Looking at the Origin: Some Insights into the General and Fermentative Microbiota of Vineyard Soils

 , , , , , , , , , and
, , , , , , , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Site Description and Weather Data
2.2. Soil Sampling
2.3. DNA Extraction and Sequencing
2.4. Bioinformatic Analysis
2.5. Functional Profiles Prediction
2.6. Statistical Analysis
3. Results and Discussion
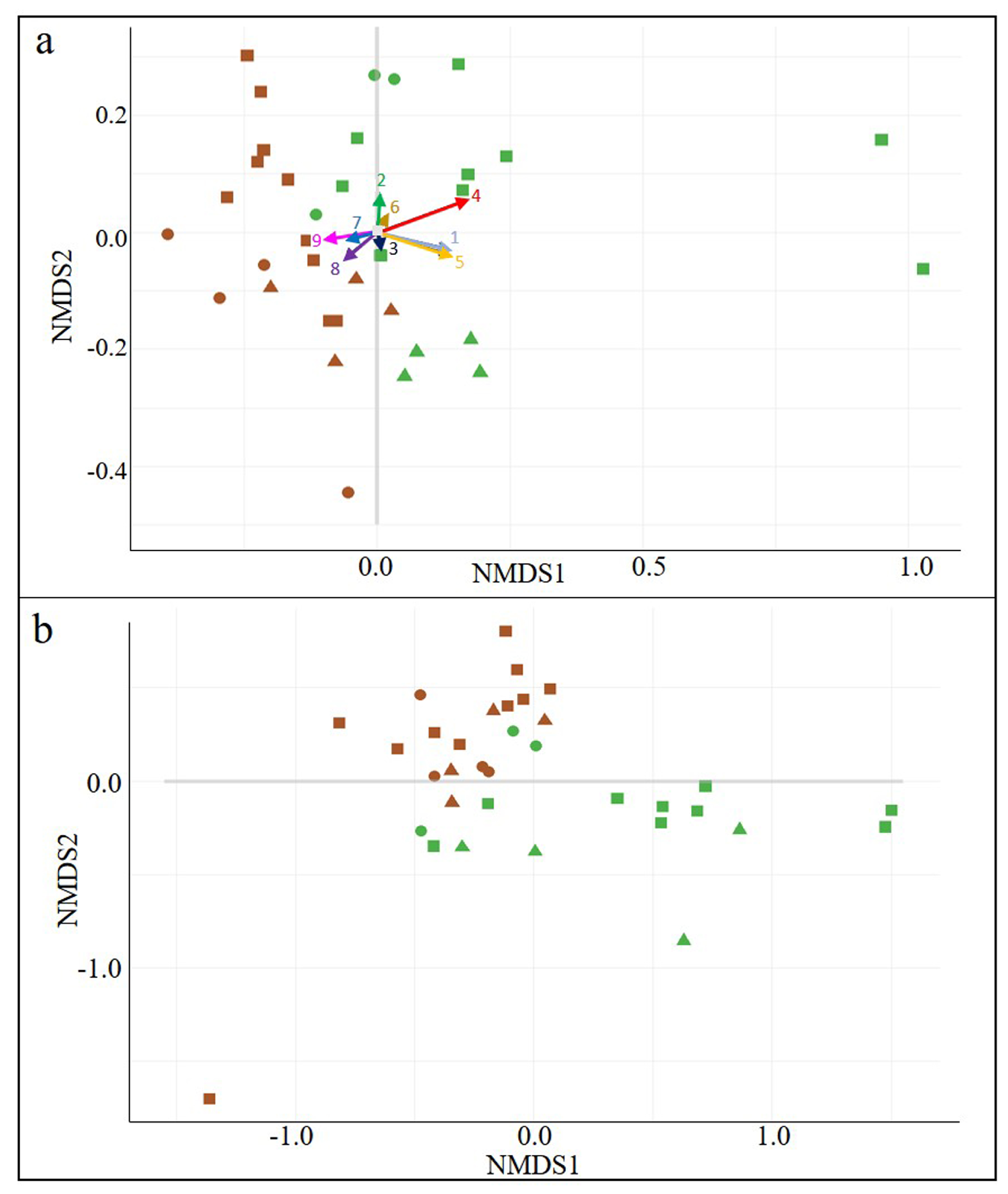
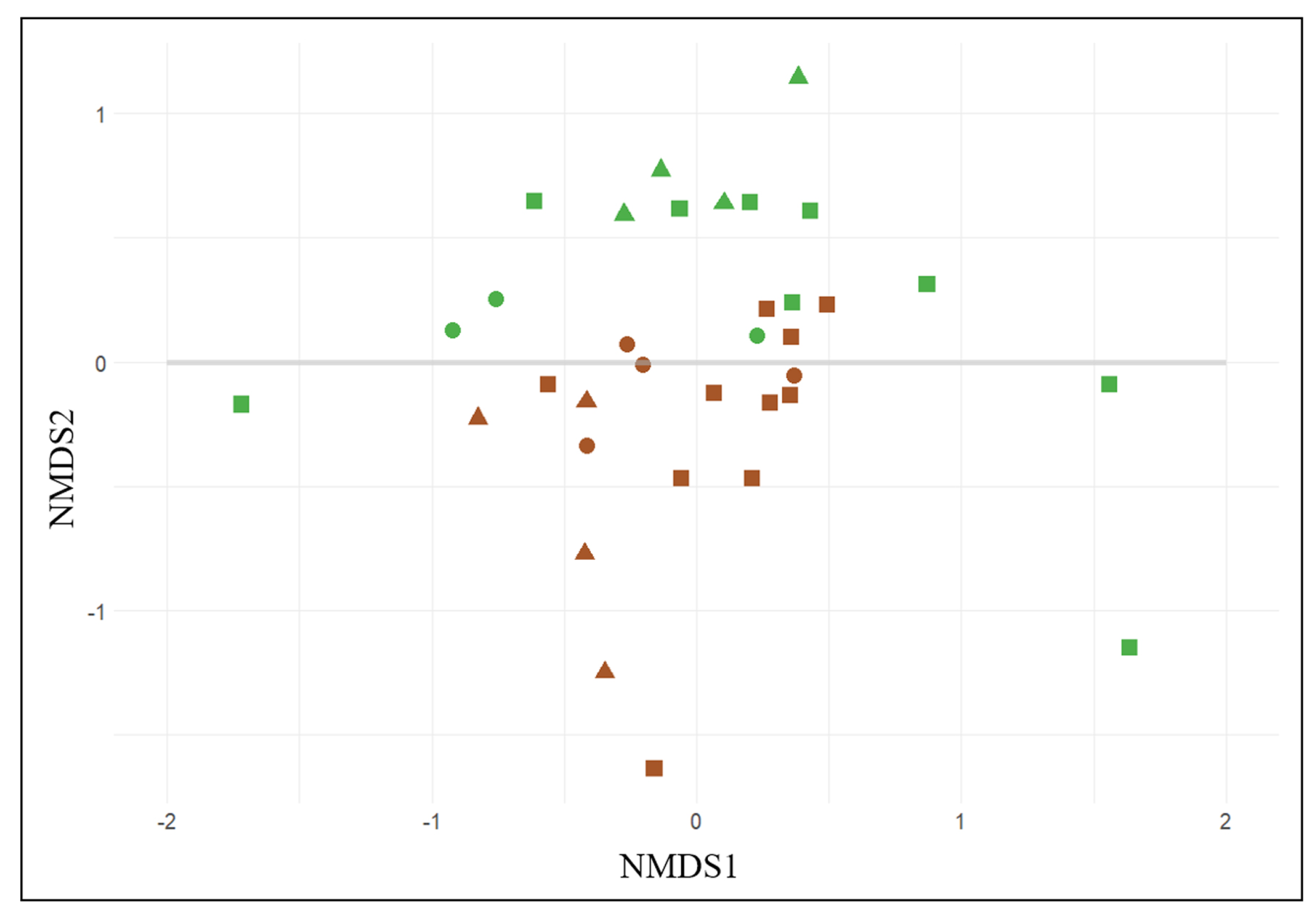
3.1. General Biodiversity
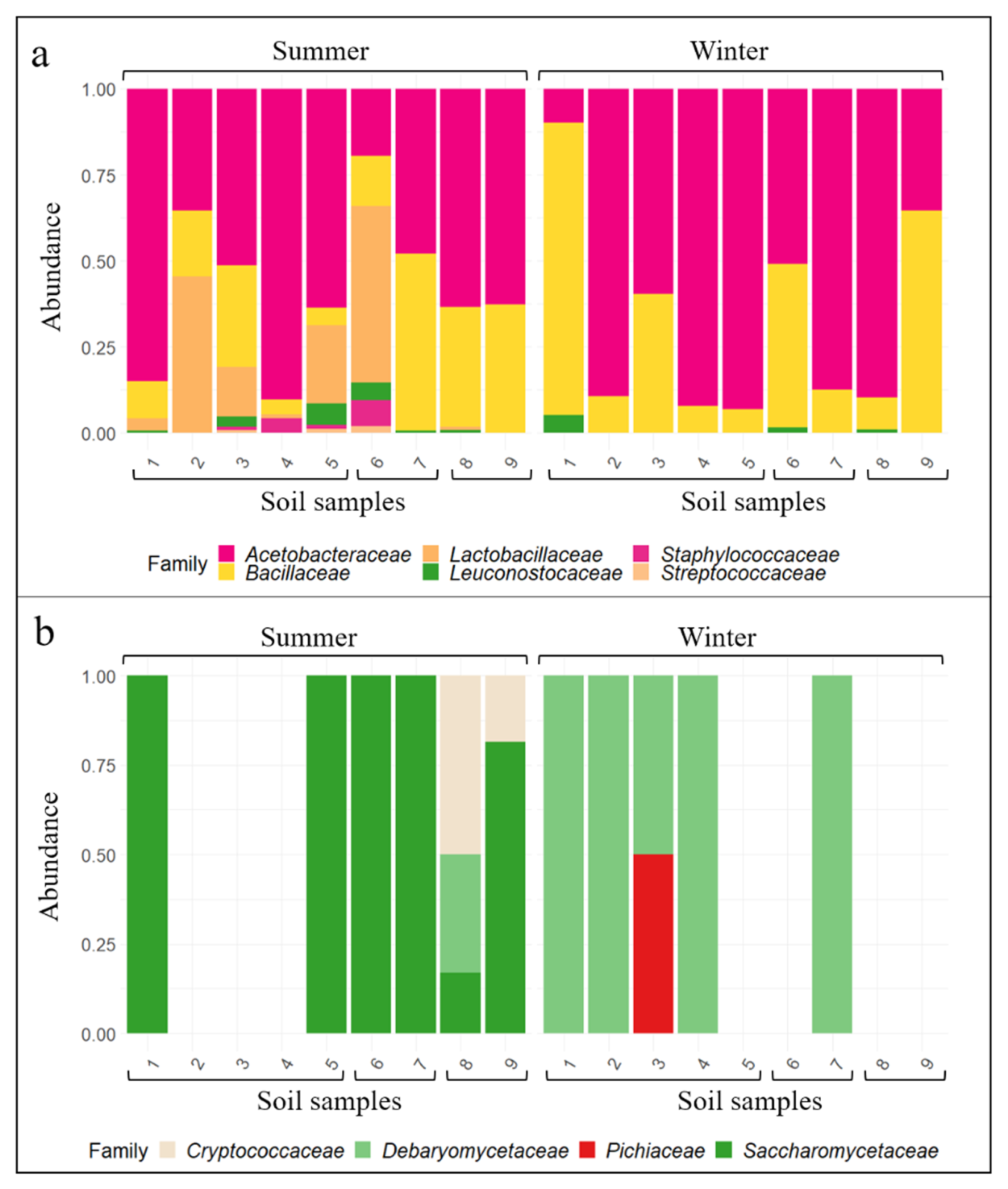
3.2. Wine-Related Microbial Diversity
4. Conclusions
Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ASV | amplicon sequence variant |
| ITS | internal transcribed spacer |
| NMDS | non-parametric multi-dimensional scale |
| PCA | principal component analysis |
| VCPRD | a quality wine psr |
| WRB | wine-related bacteria |
| WRF | wine-related fungi |
Appendix A. Soil Compositional Characteristics


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Soil Sample | Sand (%) | Clay (%) | Limestone (%) |
|---|---|---|---|
| 1 | 59.8 | 20.4 | 3.1 |
| 2 | 62.5 | 19.8 | 2.6 |
| 3 | 63.7 | 16.4 | 2.8 |
| 4 | 65.9 | 15.0 | 3.4 |
| 5 | 75.3 | 13.7 | 2.9 |
| 6 | 48.3 | 22.5 | 3.7 |
| 7 | 39.5 | 24.5 | 5.1 |
| 8 | 58.0 | 22.4 | 9.8 |
| 9 | 62.5 | 19.3 | 11.7 |
Appendix B. KEGGs and Metabolism Pathways

| KEGG | Functional Description: Name [EC] (gen) | Metabolism |
|---|---|---|
| K02274 | cytochrome c oxidase subunit I [EC:1.9.3.1] (coxA) | Aerobic Respiration |
| K00174 | 2-oxoglutarate ferredoxin oxidoreductase subunit alpha [EC:1.2.7.3] (korA) | Arnon Carbon Fixation |
| K00175 | 2-oxoglutarate ferredoxin oxidoreductase subunit beta [EC:1.2.7.3] (korB) | Arnon Carbon Fixation |
| K00244 | fumarate reductase flavoprotein subunit [EC:1.3.5.4] (frdA) | Arnon Carbon Fixation |
| K00860 | adenylylsulfate kinase [EC:2.7.1.25] (cysC) | Assimilatory Sulfate Reduction |
| K00957 | sulfate adenylyltransferase subunit 2 [EC:2.7.7.4] (cysD) | Assimilatory Sulfate Reduction |
| K00016 | l-lactate dehydrogenase [EC:1.1.1.27] (LDH, ldh) | Fermentation |
| K05816 | sn-glycerol 3-phosphate transport system ATP-binding protein [EC:3.6.3.20] (ugpC) | G3P Transporter |
| K00400 | coenzyme Methyl reductase beta subunit (mrcB) | Methanogenesis |
| K00401 | methyl coenzyme M reductase system A2 | Methanogenesis |
| K00265 | glutamate synthase (NADPH/NADH) large chain [EC:1.4.1.13, 1.4.1.14] (gltB) | Nitrogen Assimilation |
| K01915 | glutamine synthetase [EC:6.3.1.2] (glnA, GLUL) | Nitrogen Assimilation |
| K02588 | nitrogenase iron protein NifH [EC:1.18.6.1] (nifH) | Nitrogen Fixation |
| K02591 | nitrogenase molybdenum-iron protein beta chain [EC:1.18.6.1] (nifK) | Nitrogen Fixation |
| K00261 | glutamate dehydrogenase (NAD(P)+) [EC:1.4.1.3] (GLUD1 2, gdhA) | Nitrogen Mineralization |
| K00262 | glutamate dehydrogenase (NADP+) [EC:1.4.1.4] (gdhA) | Nitrogen Mineralization |
| K00260 | glutamate dehydrogenase [EC:1.4.1.2] (gudB, rocG) | Nitrogen Mineralization |
| K02567 | periplasmic nitrate reductase NapA [EC:1.7.99.4] (napA) | Nitrogen Reduction |
| K02036 | phosphate transport system ATP-binding protein [EC:3.6.3.27] (pstB) | Phosphate Transport High |
| K02038 | phosphate transport system permease protein (pstA) | Phosphate Transport High |
| K02037 | phosphate transport system permease protein (pstC) | Phosphate Transport High |
| K03430 | 2-aminoethylphosphonate-pyruvate transaminase [EC:2.6.1.37] (phnW) | Phosphonate Metabolism |
| K04750 | PhnB protein (phnB) | Phosphonate Transport |
| K02041 | phosphonate transport system ATP-binding protein [EC:3.6.3.28] (phnC) | Phosphonate Transport |
| K01011 | thiosulfate/3-mercaptopyruvate sulfurtransferase [EC:2.8.1.1, 2.8.1.2] (TST, MPST, sseA) | Sulfur Mineralitation |
Appendix C. Wine-Related Microorganism
| Kingdom | Phylum | Class | Order | Family |
|---|---|---|---|---|
| Bacteria | Firmicutes | Bacilli | Bacillales | Bacillaceae |
| Bacteria | Firmicutes | Bacilli | Bacillales | Staphylococcaceae |
| Bacteria | Firmicutes | Bacilli | Lactobacillales | Enterococcaceae |
| Bacteria | Firmicutes | Bacilli | Lactobacillales | Lactobacillaceae |
| Bacteria | Firmicutes | Bacilli | Lactobacillales | Leuconostocaceae |
| Bacteria | Firmicutes | Bacilli | Lactobacillales | Streptococcaceae |
| Bacteria | Proteobacteria | Alphaproteobacteria | Rhodospirillales | Acetobacteraceae |
| Fungi | Ascomycota | Saccharomycetes | Saccharomycetales | Debaryomycetaceae |
| Fungi | Ascomycota | Saccharomycetes | Saccharomycetales | Metschnikowiaceae |
| Fungi | Ascomycota | Saccharomycetes | Saccharomycetales | Pichiaceae |
| Fungi | Ascomycota | Saccharomycetes | Saccharomycetales | Saccharomycetaceae |
| Fungi | Basidiomycota | Tremellomycetes | Tremellales | Cryptococcaceae |
References
- Frąc, M.; Hannula, S.E.; Bełka, M.; Jędryczka, M. Fungal biodiversity and their role in soil health. Front. Microbiol. 2018, 9, 707. [Google Scholar] [CrossRef] [PubMed]
- Isbell, F.I.; Polley, H.W.; Wilsey, B.J. Biodiversity, productivity and the temporal stability of productivity: Patterns and processes. Ecol. Lett. 2009, 12, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Jactel, H.; Gritti, E.; Drössler, L.; Forrester, D.; Mason, W.; Morin, X.; Pretzsch, H.; Castagneyrol, B. Positive biodiversity–productivity relationships in forests: Climate matters. Biol. Lett. 2018, 14, 20170747. [Google Scholar] [CrossRef] [PubMed]
- Balvanera, P.; Pfisterer, A.B.; Buchmann, N.; He, J.S.; Nakashizuka, T.; Raffaelli, D.; Schmid, B. Quantifying the evidence for biodiversity effects on ecosystem functioning and services. Ecol. Lett. 2006, 9, 1146–1156. [Google Scholar] [CrossRef] [PubMed]
- Reed, H.E.; Martiny, J.B. Testing the functional significance of microbial composition in natural communities. FEMS Microbiol. Ecol. 2007, 62, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Maron, P.A.; Sarr, A.; Kaisermann, A.; Lévêque, J.; Mathieu, O.; Guigue, J.; Karimi, B.; Bernard, L.; Dequiedt, S.; Terrat, S.; et al. High microbial diversity promotes soil ecosystem functioning. Appl. Environ. Microbiol. 2018, 84, e02738-17. [Google Scholar] [CrossRef] [PubMed]
- Van Der Heijden, M.G.; Bardgett, R.D.; Van Straalen, N.M. The unseen majority: Soil microbes as drivers of plant diversity and productivity in terrestrial ecosystems. Ecol. Lett. 2008, 11, 296–310. [Google Scholar] [CrossRef]
- Allison, S.D.; Martiny, J.B. Resistance, resilience, and redundancy in microbial communities. Proc. Natl. Acad. Sci. USA 2008, 105, 11512–11519. [Google Scholar] [CrossRef]
- Bardgett, R.D.; Van Der Putten, W.H. Belowground biodiversity and ecosystem functioning. Nature 2014, 515, 505. [Google Scholar] [CrossRef]
- Maron, J.L.; Marler, M.; Klironomos, J.N.; Cleveland, C.C. Soil fungal pathogens and the relationship between plant diversity and productivity. Ecol. Lett. 2011, 14, 36–41. [Google Scholar] [CrossRef]
- Katan, J. Diseases caused by soilborne pathogens: Biology, management and challenges. J. Plant Pathol. 2017, 99, 305–315. [Google Scholar]
- Gilbert, J.A.; van der Lelie, D.; Zarraonaindia, I. Microbial terroir for wine grapes. Proc. Natl. Acad. Sci. USA 2014, 111, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.; Marquina, D. Killer toxin of Pichia membranifaciens and its possible use as a biocontrol agent against grey mould disease of grapevine. Microbiology 2004, 150, 2527–2534. [Google Scholar] [CrossRef] [PubMed]
- Karuppiah, V.; Li, T.; Vallikkannu, M.; Chen, J. Co-cultivation of Trichoderma asperellum GDFS1009 and Bacillus amyloliquefaciens 1841 causes differential gene expression and improvement in the wheat growth and biocontrol activity. Front. Microbiol. 2019, 10, 1068. [Google Scholar] [CrossRef] [PubMed]
- Zarraonaindia, I.; Owens, S.M.; Weisenhorn, P.; West, K.; Hampton-Marcell, J.; Lax, S.; Bokulich, N.A.; Mills, D.A.; Martin, G.; Taghavi, S.; et al. The soil microbiome influences grapevine-associated microbiota. MBio 2015, 6, e02527–14. [Google Scholar] [CrossRef] [PubMed]
- Cordero-Bueso, G.; Arroyo, T.; Serrano, A.; Valero, E. Remanence and survival of commercial yeast in different ecological niches of the vineyard. FEMS Microbiol. Ecol. 2011, 77, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Tello, J.; Cordero-Bueso, G.; Aporta, I.; Cabellos, J.; Arroyo, T. Genetic diversity in commercial wineries: Effects of the farming system and vinification management on wine yeasts. J. Appl. Microbiol. 2012, 112, 302–315. [Google Scholar] [CrossRef]
- Capozzi, V.; Garofalo, C.; Chiriatti, M.A.; Grieco, F.; Spano, G. Microbial terroir and food innovation: The case of yeast biodiversity in wine. Microbiol. Res. 2015, 181, 75–83. [Google Scholar] [CrossRef]
- Garofalo, C.; El Khoury, M.; Lucas, P.; Bely, M.; Russo, P.; Spano, G.; Capozzi, V. Autochthonous starter cultures and indigenous grape variety for regional wine production. J. Appl. Microbiol. 2015, 118, 1395–1408. [Google Scholar] [CrossRef]
- De Celis, M.; Ruiz, J.; Martín-Santamaría, M.; Alonso, A.; Marquina, D.; Navascués, E.; Gómez-Flechoso, M.Á.; Belda, I.; Santos, A. Diversity of Saccharomyces cerevisiae yeasts associated to spontaneous and inoculated fermenting grapes from Spanish vineyards. Lett. Appl. Microbiol. 2019, 68, 580–588. [Google Scholar] [CrossRef]
- Viel, A.; Legras, J.L.; Nadai, C.; Carlot, M.; Lombardi, A.; Crespan, M.; Migliaro, D.; Giacomini, A.; Corich, V. The geographic distribution of Saccharomyces cerevisiae isolates within three Italian neighboring winemaking regions reveals strong differences in yeast abundance, genetic diversity and industrial strain dissemination. Front. Microbiol. 2017, 8, 1595. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N.A.; Collins, T.S.; Masarweh, C.; Allen, G.; Heymann, H.; Ebeler, S.E.; Mills, D.A. Associations among wine grape microbiome, metabolome, and fermentation behavior suggest microbial contribution to regional wine characteristics. MBio 2016, 7, e00631-16. [Google Scholar] [CrossRef] [PubMed]
- Sirén, K.; Mak, S.S.T.; Fischer, U.; Hansen, L.H.; Gilbert, M.T.P. Multi-omics and potential applications in wine production. Curr. Opin. Biotechnol. 2019, 56, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Belda, I.; Zarraonaindia, I.; Perisin, M.; Palacios, A.; Acedo, A. From vineyard soil to wine fermentation: Microbiome approximations to explain the “terroir” concept. Front. Microbiol. 2017, 8, 821. [Google Scholar] [CrossRef] [PubMed]
- Raynaud, X.; Nunan, N. Spatial ecology of bacteria at the microscale in soil. PLoS ONE 2014, 9, e87217. [Google Scholar] [CrossRef] [PubMed]
- Feld, L.; Nielsen, T.K.; Hansen, L.H.; Aamand, J.; Albers, C.N. Establishment of bacterial herbicide degraders in a rapid sand filter for bioremediation of phenoxypropionate-polluted groundwater. Appl. Environ. Microbiol. 2016, 82, 878–887. [Google Scholar] [CrossRef] [PubMed]
- Albers, C.N.; Ellegaard-Jensen, L.; Hansen, L.H.; Sørensen, S.R. Bioaugmentation of rapid sand filters by microbiome priming with a nitrifying consortium will optimize production of drinking water from groundwater. Water Res. 2018, 129, 1–10. [Google Scholar] [CrossRef]
- Becares, A.A.; Fernandez, A.F. Microbiome Based Identification, Monitoring and Enhancement Of Fermentation Processes and Products. US Patent Application 15/779,531, 20 December 2018. [Google Scholar]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Callahan, B.J.; Sankaran, K.; Fukuyama, J.A.; McMurdie, P.J.; Holmes, S.P. Bioconductor workflow for microbiome data analysis: From raw reads to community analyses. F1000Research 2016, 5, 1492. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Holmes, S.P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017, 11, 2639. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Álvarez, R.; Fierer, N.; de los Ríos, A.; Casamayor, E.O.; Barberán, A. Consistent changes in the taxonomic structure and functional attributes of bacterial communities during primary succession. ISME J. 2018, 12, 1658. [Google Scholar] [CrossRef] [PubMed]
- Aßhauer, K.P.; Wemheuer, B.; Daniel, R.; Meinicke, P. Tax4Fun: Predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics 2015, 31, 2882–2884. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.; Kindt, R.; Legendre, P.; Minchin, P.; O’Hara, R.; Simpson, G.; Solymos, P.; Stevens, M.; Wagner, H. Package “vegan. ” Community Ecol. Package 2013, 12, 1–295. [Google Scholar]
- Shannon-Wiener, C.; Weaver, W.; Weater, W. The Mathematical Theory of Communication; EUA, University of Illinois Press: Champaign, IL, USA, 1949. [Google Scholar]
- Bray, J.R.; Curtis, J.T. An ordination of the upland forest communities of southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Waste not, want not: Why rarefying microbiome data is inadmissible. PLoS Comput. Biol. 2014, 10, e1003531. [Google Scholar] [CrossRef]
- Di Rienzo, J.; Casanoves, F.; Balzarina, M.; Gonzalez, L.; Tablada, M.; Robledo, C. Infostat Versión 2018. Centro de Transferencia Infostat, FCA; Universidad Nacional de Córdoba: Córdoba, Argentina, 2018. [Google Scholar]
- Stein, A.; Gerstner, K.; Kreft, H. Environmental heterogeneity as a universal driver of species richness across taxa, biomes and spatial scales. Ecol. Lett. 2014, 17, 866–880. [Google Scholar] [CrossRef]
- Heino, J.; Melo, A.S.; Bini, L.M. Reconceptualising the beta diversity-environmental heterogeneity relationship in running water systems. Freshw. Biol. 2015, 60, 223–235. [Google Scholar] [CrossRef]
- Dickens, S.; Allen, E.; Santiago, L.; Crowley, D. Exotic annuals reduce soil heterogeneity in coastal sage scrub soil chemical and biological characteristics. Soil Biol. Biochem. 2013, 58, 70–81. [Google Scholar] [CrossRef]
- Weber, C.F.; King, G.M.; Aho, K. Relative abundance of and composition within fungal orders differ between cheatgrass (Bromus tectorum) and sagebrush (Artemisia tridentata)-associated soils. PloS ONE 2015, 10, e0117026. [Google Scholar] [CrossRef] [PubMed]
- Horner-Devine, M.C.; Lage, M.; Hughes, J.B.; Bohannan, B.J. A taxa–area relationship for bacteria. Nature 2004, 432, 750–753. [Google Scholar] [CrossRef] [PubMed]
- Ranjard, L.; Dequiedt, S.; Prévost-Bouré, N.C.; Thioulouse, J.; Saby, N.; Lelievre, M.; Maron, P.; Morin, F.; Bispo, A.; Jolivet, C.; et al. Turnover of soil bacterial diversity driven by wide-scale environmental heterogeneity. Nat. Commun. 2013, 4, 1434. [Google Scholar] [CrossRef] [PubMed]
- Peay, K.G.; Baraloto, C.; Fine, P.V. Strong coupling of plant and fungal community structure across western Amazonian rainforests. ISME J. 2013, 7, 1852. [Google Scholar] [CrossRef] [PubMed]
- Venter, Z.S.; Jacobs, K.; Hawkins, H.J. The impact of crop rotation on soil microbial diversity: A meta-analysis. Pedobiologia 2016, 59, 215–223. [Google Scholar] [CrossRef]
- He, J.; Tedersoo, L.; Hu, A.; Han, C.; He, D.; Wei, H.; Jiao, M.; Anslan, S.; Nie, Y.; Jia, Y.; et al. Greater diversity of soil fungal communities and distinguishable seasonal variation in temperate deciduous forests compared with subtropical evergreen forests of eastern China. FEMS Microbiol. Ecol. 2017, 93. [Google Scholar] [CrossRef]
- Zhou, J.; Xia, B.; Huang, H.; Palumbo, A.V.; Tiedje, J.M. Microbial diversity and heterogeneity in sandy subsurface soils. Appl. Environ. Microbiol. 2004, 70, 1723–1734. [Google Scholar] [CrossRef]
- Koranda, M.; Kaiser, C.; Fuchslueger, L.; Kitzler, B.; Sessitsch, A.; Zechmeister-Boltenstern, S.; Richter, A. Seasonal variation in functional properties of microbial communities in beech forest soil. Soil Biol. Biochem. 2013, 60, 95–104. [Google Scholar] [CrossRef]
- Kaiser, C.; Fuchslueger, L.; Koranda, M.; Gorfer, M.; Stange, C.F.; Kitzler, B.; Rasche, F.; Strauss, J.; Sessitsch, A.; Zechmeister-Boltenstern, S.; et al. Plants control the seasonal dynamics of microbial N cycling in a beech forest soil by belowground C allocation. Ecology 2011, 92, 1036–1051. [Google Scholar] [CrossRef]
- Gupta, V.V.S.R.; Bramley, R.G.V.; Greenfield, P.; Yu, J.; Herderich, M.J. Vineyard Soil Microbiome Composition Related to Rotundone Concentration in Australian Cool Climate ‘Peppery’ Shiraz Grapes. Front. Microbiol. 2019, 10, 1607. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Kelso, J. High-throughput DNA sequencing–concepts and limitations. Bioessays 2010, 32, 524–536. [Google Scholar] [CrossRef] [PubMed]
- Cappello, M.S.; Zapparoli, G.; Logrieco, A.; Bartowsky, E.J. Linking wine lactic acid bacteria diversity with wine aroma and flavour. Int. J. Food Microbiol. 2017, 243, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Sponholz, W.R. Wine spoilage by microorganisms. In Wine Microbiology and Biotechnology; Taylor & Francis: Abingdon, UK, 1993; pp. 395–420. [Google Scholar]
- Bartowsky, E.J.; Henschke, P.A. Acetic acid bacteria spoilage of bottled red wine—A review. Int. J. Food Microbiol. 2008, 125, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Chou, M.; Vanden, H.J.; Bell, T.; Panke-Buisse, K.; J, K.K. Vineyard under-vine floor management alters soil microbial composition, while the fruit microbiome shows no corresponding shifts. Sci. Rep. 2018, 8, e00631-16. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.I.; Lombard, P.B. Environmental and Management Practices Affecting Grape Composition and Wine Quality—A Review. Am. J. Enol. Viticult. 1993, 44, 409–430. [Google Scholar]
- Fleet, G.H. Wine Microbiology and Biotechnology; CRC Press: Boca Raton, FL, USA, 1993. [Google Scholar]
- König, H.; Unden, G.; Fröhlich, J. Biology of Microorganisms on Grapes, in Must and in Wine; Springer: Berlin, Germany, 2009. [Google Scholar]
- Capozzi, V.; Ladero, V.; Beneduce, L.; Fernández, M.; Alvarez, M.A.; Benoit, B.; Laurent, B.; Grieco, F.; Spano, G. Isolation and characterization of tyramine-producing Enterococcus faecium strains from red wine. Food Microbiol. 2011, 28, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Benavent-Gil, Y.; Berbegal, C.; Lucio, O.; Pardo, I.; Ferrer, S. A new fear in wine: Isolation of Staphylococcus epidermidis histamine producer. Food Control 2016, 62, 142–149. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alonso, A.; de Celis, M.; Ruiz, J.; Vicente, J.; Navascués, E.; Acedo, A.; Ortiz-Álvarez, R.; Belda, I.; Santos, A.; Gómez-Flechoso, M.Á.; et al. Looking at the Origin: Some Insights into the General and Fermentative Microbiota of Vineyard Soils. Fermentation 2019, 5, 78. https://doi.org/10.3390/fermentation5030078
Alonso A, de Celis M, Ruiz J, Vicente J, Navascués E, Acedo A, Ortiz-Álvarez R, Belda I, Santos A, Gómez-Flechoso MÁ, et al. Looking at the Origin: Some Insights into the General and Fermentative Microbiota of Vineyard Soils. Fermentation. 2019; 5(3):78. https://doi.org/10.3390/fermentation5030078
Chicago/Turabian StyleAlonso, Alejandro, Miguel de Celis, Javier Ruiz, Javier Vicente, Eva Navascués, Alberto Acedo, Rüdiger Ortiz-Álvarez, Ignacio Belda, Antonio Santos, María Ángeles Gómez-Flechoso, and et al. 2019. "Looking at the Origin: Some Insights into the General and Fermentative Microbiota of Vineyard Soils" Fermentation 5, no. 3: 78. https://doi.org/10.3390/fermentation5030078
APA StyleAlonso, A., de Celis, M., Ruiz, J., Vicente, J., Navascués, E., Acedo, A., Ortiz-Álvarez, R., Belda, I., Santos, A., Gómez-Flechoso, M. Á., & Marquina, D. (2019). Looking at the Origin: Some Insights into the General and Fermentative Microbiota of Vineyard Soils. Fermentation, 5(3), 78. https://doi.org/10.3390/fermentation5030078