

Thermodynamic Stability and Electronic Properties of Graphene Nanoflakes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
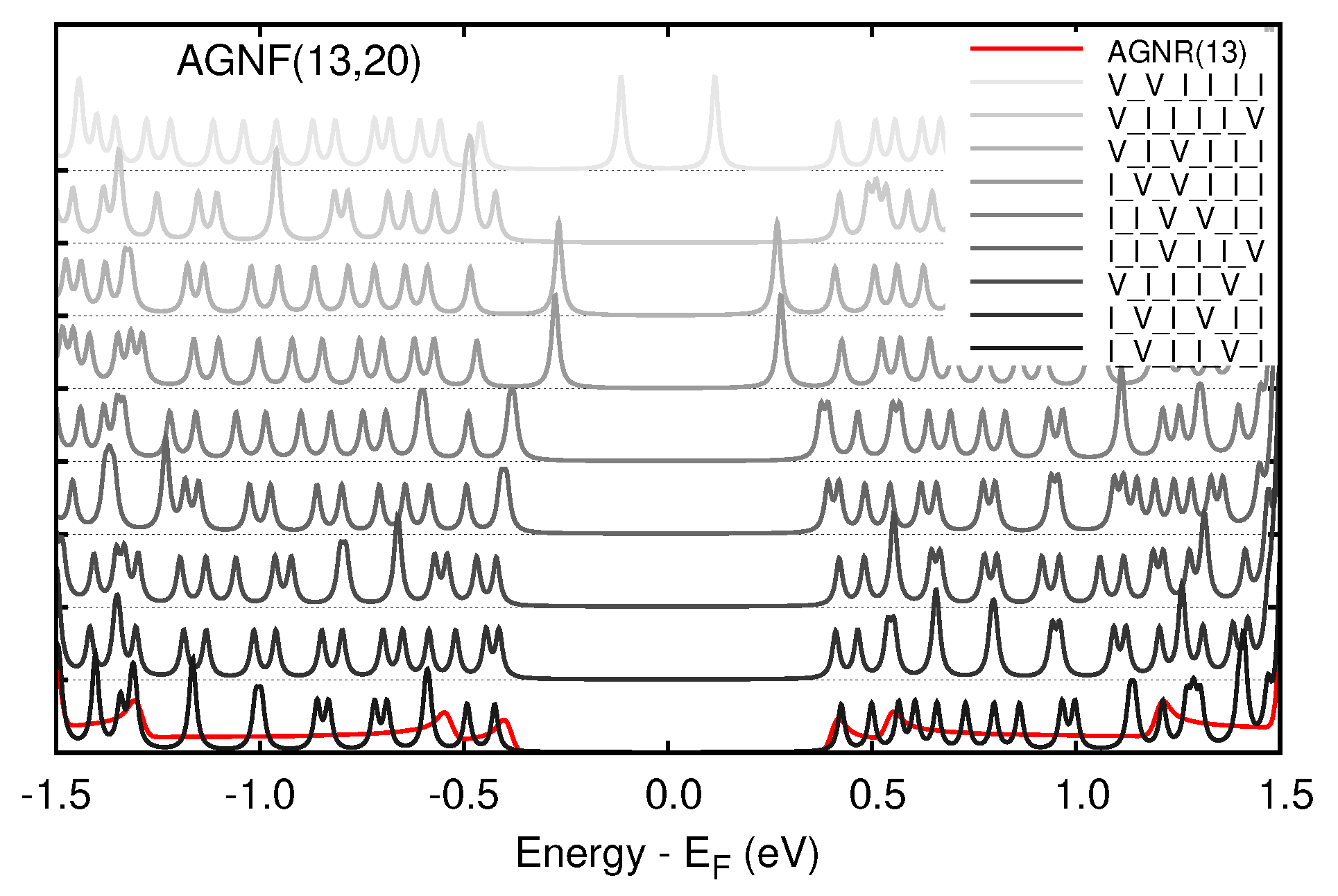
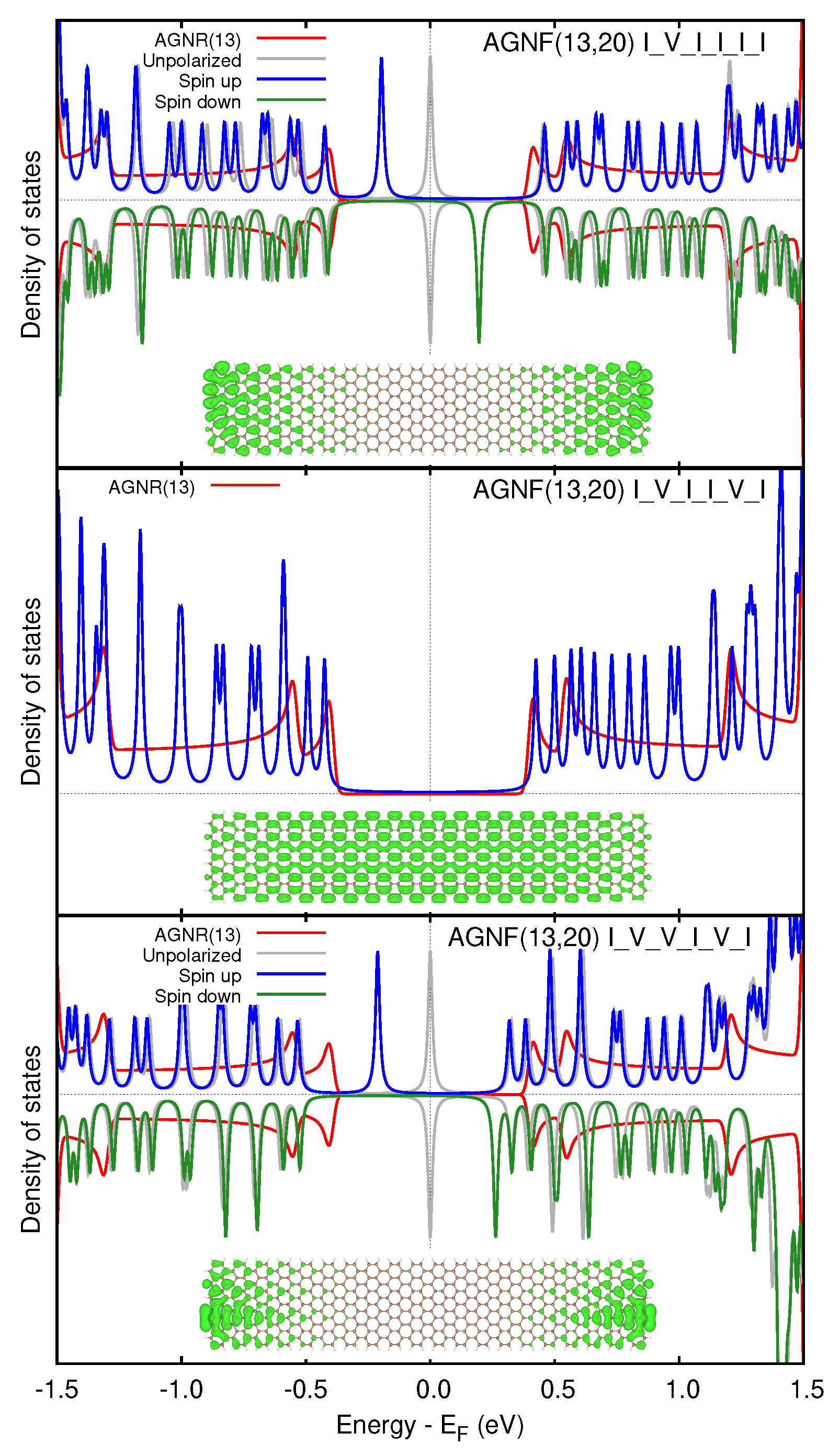
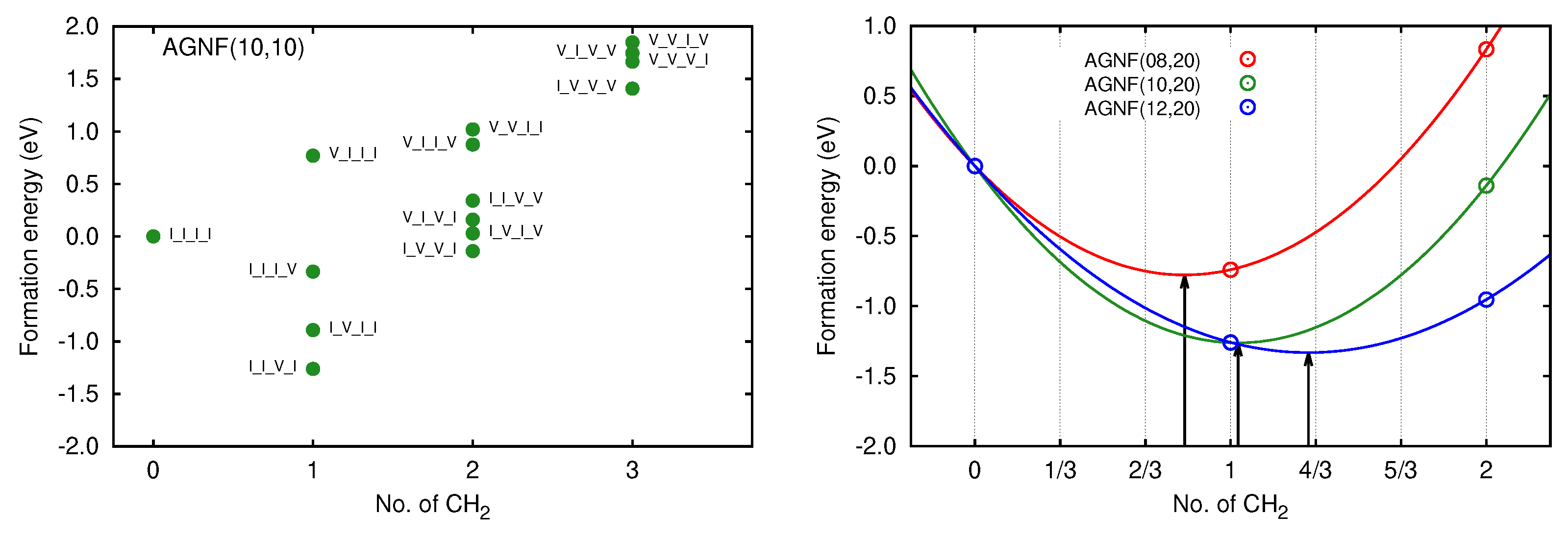
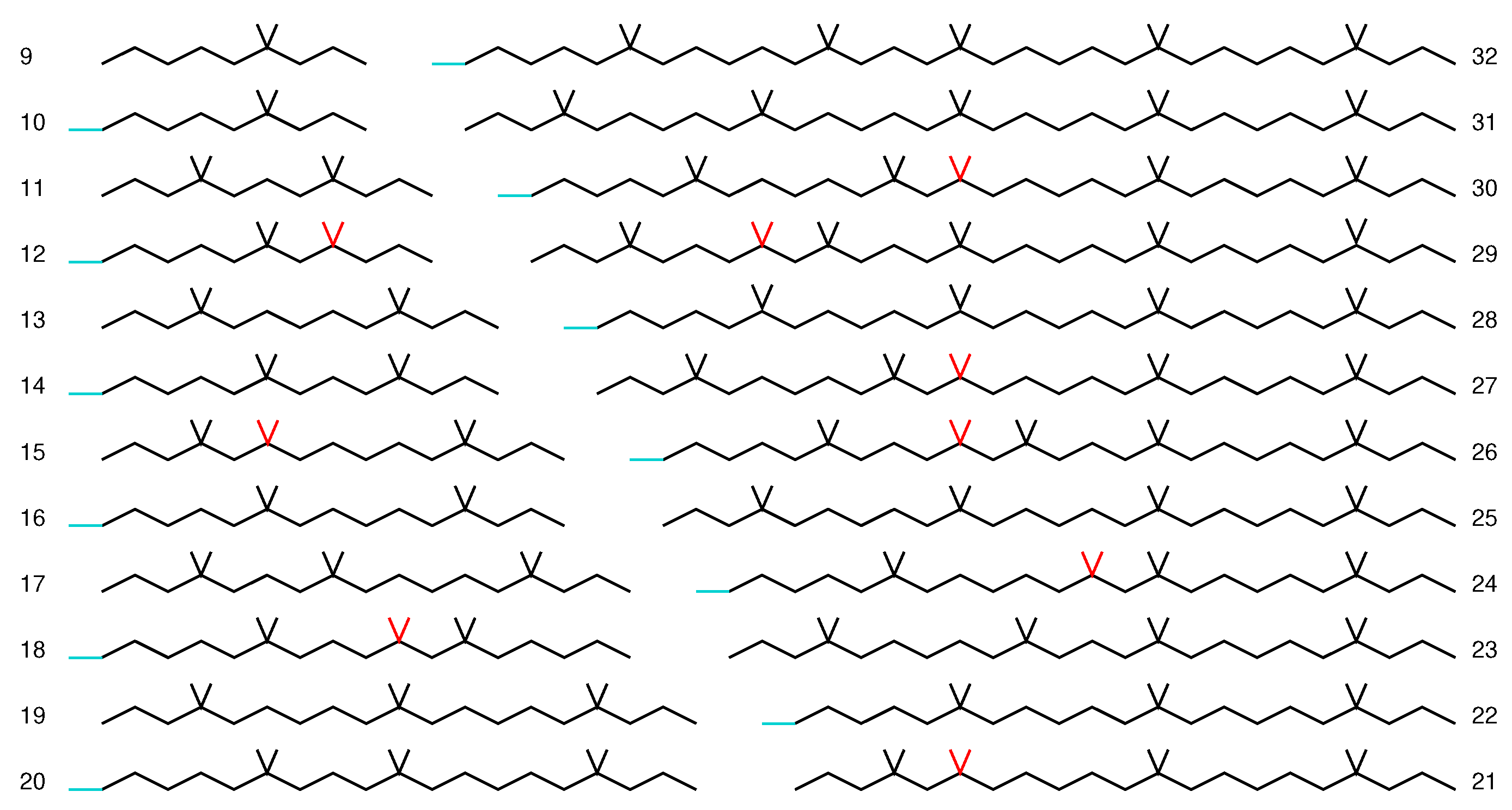
3. Results
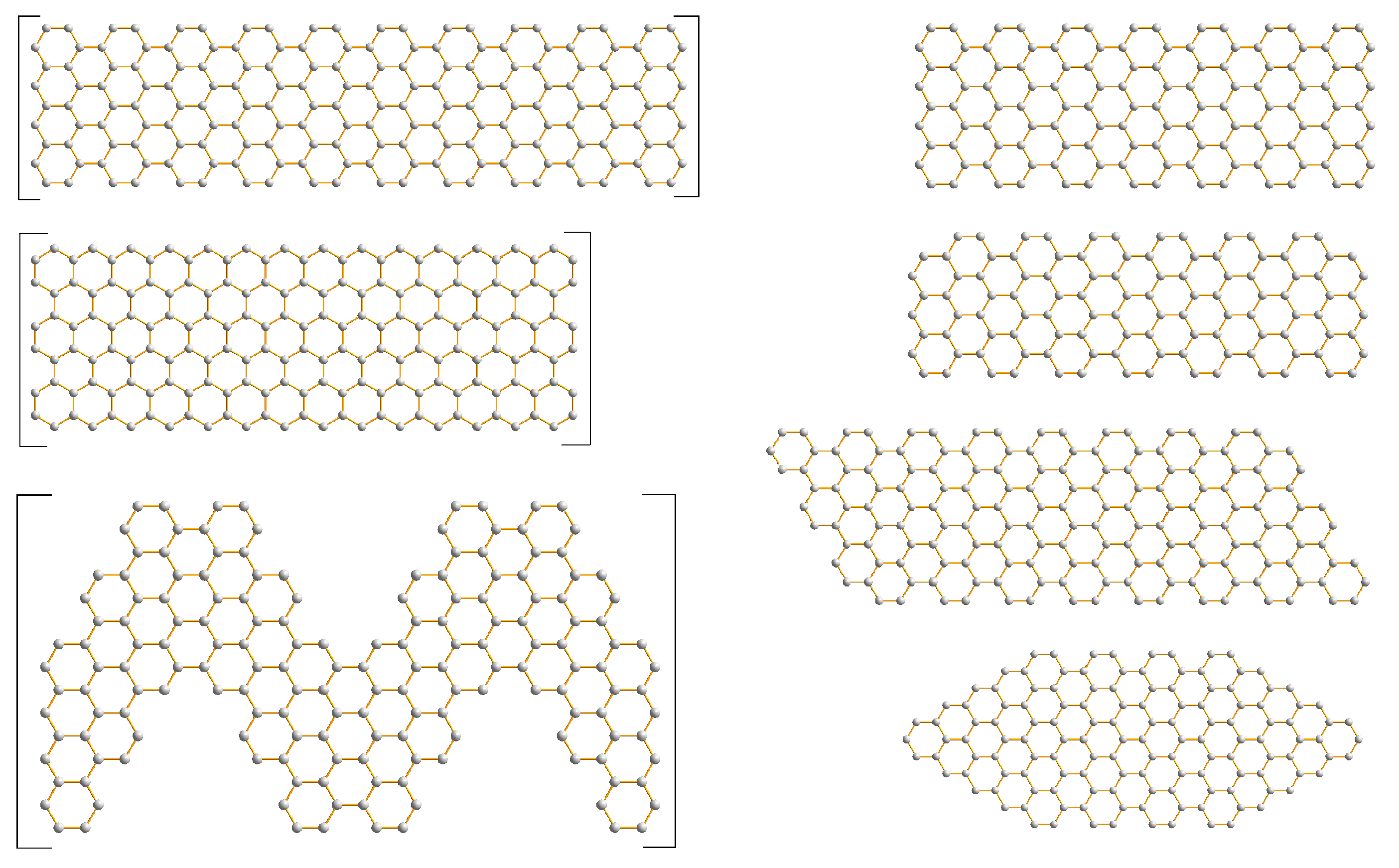
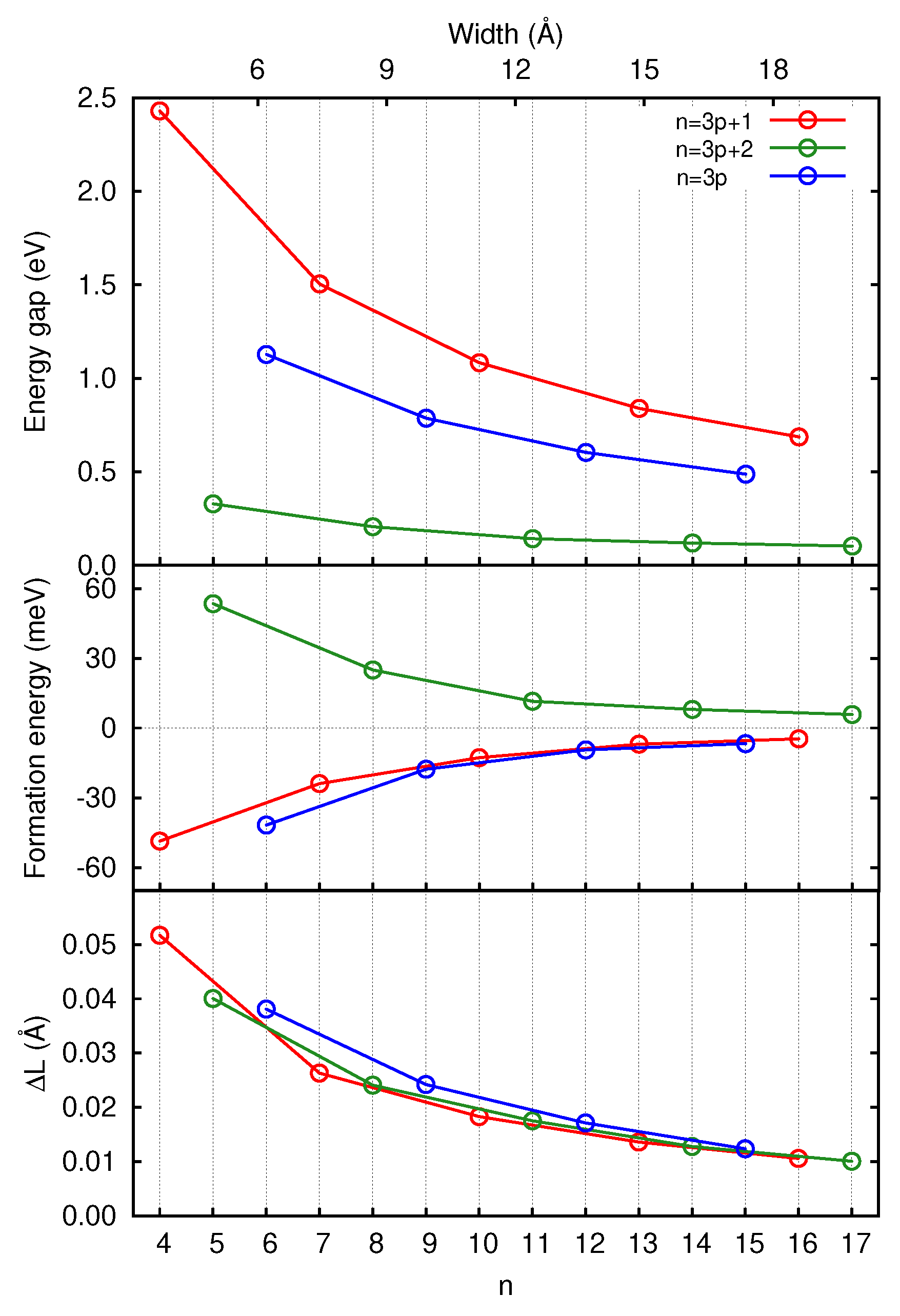
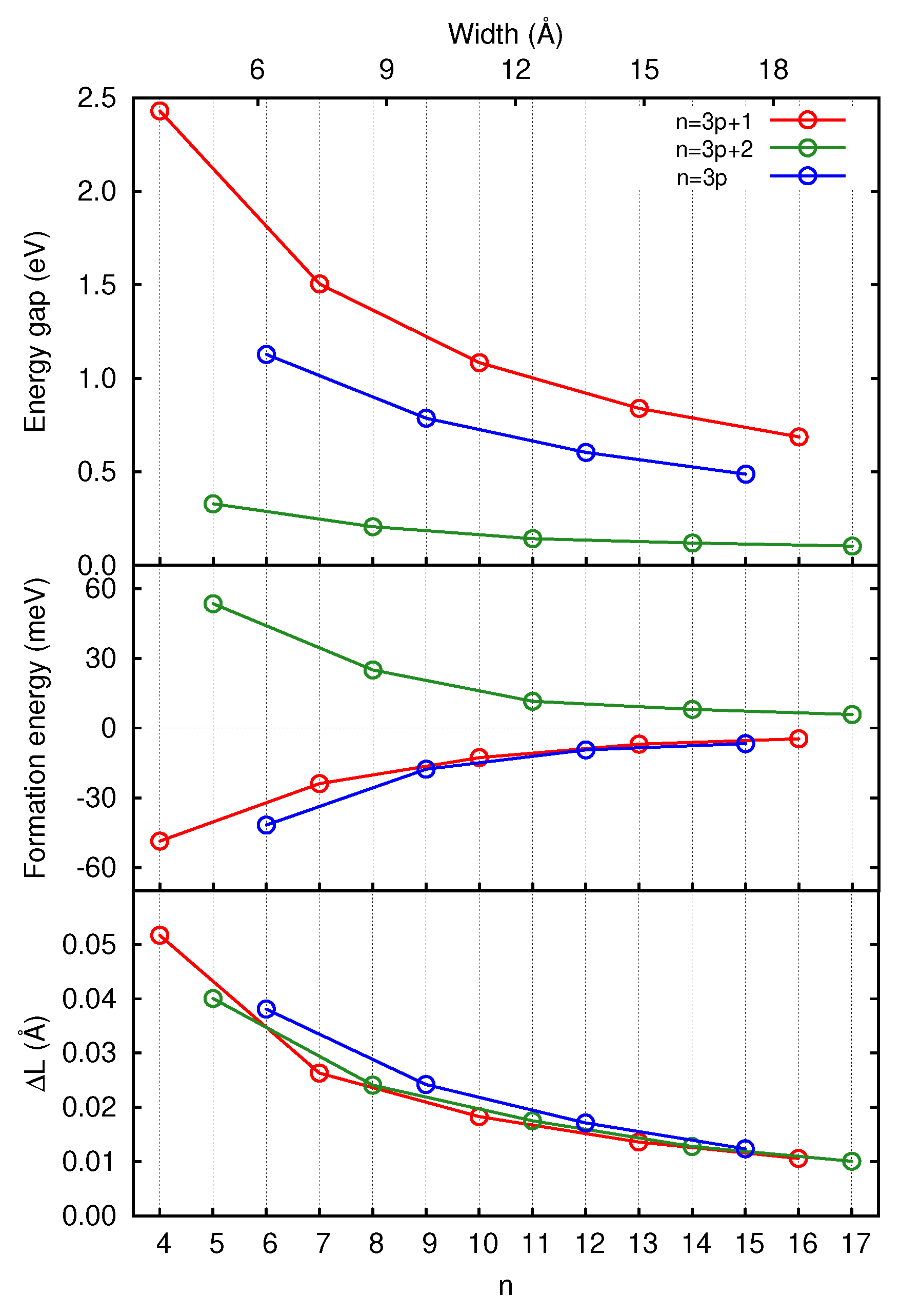
3.1. Graphene and Infinite Ribbons
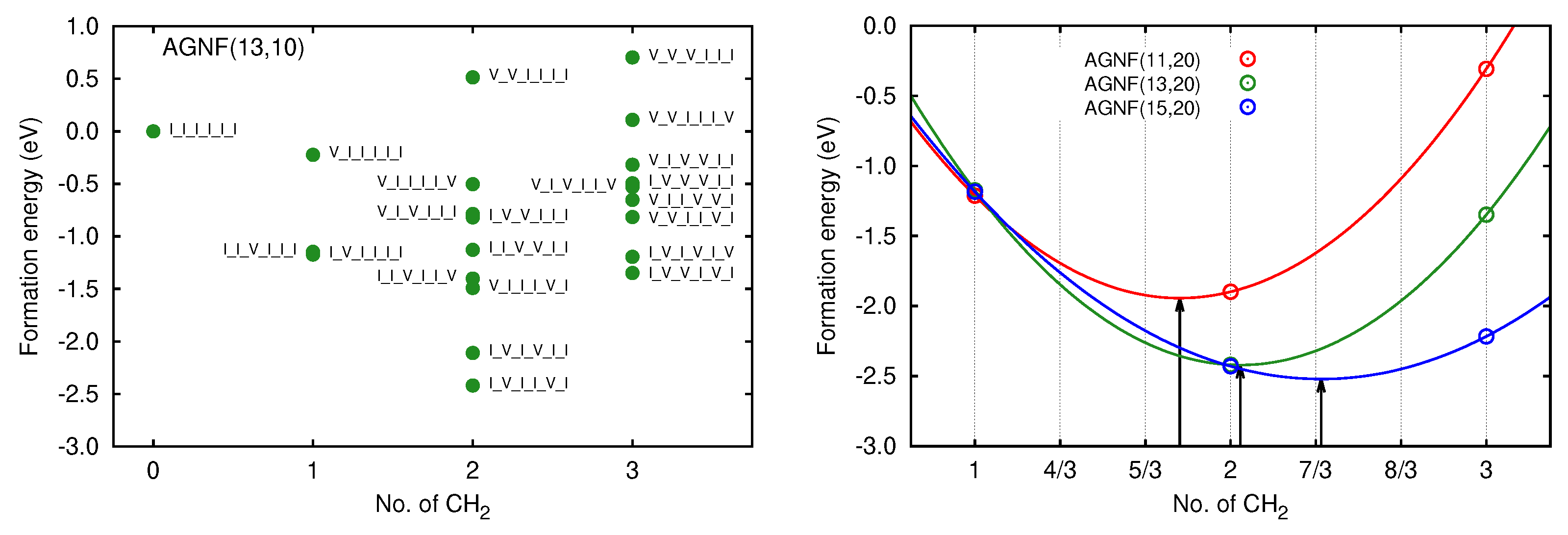
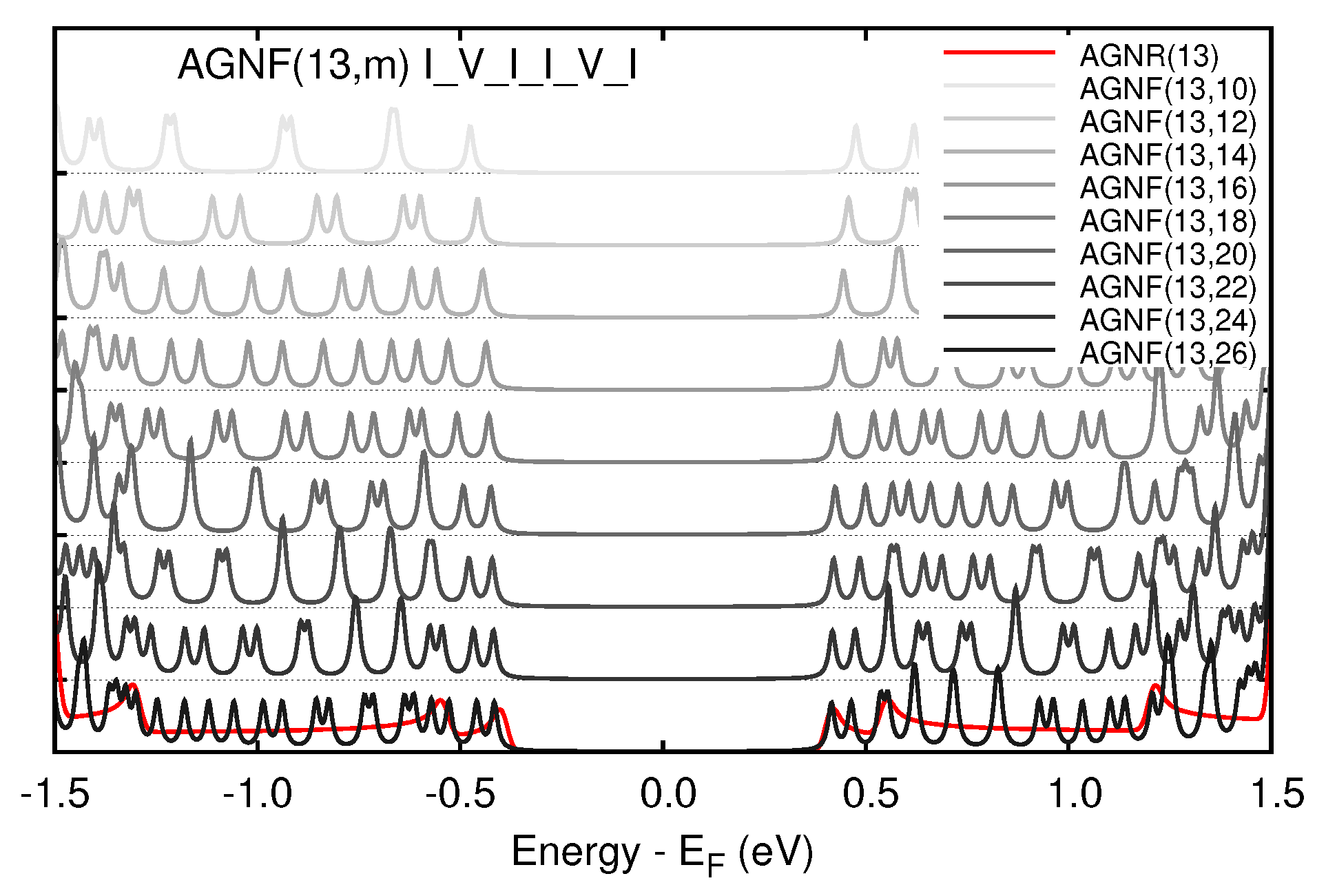
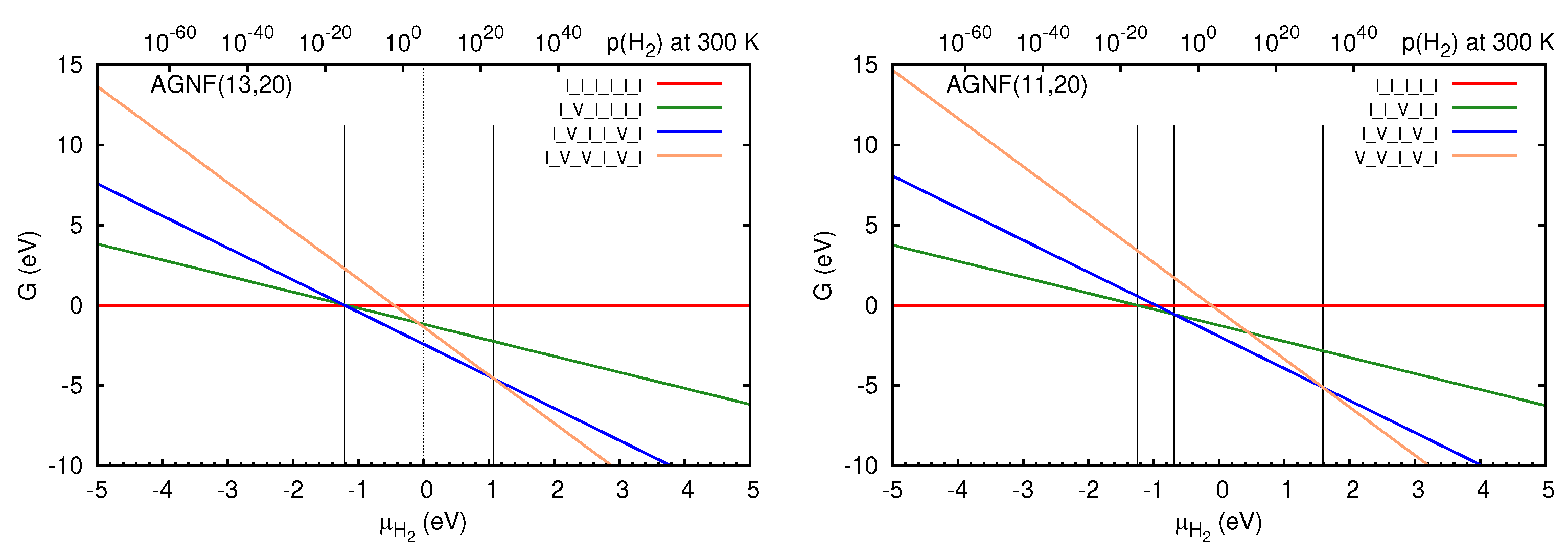
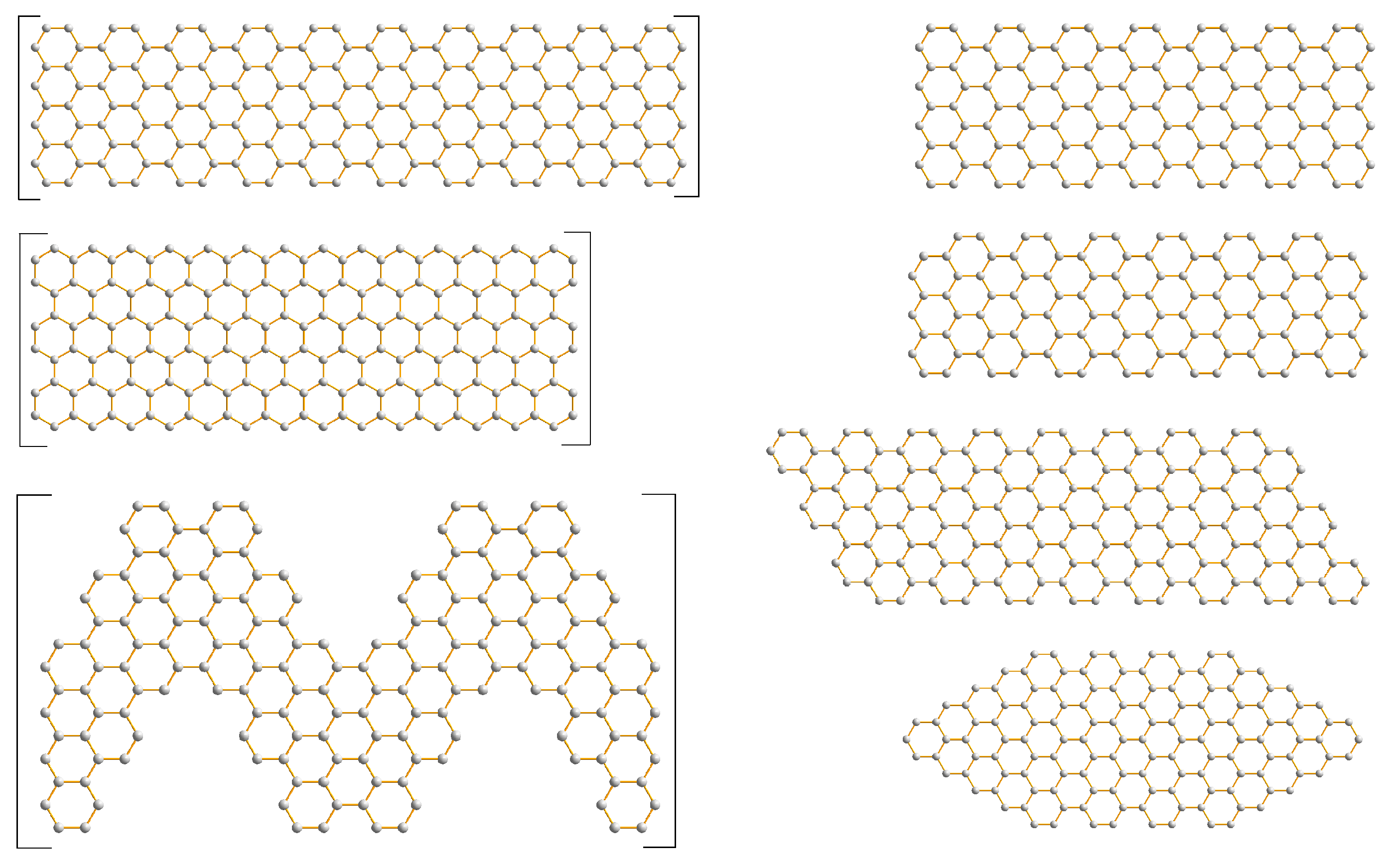
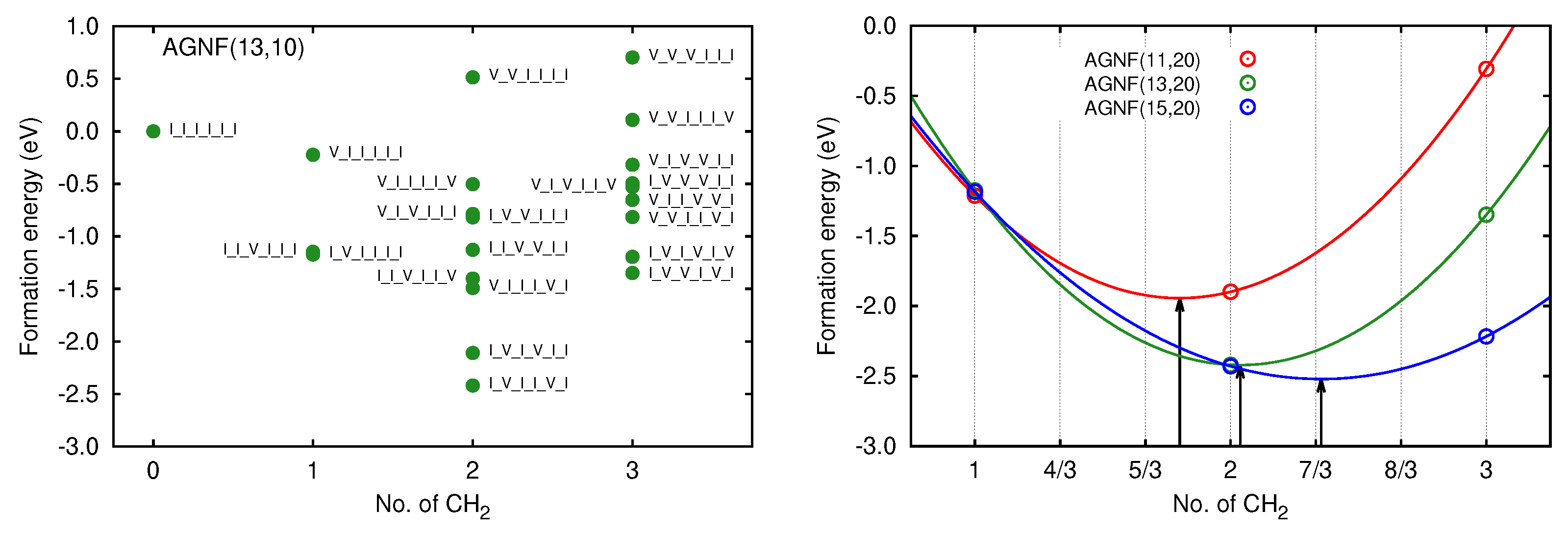
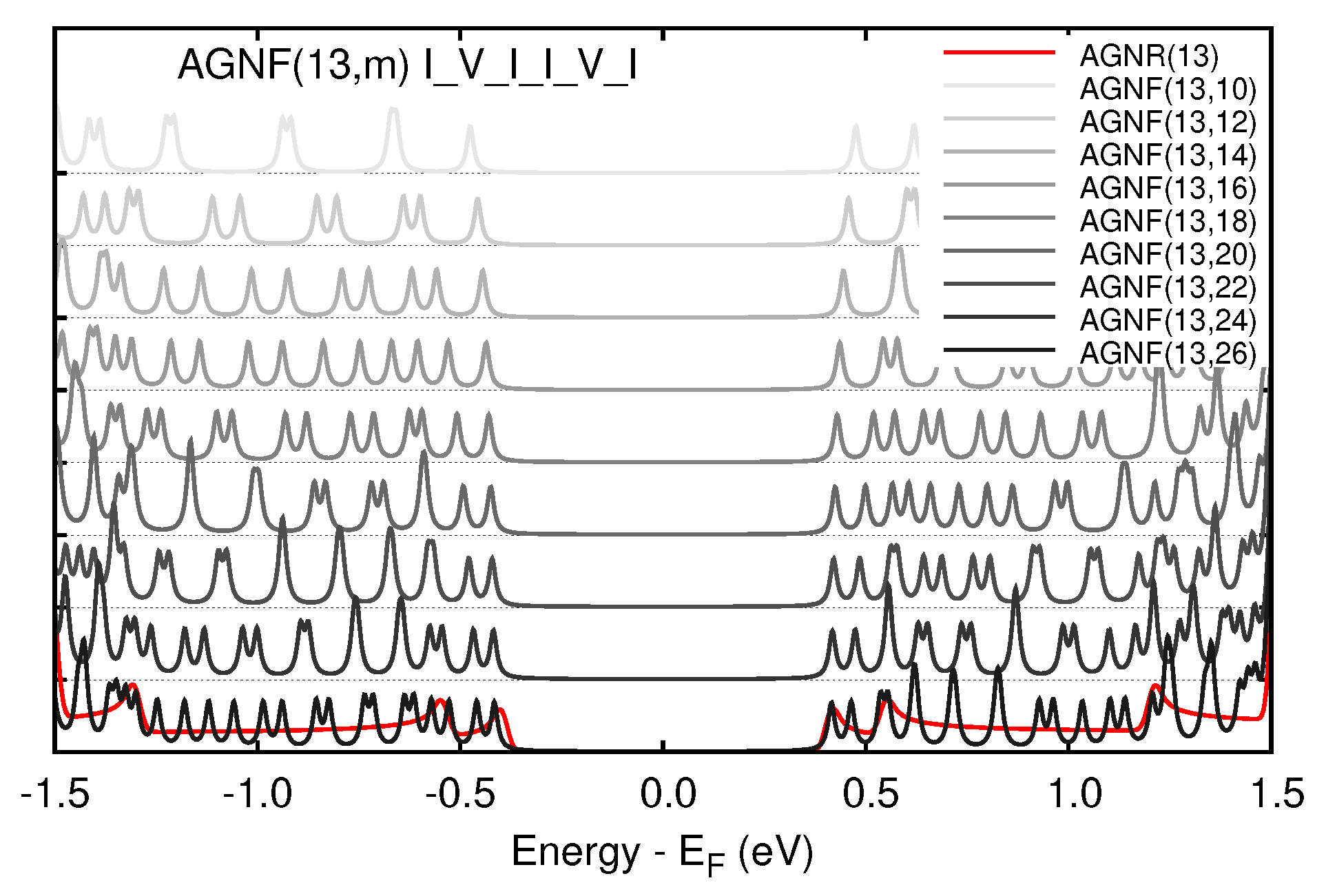
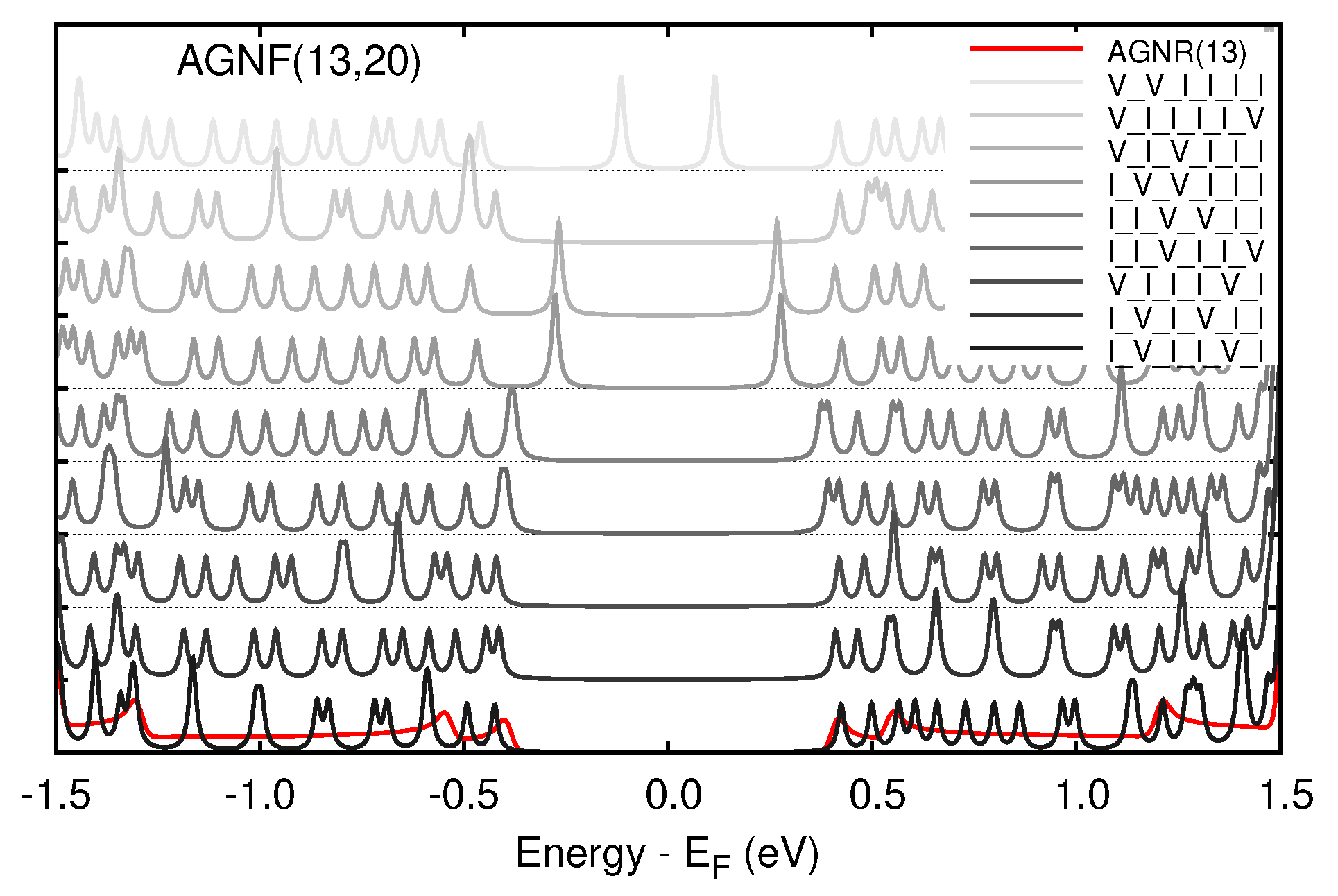
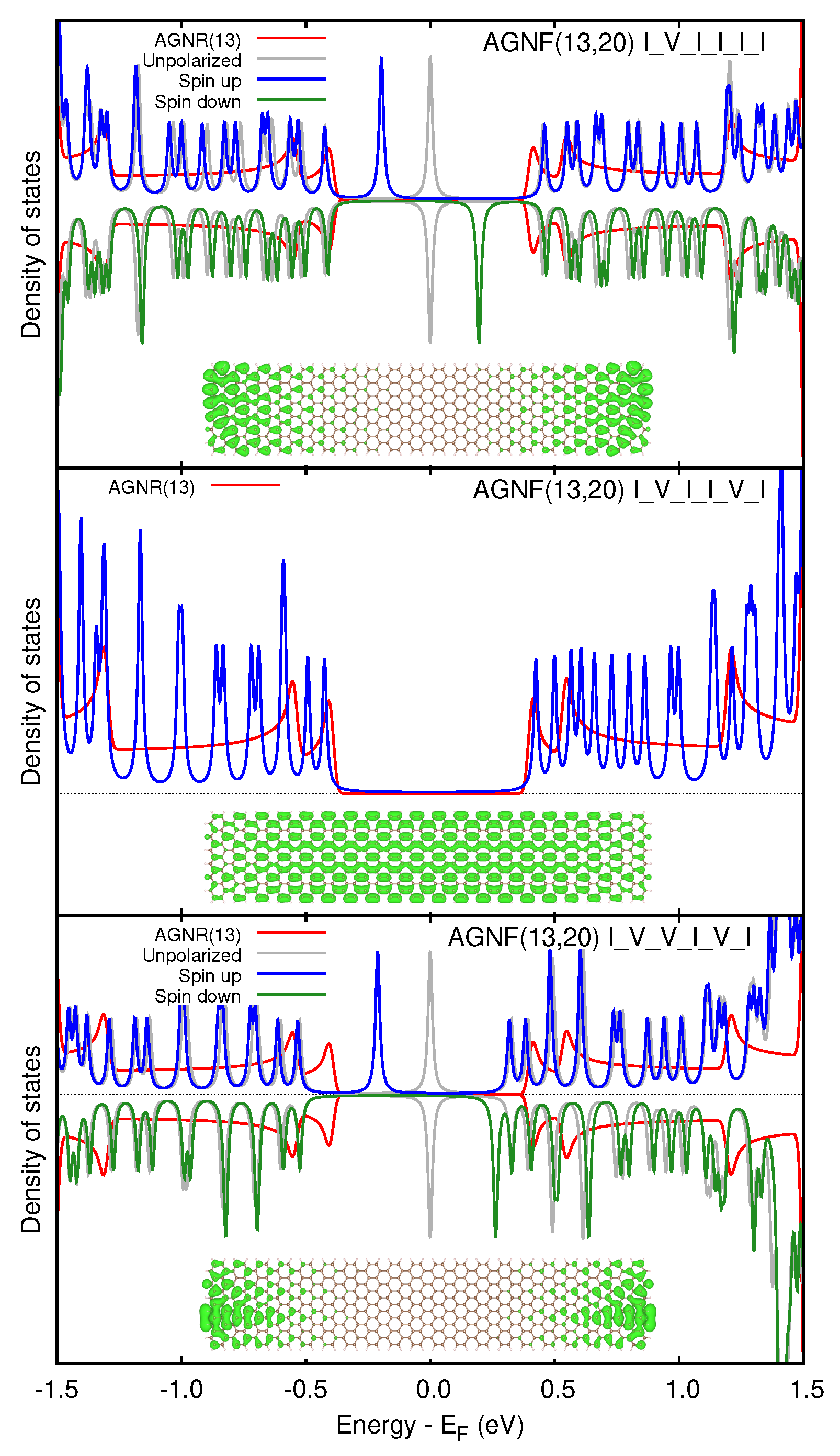
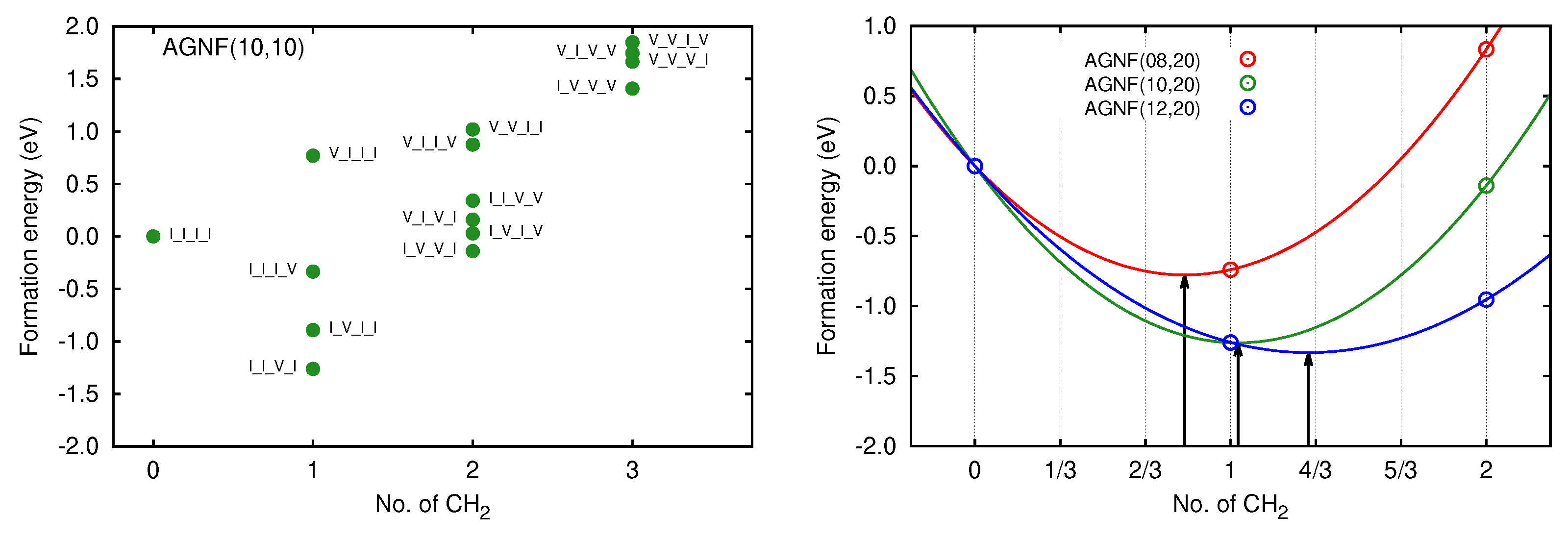
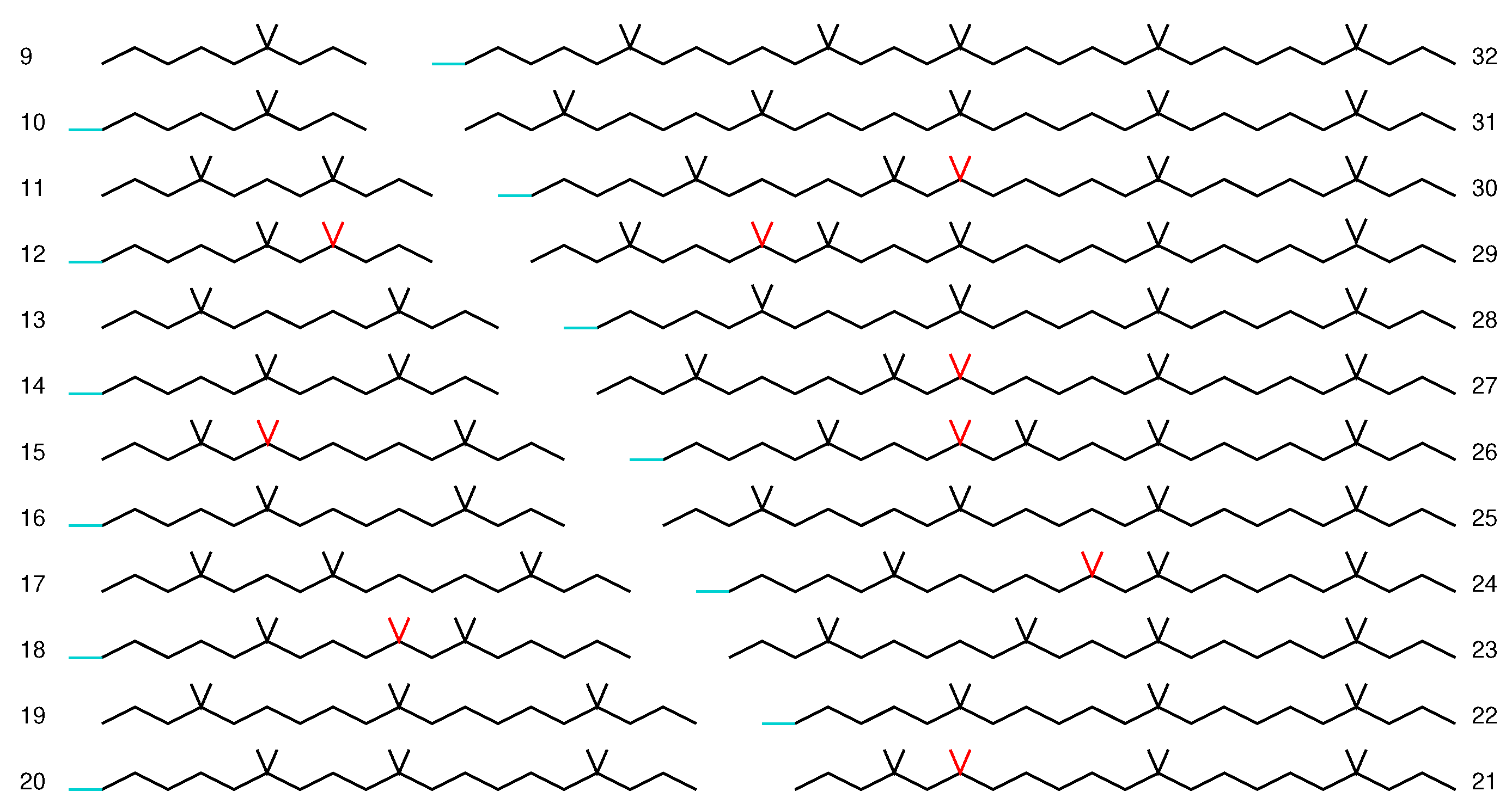
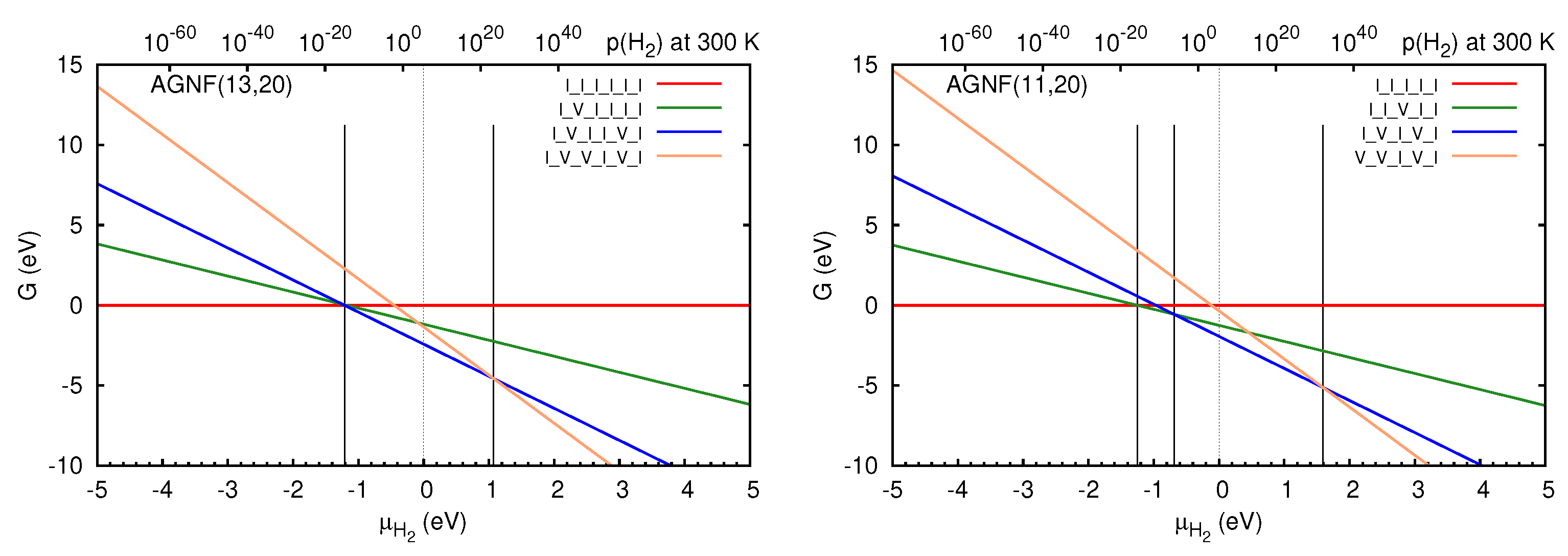
3.2. Graphene Nanoflakes
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dutta, S.; Pati, S.K. Novel properties of graphene nanoribbons: A review. J. Mater. Chem. 2010, 20, 8207–8223. [Google Scholar] [CrossRef]
- Wang, H.; Wang, H.S.; Ma, C.; Chen, L.; Jiang, C.; Chen, C.; Xie, X.; Li, A.P.; Wang, X. Graphene nanoribbons for quantum electronics. Nat. Rev. Phys. 2021, 3, 791–802. [Google Scholar] [CrossRef]
- Houtsma, R.S.K.; de la Rie, J.; Stohr, M. Atomically precise graphene nanoribbons: Interplay of structural and electronic properties. Chem. Soc. Rev. 2021, 50, 6541–6568. [Google Scholar] [CrossRef] [PubMed]
- Cocchi, C.; Prezzi, D.; Ruini, A.; Caldas, M.J.; Molinari, E. Electronics and Optics of Graphene Nanoflakes: Edge Functionalization and Structural Distortions. J. Phys. Chem. C 2012, 116, 17328–17335. [Google Scholar] [CrossRef]
- Gotz, A.; Wang, X.Y.; Ruini, A.; Zheng, W.; Soltani, P.; Graf, R.; Tries, A.; Li, J.; Palma, C.A.; Molinari, E.; et al. Band structure modulation by methoxy-functionalization of graphene nanoribbons. J. Mater. Chem. C 2022, 10, 4173–4181. [Google Scholar] [CrossRef]
- Martini, L.; Chen, Z.; Mishra, N.; Barin, G.B.; Fantuzzi, P.; Ruffieux, P.; Fasel, R.; Feng, X.; Narita, A.; Coletti, C.; et al. Structure-dependent electrical properties of graphene nanoribbon devices with graphene electrodes. Carbon 2019, 146, 36–43. [Google Scholar] [CrossRef]
- Gu, Y.; Qiu, Z.; Mullen, K. Nanographenes and Graphene Nanoribbons as Multitalents of Present and Future Materials Science. J. Am. Chem. Soc. 2022, 144, 11499–11524. [Google Scholar] [CrossRef]
- Fedotov, P.V.; Obraztsova, E.D. Near infrared photoluminescence of the bottom-up produced 7-armchair graphene nanoribbons. Appl. Phys. Lett. 2023, 122, 013101. [Google Scholar] [CrossRef]
- Liu, Z.; Fu, S.; Liu, X.; Narita, A.; Samorì, P.; Bonn, M.; Wang, H.I. Small Size, Big Impact: Recent Progress in Bottom-Up Synthesized Nanographenes for Optoelectronic and Energy Applications. Adv. Sci. 2022, 9, 2106055. [Google Scholar] [CrossRef]
- Tan, Y.Z.; Osella, S.; Liu, Y.; Yang, B.; Beljonne, D.; Feng, X.; Müllen, K. Sulfur-Annulated Hexa-peri-hexabenzocoronene Decorated with Phenylthio Groups at the Periphery. Angew. Chem. 2015, 127, 2970–2974. [Google Scholar] [CrossRef]
- Tan, Y.Z.; Yang, B.; Parvez, K.; Narita, A.; Osella, S.; Beljonne, D.; Feng, X.; Müllen, K. Atomically precise edge chlorination of nanographenes and its application in graphene nanoribbons. Nat. Commun. 2013, 4, 2646. [Google Scholar] [CrossRef] [PubMed]
- Wassmann, T.; Seitsonen, A.P.; Saitta, M.A.; Lazzeri, M.; Mauri, F. Structure, stability, edge states, and aromaticity of graphene ribbons. Phys. Rev. Lett. 2008, 101, 096402. [Google Scholar] [CrossRef] [PubMed]
- Soler, J.M.; Artacho, E.; Gale, J.D.; García, A.; Junquera, J.; Ordejón, P.; Sánchez-Portal, D. The SIESTA method for ab initio order-N materials simulation. J. Phys. Condens. Matter 2002, 14, 2745. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Troullier, N.; Martins, J.L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 1991, 43, 1993. [Google Scholar] [CrossRef] [PubMed]
- Chase, M.W., Jr. NIST-JANAF Thermochemical Tables; American Chemical Society: Washington, DC, USA, 1998; Volume 9, pp. 1–1951. [Google Scholar]
- Lynch, R.W.; Drickamer, H.G. Effect of High Pressure on the Lattice Parameters of Diamond, Graphite, and Hexagonal Boron Nitride. J. Chem. Phys. 1966, 44, 181–184. [Google Scholar] [CrossRef]
- Hembacher, S.; Giessibl, F.J.; Mannhart, J.; Quate, C.F. Revealing the hidden atom in graphite by low-temperature atomic force microscopy. Proc. Natl. Acad. Sci. USA 2003, 22, 12539–12542. [Google Scholar] [CrossRef]
- Dappe, Y.J.; Oszwaldowski, R.; Pou, P.; Ortega, J.; Pérez, R.; Flores, F. Local-orbital occupancy formulation of density functional theory: Application to Si, C, and graphene. Phys. Rev. B 2006, 73, 235124. [Google Scholar] [CrossRef]
- Barone, V.; Hod, O.; Scuseria, G.E. Electronic Structure and Stability of Semiconducting Graphene Nanoribbons. Nano Lett. 2006, 6, 2748–2754. [Google Scholar] [CrossRef]
- Koskinen, P.; Malola, S.; Häkkinen, H. Self-Passivating Edge Reconstructions of Graphene. Phys. Rev. Lett. 2008, 101, 115502. [Google Scholar] [CrossRef]
- Shin, H.; Kang, S.; Koo, J.; Lee, H.; Kim, J.; Kwon, Y. Cohesion energetics of carbon allotropes: Quantum Monte Carlo study. J. Chem. Phys. 2014, 140, 114702. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. First-principles study of the adsorption of atomic H on Ni (111), (100) and (110). Surf. Sci. 2000, 459, 287. [Google Scholar] [CrossRef]
- Huber, K.P. American Institute of Physics Handbook; McGraw-Hill: New York, NY, USA, 1979. [Google Scholar]
- Abbas, A.N.; Liu, G.; Narita, A.; Orosco, M.; Feng, X.; Müllen, K.; Zhou, C. Deposition, Characterization, and Thin-Film-Based Chemical Sensing of Ultra-long Chemically Synthesized Graphene Nanoribbons. J. Am. Chem. Soc. 2014, 136, 7555–7558. [Google Scholar] [CrossRef] [PubMed]
- El Gemayel, M.; Narita, A.; Dössel, L.F.; Sundaram, R.S.; Kiersnowski, A.; Pisula, W.; Hansen, M.R.; Ferrari, C.; Orgiu, E.; Feng, X.; et al. Graphene nanoribbon blends with P3HT for organic electronics. Nanoscale 2014, 6, 6301–6314. [Google Scholar] [CrossRef] [PubMed]
- Candini, A.; Martini, L.; Chen, Z.; Mishra, N.; Convertino, D.; Coletti, C.; Narita, A.; Feng, X.; Müllen, K.; Affronte, M. High Photoresponsivity in Graphene Nanoribbon Field-Effect Transistor Devices Contacted with Graphene Electrodes. J. Phys. Chem. C 2017, 121, 10620–10625. [Google Scholar] [CrossRef]
- Ma, C.; Xiao, Z.; Puretzky, A.A.; Wang, H.; Mohsin, A.; Huang, J.; Liang, L.; Luo, Y.; Lawrie, B.J.; Gu, G.; et al. Engineering edge states of graphene nanoribbons for narrow-band photoluminescence. ACS Nano 2020, 14, 5090–5098. [Google Scholar] [CrossRef]
- Cai, J.; Ruffieux, P.; Jaafar, R.; Bieri, M.; Braun, T.; Blankenburg, S.; Muoth, M.; Seitsonen, A.P.; Saleh, M.; Feng, X.; et al. Atomically precise bottom-up fabrication of graphene nanoribbons. Nature 2010, 466, 470–473. [Google Scholar] [CrossRef]
- Huang, H.; Wei, D.; Sun, J.; Wong, S.L.; Feng, Y.P.; Neto, A.H.C.; Wee, A.T.S. Spatially Resolved Electronic Structures of Atomically Precise Armchair Graphene Nanoribbons. Sci. Rep. 2012, 2, 983. [Google Scholar] [CrossRef]
- Vo, T.H.; Shekhirev, M.; Kunkel, D.A.; Morton, M.D.; Berglund, E.; Kong, L.; Wilson, P.M.; Dowben, P.A.; Enders, A.; Sinitskii, A. Large-scale solution synthesis of narrow graphene nanoribbons. Nat. Commun. 2014, 5, 3189. [Google Scholar] [CrossRef]
- Narita, A.; Feng, X.; Müllen, K. Bottom-Up Synthesis of Chemically Precise Graphene Nanoribbons. Chem. Rec. 2015, 15, 295–309. [Google Scholar] [CrossRef]
- Abdurakhmanova, N.; Amsharov, N.; Stepanow, S.; Jansen, M.; Kern, K.; Amsharov, K. Synthesis of wide atomically precise graphene nanoribbons from para-oligophenylene based molecular precursor. Carbon 2014, 77, 1187–1190. [Google Scholar] [CrossRef]
- Basagni, A.; Sedona, F.; Pignedoli, C.A.; Cattelan, M.; Nicolas, L.; Casarin, M.; Sambi, M. Molecules–Oligomers–Nanowires–Graphene Nanoribbons: A Bottom-Up Stepwise On-Surface Covalent Synthesis Preserving Long-Range Order. J. Am. Chem. Soc. 2015, 137, 1802–1808. [Google Scholar] [CrossRef] [PubMed]
- Narita, A.; Wang, X.Y.; Feng, X.; Mullen, K. New Advances in Nanographene Chemistry. Chem. Soc. Rev. 2015, 44, 6616–6643. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.W.; Cohen, M.L.; Louie, S.G. Energy gaps in graphene nanoribbons. Phys. Rev. Lett. 2006, 97, 216803. [Google Scholar] [CrossRef]
- Shemella, P.; Zhang, Y.; Mailman, M.; Ajayan, P.M.; Nayak, S.K. Energy gaps in zero-dimensional graphene nanoribbons. Appl. Phys. Lett. 2007, 91, 042101. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soave, R.; Cargnoni, F.; Trioni, M.I. Thermodynamic Stability and Electronic Properties of Graphene Nanoflakes. C 2024, 10, 5. https://doi.org/10.3390/c10010005
Soave R, Cargnoni F, Trioni MI. Thermodynamic Stability and Electronic Properties of Graphene Nanoflakes. C. 2024; 10(1):5. https://doi.org/10.3390/c10010005
Chicago/Turabian StyleSoave, Raffaella, Fausto Cargnoni, and Mario Italo Trioni. 2024. "Thermodynamic Stability and Electronic Properties of Graphene Nanoflakes" C 10, no. 1: 5. https://doi.org/10.3390/c10010005
APA StyleSoave, R., Cargnoni, F., & Trioni, M. I. (2024). Thermodynamic Stability and Electronic Properties of Graphene Nanoflakes. C, 10(1), 5. https://doi.org/10.3390/c10010005