permissive Condition Growth Kinetics Characterization
2.7. Icewine Fermentations and Samplings
2.8. Metabolite Analysis
2.9. Statistical Analysis
3. Results
3.1. Development of a CRISPR-Cas9 Genome Editing Protocol for K1-V1116
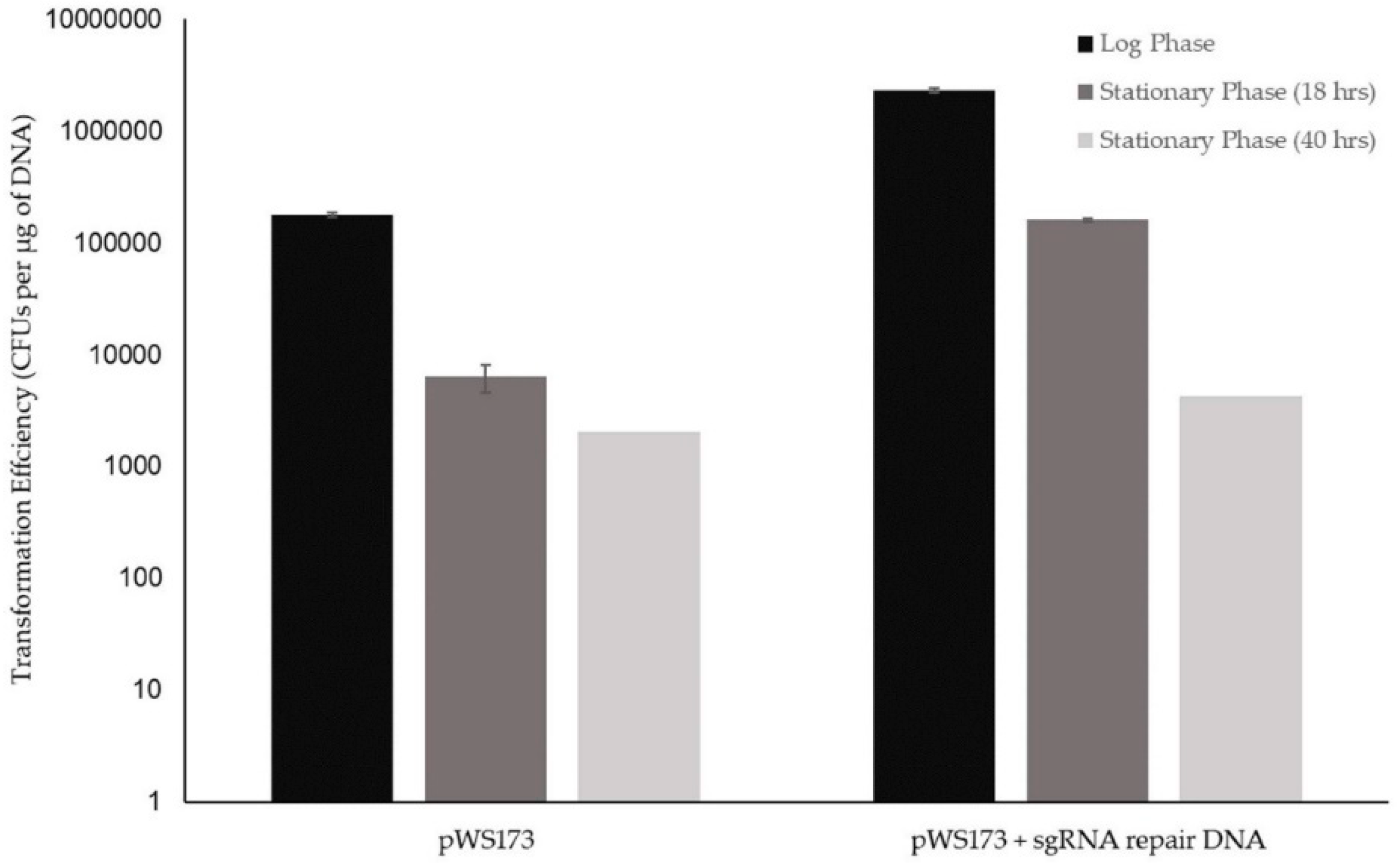
3.1.1. K1-V1116 Drug Sensitivity Testing
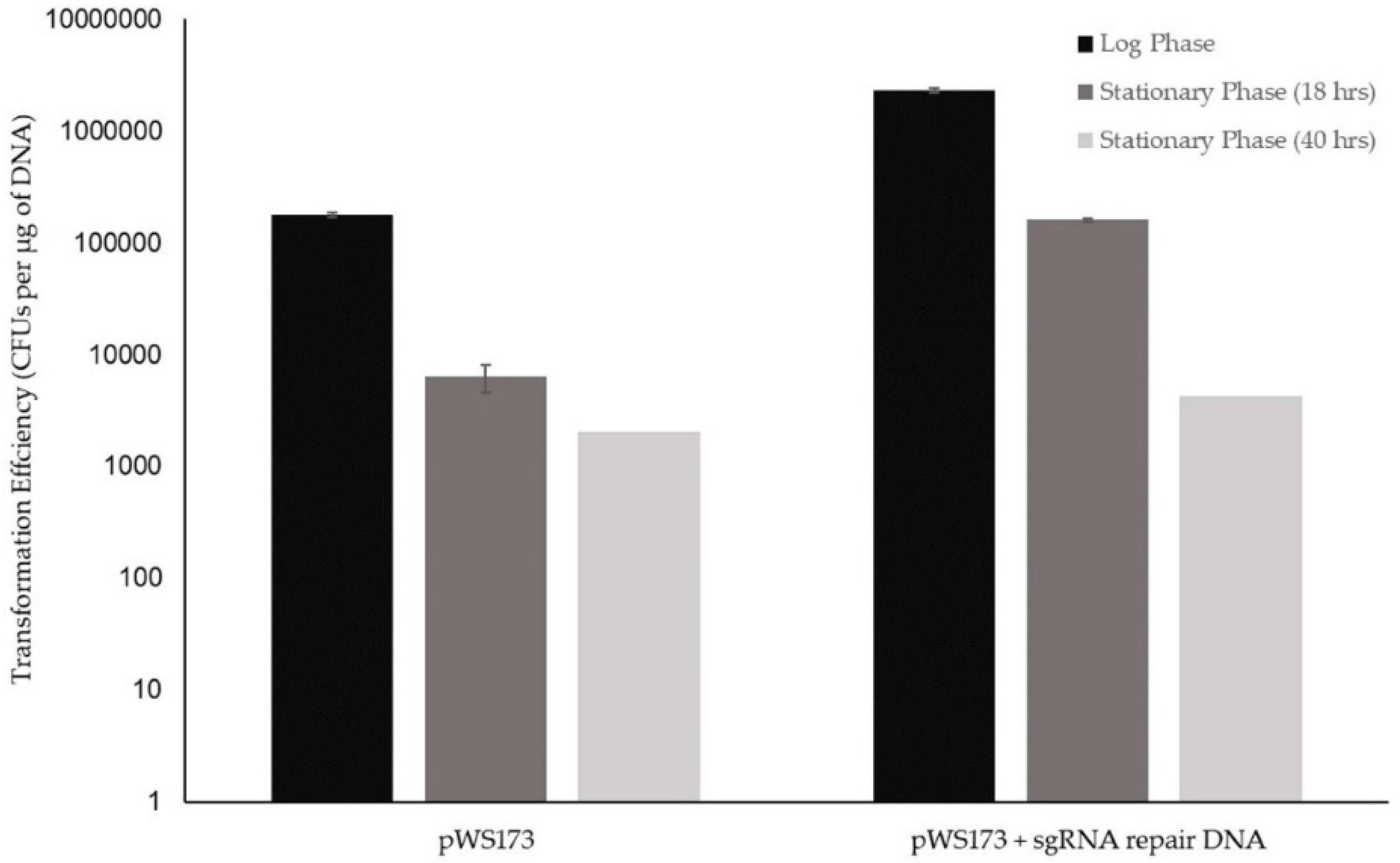
3.1.2. Initial K1-V1116 Transformations
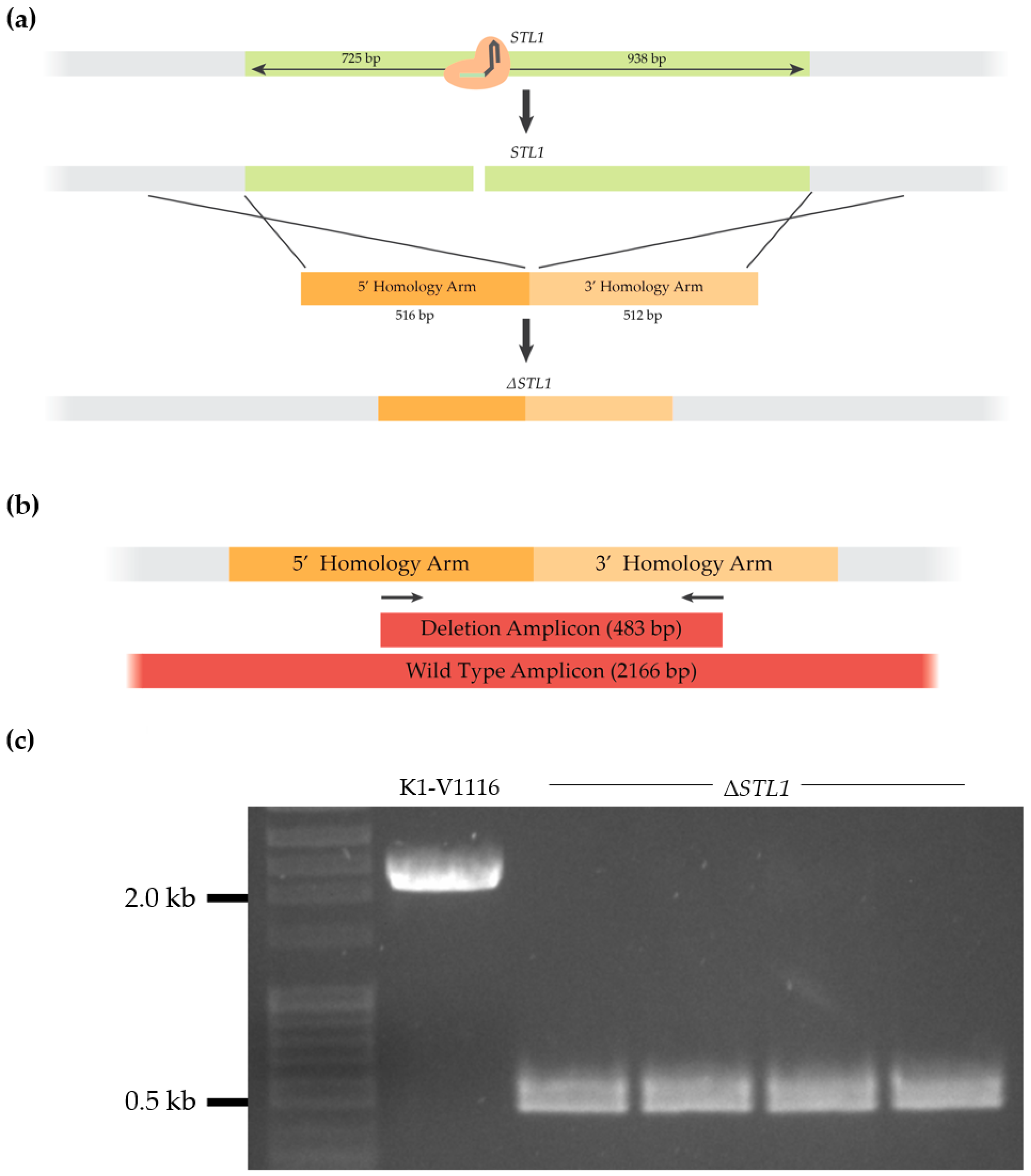
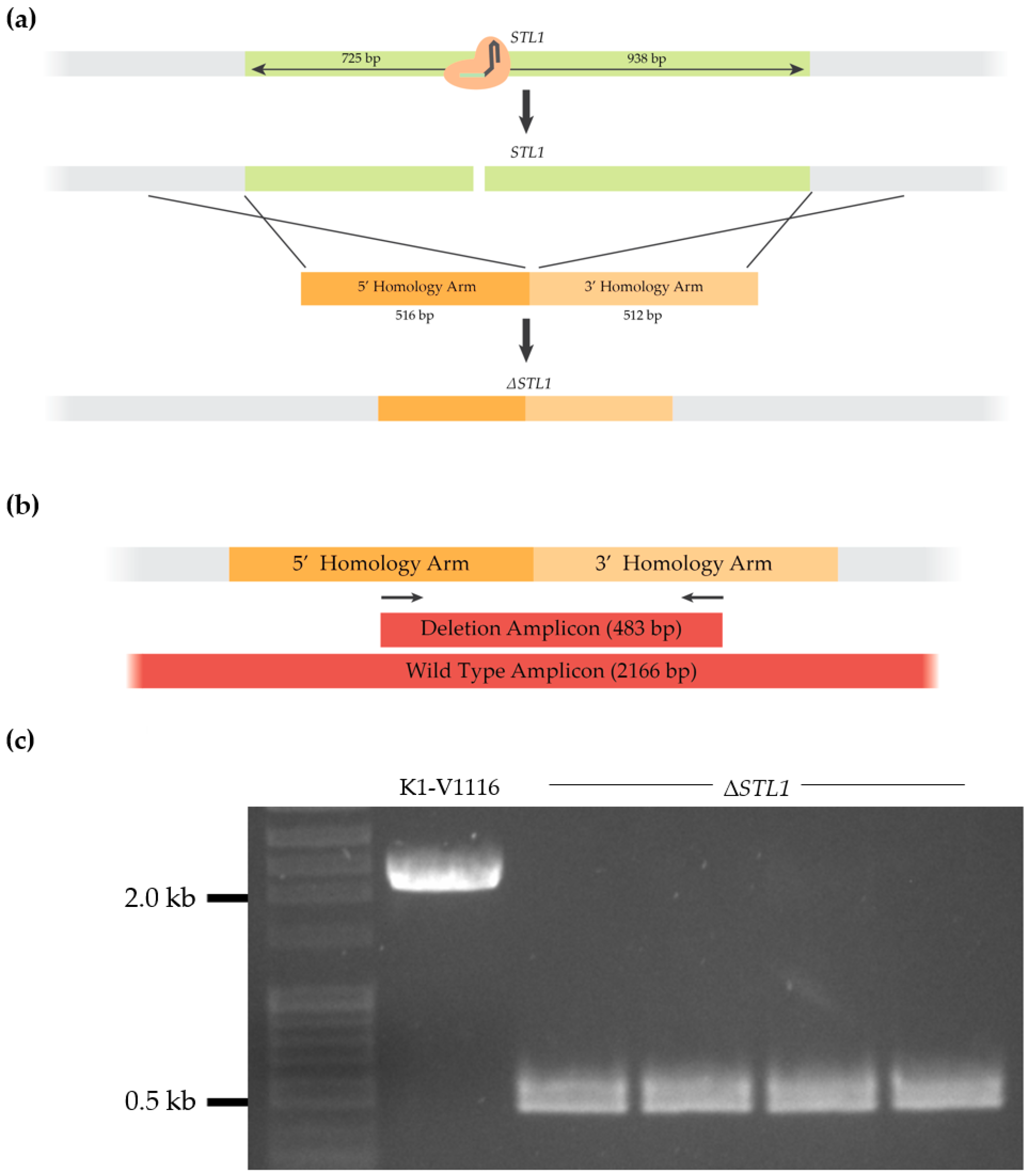
3.1.3. Construction of a K1-V1116 ∆STL1 Strain
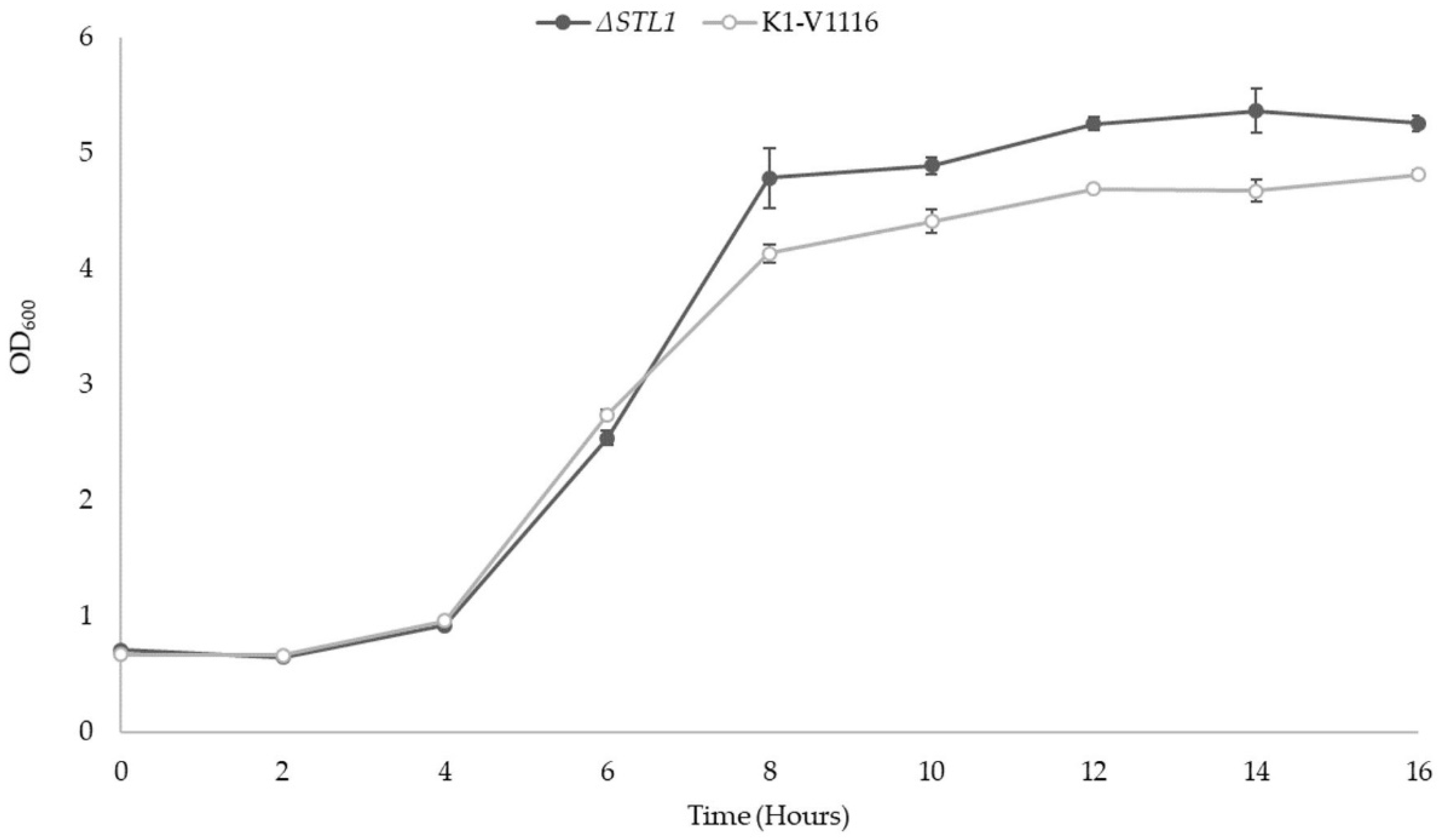
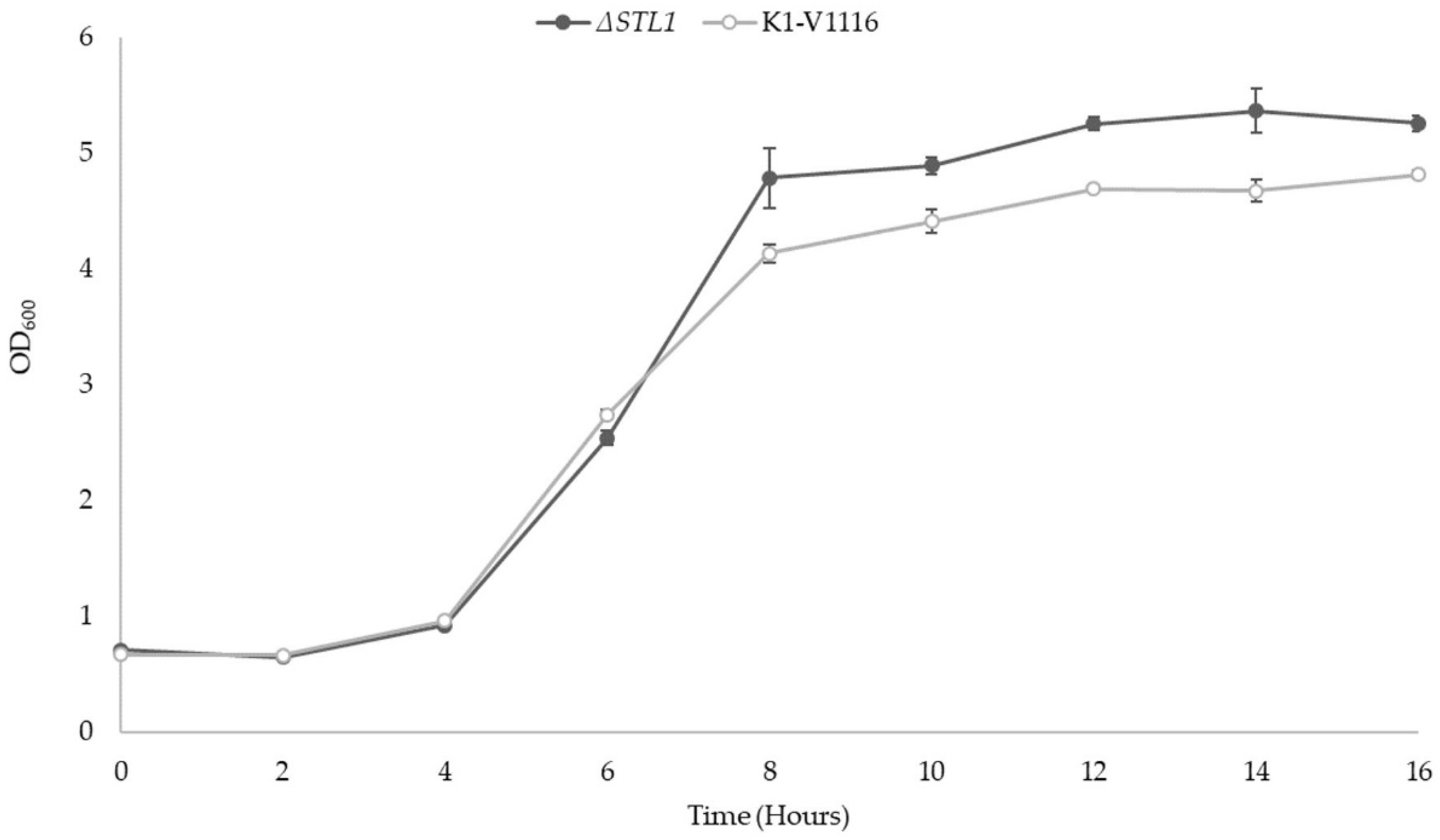
3.2. ∆STL1 Growth Characterization in Permissive Conditions and Icewine Fermentations
3.2.1. ∆STL1 Growth Kinetics in Permissive Conditions
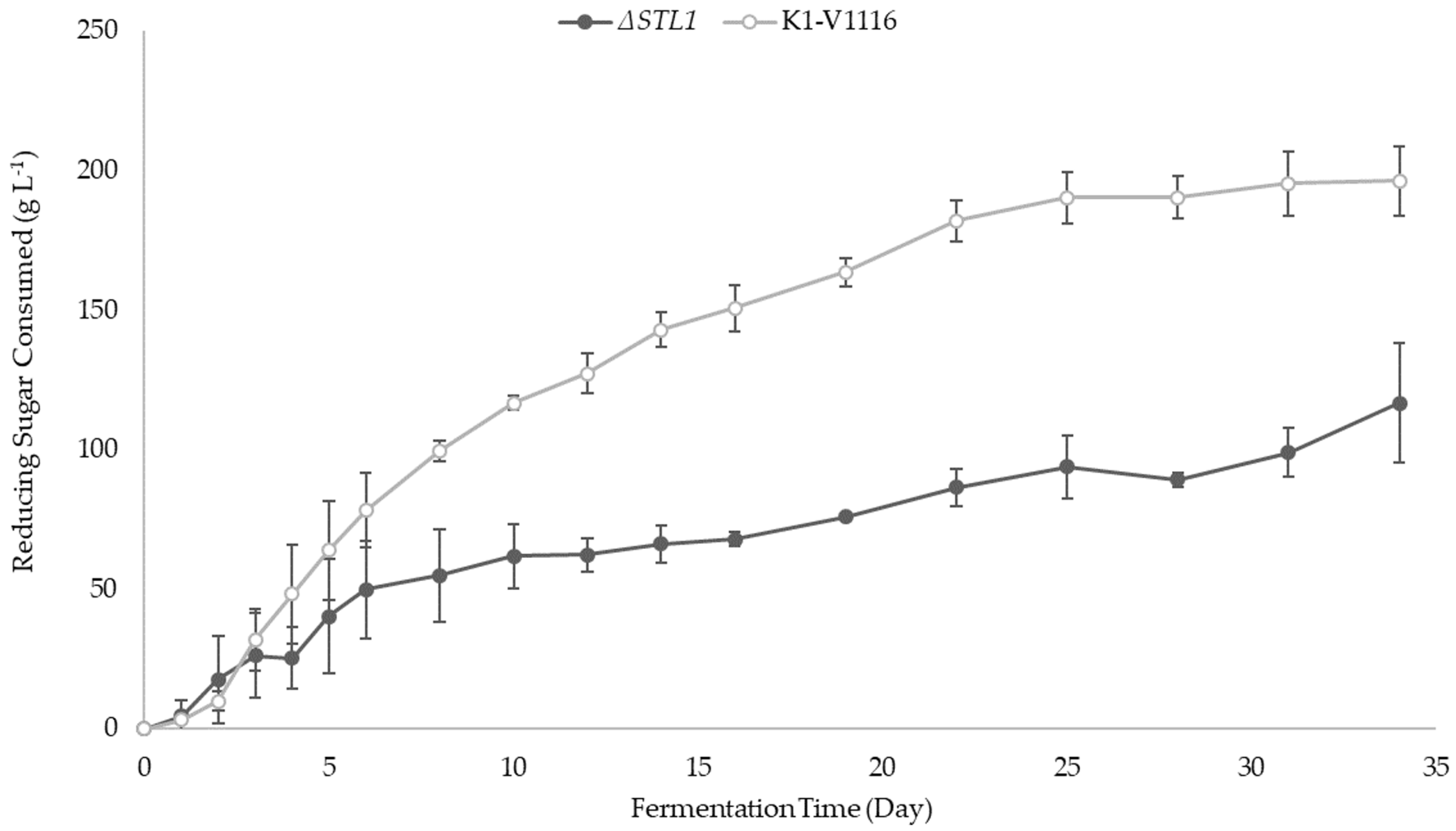
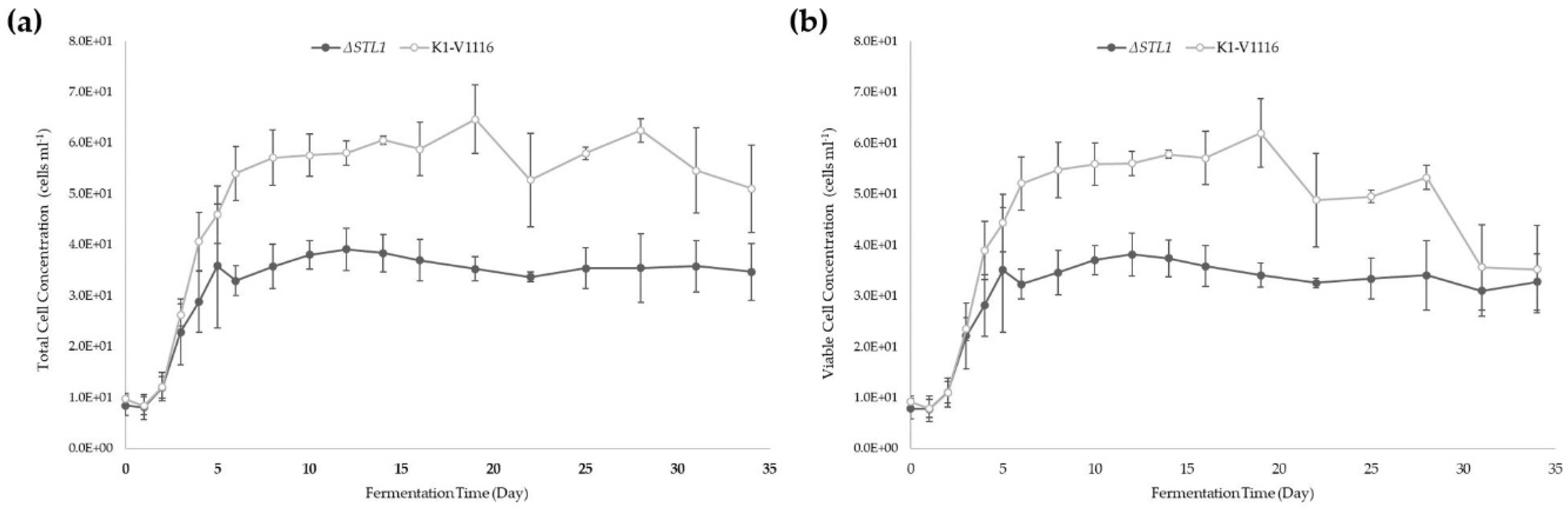
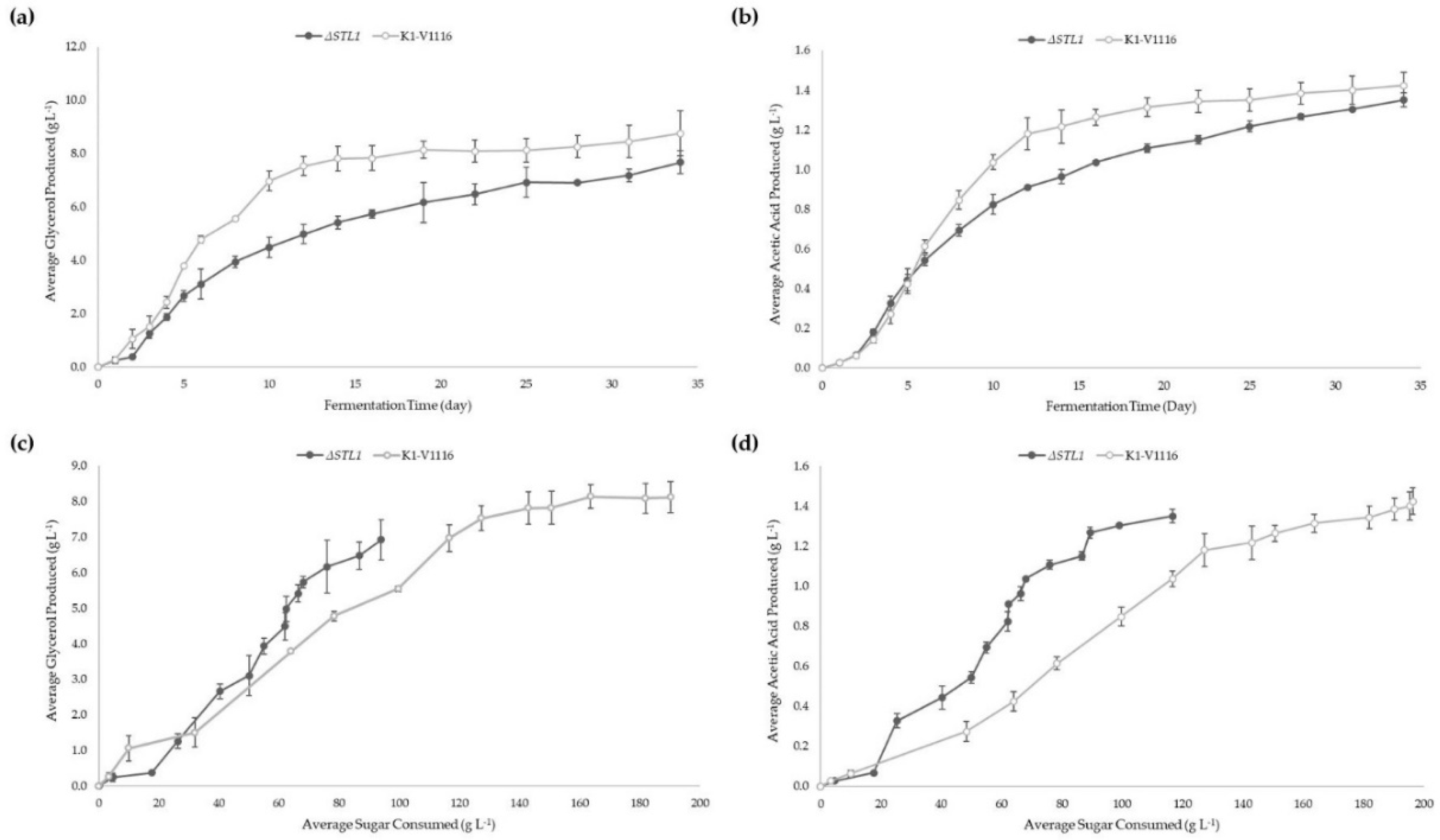
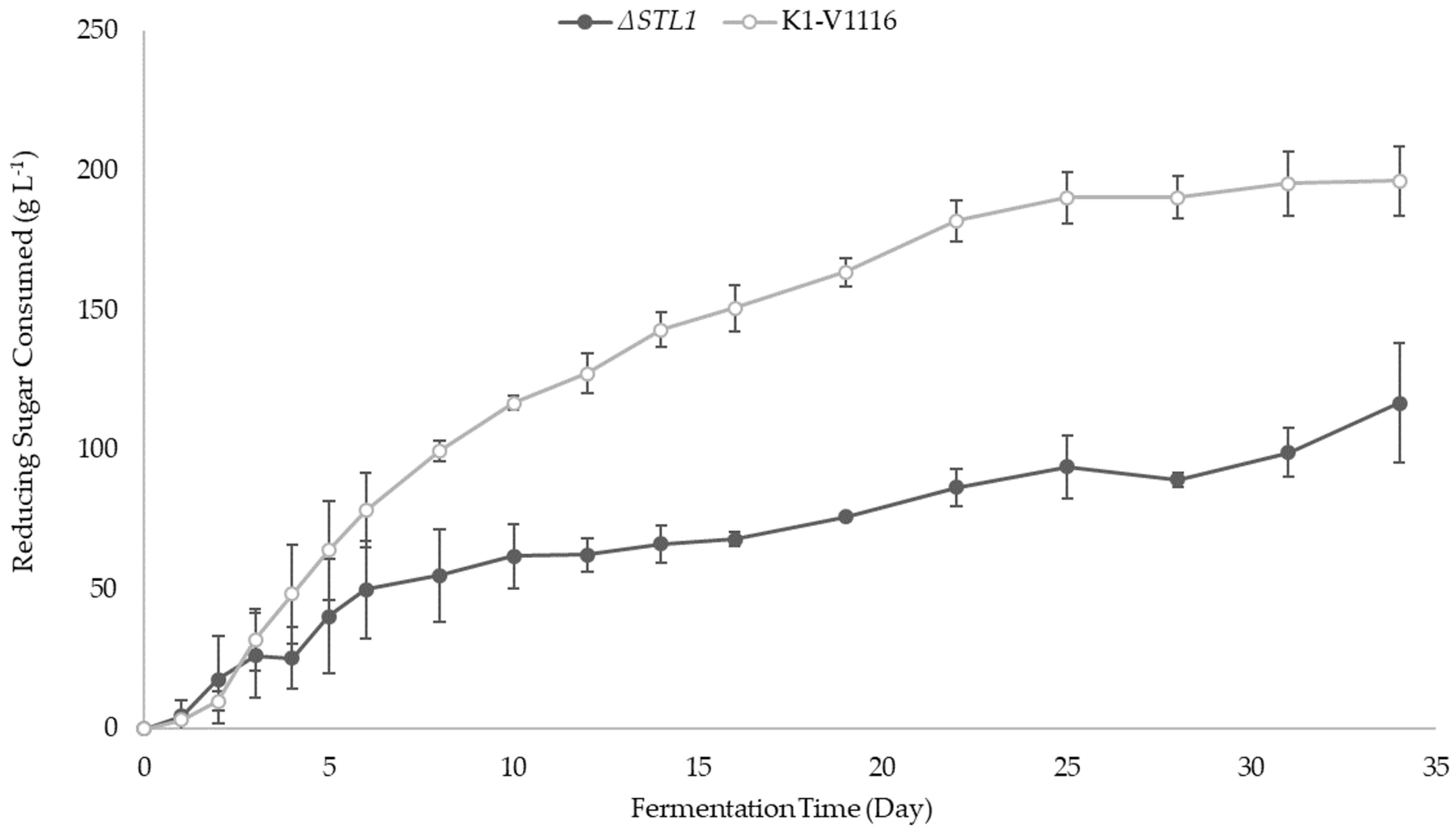
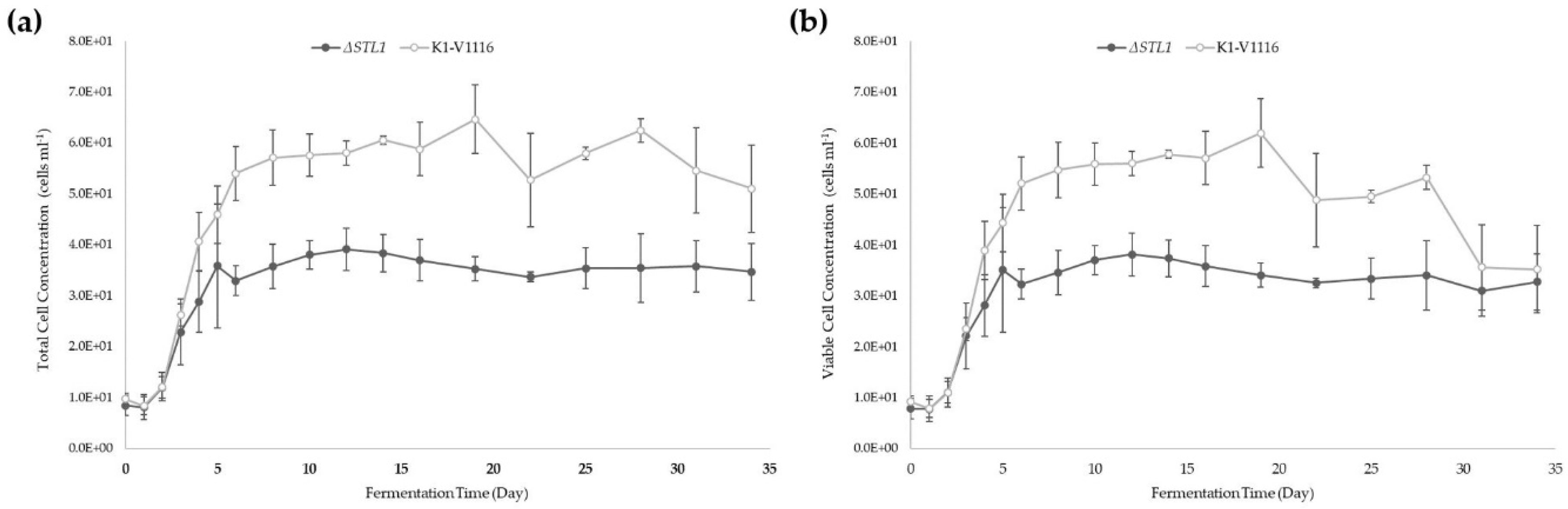
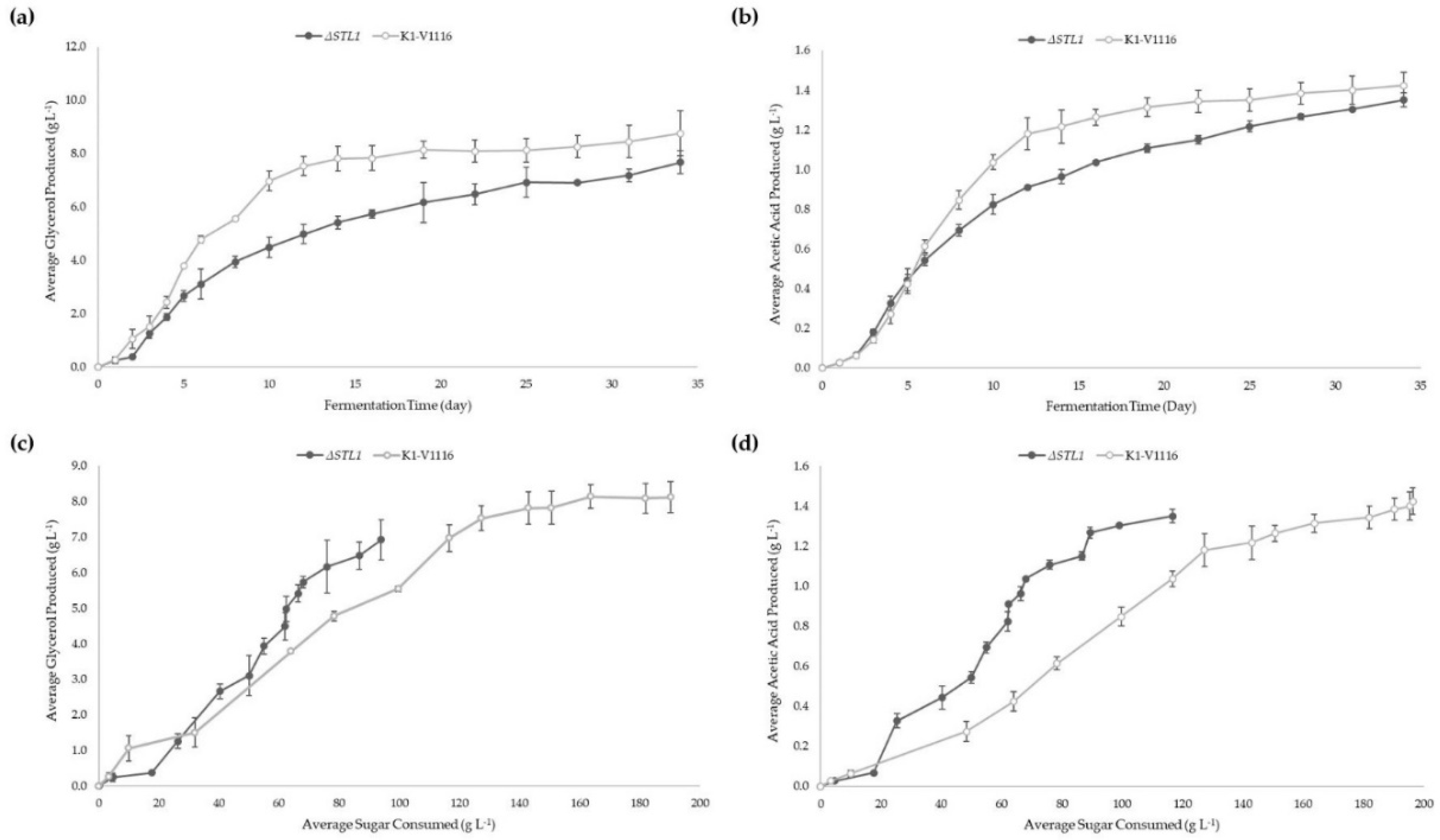
3.2.2. ∆STL1 Icewine Fermentation
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protospacer Sequence | Protospacer Adjacent Motif |
|---|---|
| GGTAGGAACACTAGACGACG | CGG |
| Primer Name | Sequence |
|---|---|
| pWS173 sgRNA FW | ATCAACAACAGAGGACATATGCCCTACCTCCATG |
| pWS173 sgRNA RV | ATCCTGCACTCATCTACTACCCGCATCCC |
| STL1 sgRNA FW | GCTATTTCTAGCTCTAAAACCGTCGTCTAGTGTTCCTACCAAAGTCCCATTCGCCACCCG |
| STL1 sgRNA RV | CGGGTGGCGAATGGGACTTTGGTAGGAACACTAGACGACGGTTTTAGAGCTAGAAATAGC |
| No target sgRNA FW | GCCTTATTTTAACTTGCTATTTCTAGCTCTAAAACAAAGTCCCATTCGCCACCCG |
| No target sgRNA RV | GCTGGGCAACACCTTCGGGTGGCGAATGGGACTTTGTTTTAGAGCTAGAAATAGC |
| STL1 5’ homology arm FW | CGCGGGGTATTCGGCGCTACG |
| STL1 5’ homology arm RV | GTGTATGAATCAACCCTCAAAATTTGCTTTATCGTGGTCTAAAACTTTCTATGTTCTAC |
| STL1 3’ homology arm FW | TAAATAAAAAATAGAAAAGTAGAACATAGAAAGTTTTAGACCACGATAAAGCAAATTTTG |
| STL1 3’ homology arm RV | GCAGTGTTCGAAAGTTGGTGGCTTTTTGTATGTTGC |
| STL1 genotyping FW | CAGAATTGCAGTTCAGGAGTAGTCACAC |
| STL1 genotyping RV | CGCTCCAATGTAATAACACGTCCACGTTATC |
| pWS173 KanR genotyping FW | GGATGGCAAGATCCTGGTATCGG |
| pWS173 KanR genotyping RV | ACTGAATCGGGAGGATCTGCTG |
References
- Inglis, D.L.; Pickering, G.P. Vintning on thin ice—The making of Canada’s iconic dessert wine. In The World of Niagara Wine; Ripmeester, M., Mackintosh, P., Fullerton, C., Eds.; Wilfrid Laurier University Press: Waterloo, ON, Canada, 2013; pp. 229–248. [Google Scholar]
- Kontkanen, D.; Inglis, D.L.; Pickering, G.J.; Reynolds, A. Effect of Yeast Inoculation Rate, Acclimatization, and Nutrient Addition on Icewine Fermentation. Am. J. Enol. Vitic. 2004, 55, 363–370. [Google Scholar]
- Pigeau, G.M.; Inglis, D.L. Upregulation of ALD3 and GPD1 in Saccharomyces cerevisiae during Icewine fermentation. J. Appl. Microbiol. 2005, 99, 112–125. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Heit, C.; Inglis, D.L. Cytosolic Redox Status of Wine Yeast (Saccharomyces cerevisiae) under Hyperosmotic Stress during Icewine Fermentation. Fermentation 2017, 3, 61. [Google Scholar] [CrossRef]
- Björkqvist, S.; Ansell, R.; Adler, L.; Lidén, G. Physiological response to anaerobicity of glycerol-3-phosphate dehydrogenase mutants of Saccharomyces cerevisiae. Appl. Environ. Microbiol. 1997, 63, 128–132. [Google Scholar]
- Bakker, B.M.; Overkamp, K.M.; van Maris, A.J.A.; Kötter, P.; Luttik, M.A.H.; van Dijken, J.P.; Pronk, J.T. Stoichiometry and compartmentation of NADH metabolism in Saccharomyces cerevisiae. FEMS Microbiol. Rev. 2001, 25, 15–37. [Google Scholar] [CrossRef]
- Cronwright, G.R.; Rohwer, J.M.; Prior, B.A. Metabolic Control Analysis of Glycerol Synthesis in Saccharomyces cerevisiae. Appl. Environ. Microbiol. 2002, 68, 4448–4456. [Google Scholar] [CrossRef]
- Blomberg, A.; Adler, L. Roles of glycerol and glycerol-3-phosphate dehydrogenase (NAD +) in acquired osmotolerance of Saccharomyces cerevisiae. J. Bacteriol. 1989, 171, 1087–1092. [Google Scholar] [CrossRef]
- Aranda, A.; del Olmo, M.L. Response to acetaldehyde stress in the yeast Saccharomyces cerevisiae involves a strain-dependent regulation of several ALD genes and is mediated by the general stress response pathway. Yeast 2003, 20, 747–759. [Google Scholar] [CrossRef]
- Hohmann, S. An integrated view on a eukaryotic osmoregulation system. Curr. Genet. 2015, 61, 373–382. [Google Scholar] [CrossRef]
- Ferreira, C.; van Voorst, F.; Martins, A.; Neves, L.; Oliveira, R.; Kielland-Brandt, M.C.; Lucas, C.; Brandt, A. A Member of the Sugar Transporter Family, Stl1p Is the Glycerol/H+ Symporter in Saccharomyces cerevisiae. Mol. Biol. Cell 2005, 16, 2068–2076. [Google Scholar] [CrossRef]
- Nelissen, B.; De Wachter, R.; Goffeau, A. Classification of all putative permeases and other membrane plurispanners of the major facilitator superfamily encoded by the complete genome of Saccharomyces cerevisiae. FEMS Microbiol. Rev. 1997, 21, 113–134. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.; Lucas, C. Glucose repression over Saccharomyces cerevisiae glycerol/H+ symporter gene STL1 is overcome by high temperature. FEBS Lett. 2007, 581, 1923–1927. [Google Scholar] [CrossRef] [PubMed]
- Noti, O.; Vaudano, E.; Pessione, E.; Garcia-Moruno, E. Short-term response of different Saccharomyces cerevisiae strains to hyperosmotic stress caused by inoculation in grape must: RT-qPCR study and metabolite analysis. Food Microbiol. 2015, 52, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Torrado, R.; Oliveira, B.M.; Zemančíková, J.; Sychrová, H.; Querol, A. Alternative Glycerol Balance Strategies among Saccharomyces Species in Response to Winemaking Stress. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef]
- Heit, C.; Martin, S.J.; Yang, F.; Inglis, D.L. Osmoadaptation of wine yeast (Saccharomyces cerevisiae) during Icewine fermentation leads to high levels of acetic acid. J. Appl. Microbiol. 2018, 124, 1506–1520. [Google Scholar] [CrossRef] [PubMed]
- Hauser, N.C.; Fellenberg, K.; Gil, R.; Bastuck, S.; Hoheisel, J.D.; Pérez-Ortín, J.E. Whole Genome Analysis of a Wine Yeast Strain. Comp. Funct. Genomics 2001, 2, 69–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiménez-Martí, E.; Gomar-Alba, M.; Palacios, A.; Ortiz-Julien, A.; del Olmo, M. Towards an understanding of the adaptation of wine yeasts to must: Relevance of the osmotic stress response. Appl. Microbiol. Biotechnol. 2011, 89, 1551–1561. [Google Scholar] [CrossRef]
- Jiménez-Martí, E.; Zuzuarregui, A.; Gomar-Alba, M.; Gutiérrez, D.; Gil, C.; del Olmo, M. Molecular response of Saccharomyces cerevisiae wine and laboratory strains to high sugar stress conditions. Int. J. Food Microbiol. 2011, 145, 211–220. [Google Scholar] [CrossRef]
- Pronk, J.T. Auxotrophic Yeast Strains in Fundamental and Applied Research. Appl. Environ. Microbiol. 2002, 68, 2095–2100. [Google Scholar] [CrossRef] [Green Version]
- Le Borgne, S. Genetic Engineering of Industrial Strains of Saccharomyces cerevisiae. In Recombinant Gene Expression; Lorence, A., Ed.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 451–465. [Google Scholar] [CrossRef]
- Stovicek, V.; Holkenbrink, C.; Borodina, I. CRISPR/Cas system for yeast genome engineering: Advances and applications. FEMS Yeast Res. 2017, 17. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, J.; Wang, Z.; Zhang, Y.; Shi, S.; Nielsen, J.; Liu, Z. A gRNA-tRNA array for CRISPR-Cas9 based rapid multiplexed genome editing in Saccharomyces cerevisiae. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Vigentini, I.; Gebbia, M.; Belotti, A.; Foschino, R.; Roth, F.P. CRISPR/Cas9 System as a Valuable Genome Editing Tool for Wine Yeasts with Application to Decrease Urea Production. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Shaw, W. Quick and Easy CRISPR Engineering in Saccharomyces cerevisiae. 2018. Available online: https://www.benchling.com/2018/11/06/quick-and-easy-crispr-engineering-in-saccharomyces-cerevisiae/ (accessed on 10 November 2018).
- Novo, M.; Bigey, F.; Beyne, E.; Galeote, V.; Gavory, F.; Mallet, S.; Cambon, B.; Legras, J.-L.; Wincker, P.; Casaregola, S.; et al. Eukaryote-to-eukaryote gene transfer events revealed by the genome sequence of the wine yeast Saccharomyces cerevisiae EC1118. Proc. Natl. Acad. Sci. USA 2009, 106, 16333–16338. [Google Scholar] [CrossRef]
- DiCarlo, J.E.; Norville, J.E.; Mali, P.; Rios, X.; Aach, J.; Church, G.M. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 2013, 41, 4336–4343. [Google Scholar] [CrossRef]
- Zoecklein, B.W.; Fugelsang, K.C.; Gump, B.H. Laboratory Procedures. In Wine Analysis and Production, 1st ed.; Chapman and Hall, International Thomson Publishing: New York, NY, USA, 1995; pp. 374–378. [Google Scholar]
- Grey, M.; Brendel, M. A ten-minute protocol for transforming Saccharomyces cerevisiae by electroporation. Curr. Genet. 1992, 22, 335–336. [Google Scholar] [CrossRef]
- Tripp, J.D.; Lilley, J.L.; Wood, W.N.; Lewis, L.K. Enhancement of plasmid DNA transformation efficiencies in early stationary phase yeast cell cultures. Yeast 2013, 30, 191–200. [Google Scholar] [CrossRef]
- Kuijpers, N.G.; Solis-Escalante, D.; Bosman, L.; van den Broek, M.; Pronk, J.T.; Daran, J.-M.; Daran-Lapujade, P. A versatile, efficient strategy for assembly of multi-fragment expression vectors in Saccharomyces cerevisiae using 60 bp synthetic recombination sequences. Microb. Cell Factories 2013, 12, 47. [Google Scholar] [CrossRef]
- Förster, J.; Famili, I.; Fu, P.; Palsson, B.Ø.; Nielsen, J. Genome-Scale Reconstruction of the Saccharomyces cerevisiae Metabolic Network. Genome Res. 2003, 13, 244–253. [Google Scholar] [CrossRef]
- Nielsen, J. It Is All about Metabolic Fluxes. J. Bacteriol. 2003, 185, 7031–7035. [Google Scholar] [CrossRef]
- Miermont, A.; Waharte, F.; Hu, S.; McClean, M.N.; Bottani, S.; Léon, S.; Hersen, P. Severe osmotic compression triggers a slowdown of intracellular signaling, which can be explained by molecular crowding. Proc. Natl. Acad. Sci. USA 2013, 110, 5725–5730. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.P. Biological principles for the effects of ethanol. Enzym. Microb. Technol. 1989, 11, 130–153. [Google Scholar] [CrossRef]
- Bisson, L.F. Stuck and Sluggish Fermentations. Am. J. Enol. Vitic. 1999, 50, 107–119. [Google Scholar]
- Monteiro, B.; Ferraz, P.; Barroca, M.; da Cruz, S.H.; Collins, T.; Lucas, C. Conditions promoting effective very high gravity sugarcane juice fermentation. Biotechnol. Biofuels 2018, 11, 251. [Google Scholar] [CrossRef] [PubMed]
- Lages, F.; Silva-Graça, M.; Lucas, C. Active glycerol uptake is a mechanism underlying halotolerance in yeasts: A study of 42 species. Microbiology 1999, 145, 2577–2585. [Google Scholar] [CrossRef]
- Dušková, M.; Ferreira, C.; Lucas, C.; Sychrová, H. Two glycerol uptake systems contribute to the high osmotolerance of Zygosaccharomyces rouxii. Mol. Microbiol. 2015, 97, 541–559. [Google Scholar] [CrossRef]
| Parameter | Icewine Juice | Wine from K1-V1116 | Wine from K1-V1116 ΔSTL1 |
|---|---|---|---|
| Reducing Sugars (g L−1) | 447 ± 19 a | 251 ± 11 c | 333 ± 19 b |
| pH | 3.79 ± 0.04 b | 3.90 ± 0.02 a | 3.84 ± 0.08 a,b |
| Titratable Acidity (g L−1) | 7.0 ± 0.1 b | 8.6 ± 0.2 a | 8.6 ± 0.3 a |
| Yeast Assimilable Nitrogen (mg N L −1) | 441 ± 2 a | 345 ± 13 c | 396 ± 14 b |
| Glycerol (g L−1) | 8.67 ± 0.32 b | 17.42 ± 1.12 a | 16.36 ± 0.32 a |
| Acetic Acid (g L−1) | 0.06 ± 0.01 b | 1.48 ± 0.07 a | 1.39 ± 0.05 a |
| Ethanol (% v/v) | 1.3 ± 0.0 c | 12.3 ± 0.6 a | 6.7 ± 0.6 b |
| Parameter | K1-V1116 | K1-V1116 ΔSTL1 |
|---|---|---|
| Reducing Sugar consumed (g L−1) | 196 ± 13 ** | 117 ± 21 ** |
| Yeast Assimilable Nitrogen consumed (mg N L−1) | 92 ± 17 * | 44 ± 12 * |
| Glycerol produced (g L−1) | 8.76 ± 0.84 | 7.68 ± 0.44 |
| Acetic Acid produced (g L−1) | 1.42 ± 0.07 | 1.35 ± 0.07 |
| Normalized Glycerol production (mg glycerol per g sugar consumed) | 45 ± 3 * | 67 ± 12 * |
| Normalized Acetic Acid production (mg acetic acid per g sugar consumed) | 7 ± 0 ** | 12 ± 2 ** |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muysson, J.; Miller, L.; Allie, R.; Inglis, D.L. The Use of CRISPR-Cas9 Genome Editing to Determine the Importance of Glycerol Uptake in Wine Yeast During Icewine Fermentation. Fermentation 2019, 5, 93. https://doi.org/10.3390/fermentation5040093
Muysson J, Miller L, Allie R, Inglis DL. The Use of CRISPR-Cas9 Genome Editing to Determine the Importance of Glycerol Uptake in Wine Yeast During Icewine Fermentation. Fermentation. 2019; 5(4):93. https://doi.org/10.3390/fermentation5040093
Chicago/Turabian StyleMuysson, Jared, Laurianne Miller, Robert Allie, and Debra L. Inglis. 2019. "The Use of CRISPR-Cas9 Genome Editing to Determine the Importance of Glycerol Uptake in Wine Yeast During Icewine Fermentation" Fermentation 5, no. 4: 93. https://doi.org/10.3390/fermentation5040093
APA StyleMuysson, J., Miller, L., Allie, R., & Inglis, D. L. (2019). The Use of CRISPR-Cas9 Genome Editing to Determine the Importance of Glycerol Uptake in Wine Yeast During Icewine Fermentation. Fermentation, 5(4), 93. https://doi.org/10.3390/fermentation5040093