Evaluation of the Interplay between the ADAR Editome and Immunotherapy in Melanoma

 , ,
, ,  ,
,  ,
,  , , ,
, , ,  , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Cell Lines
2.2. Bioinformatics and Statistics
3. Results
3.1. Expression of the ADAR Genes before IT Versus in Relapsing Tumours
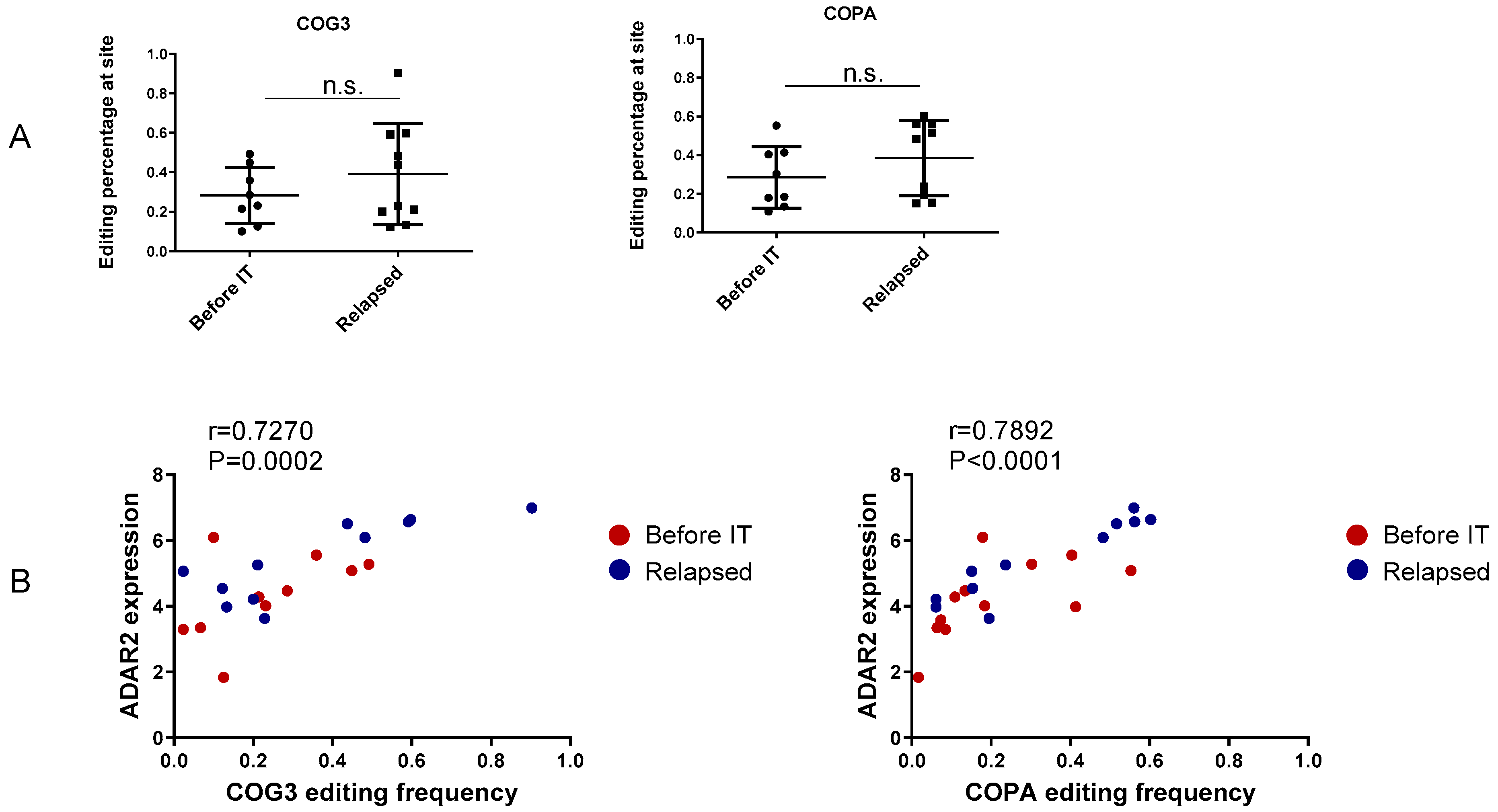
3.2. Recoding Editing
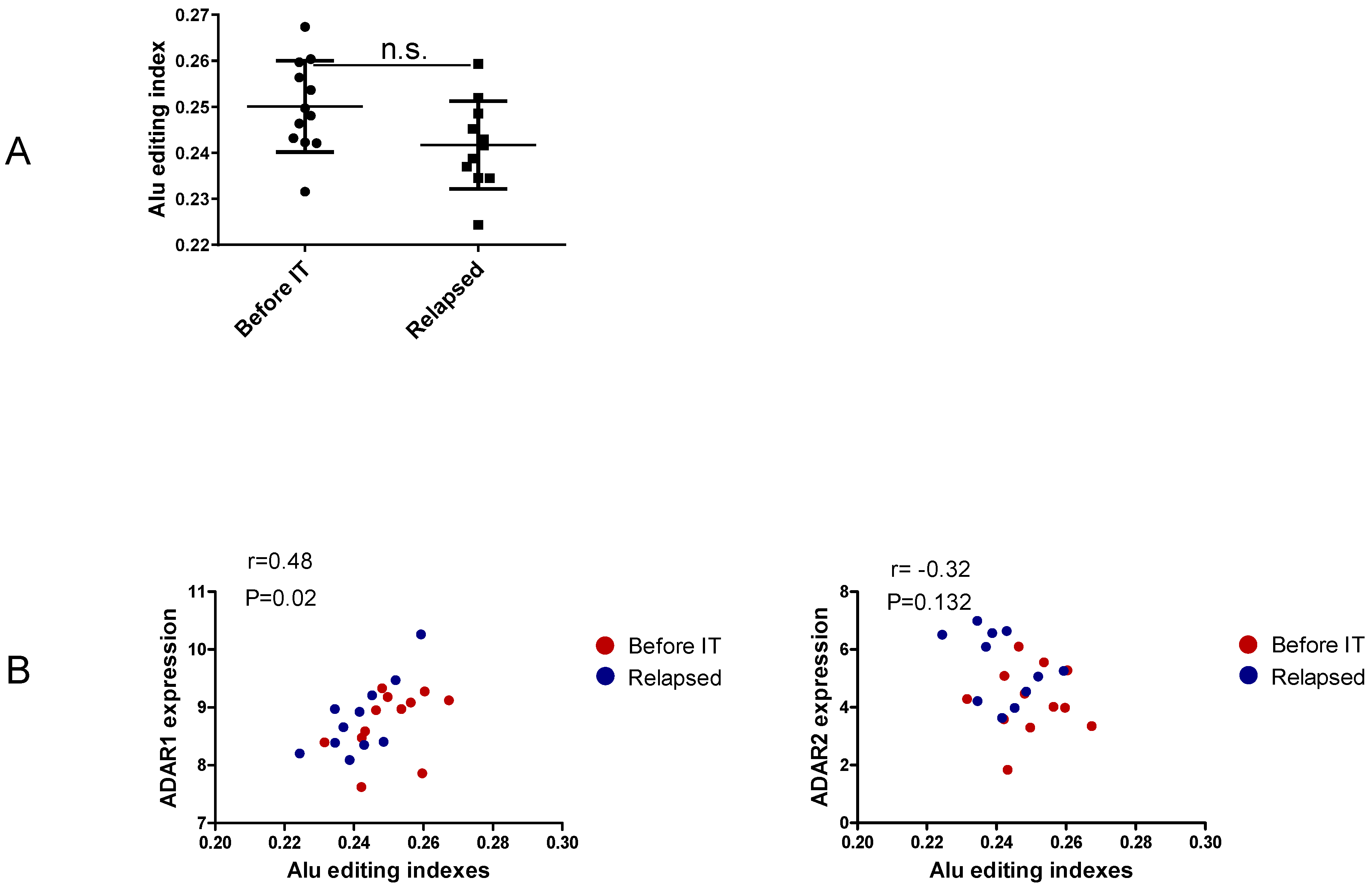
3.3. Non-Recoding Editing in Alu Elements
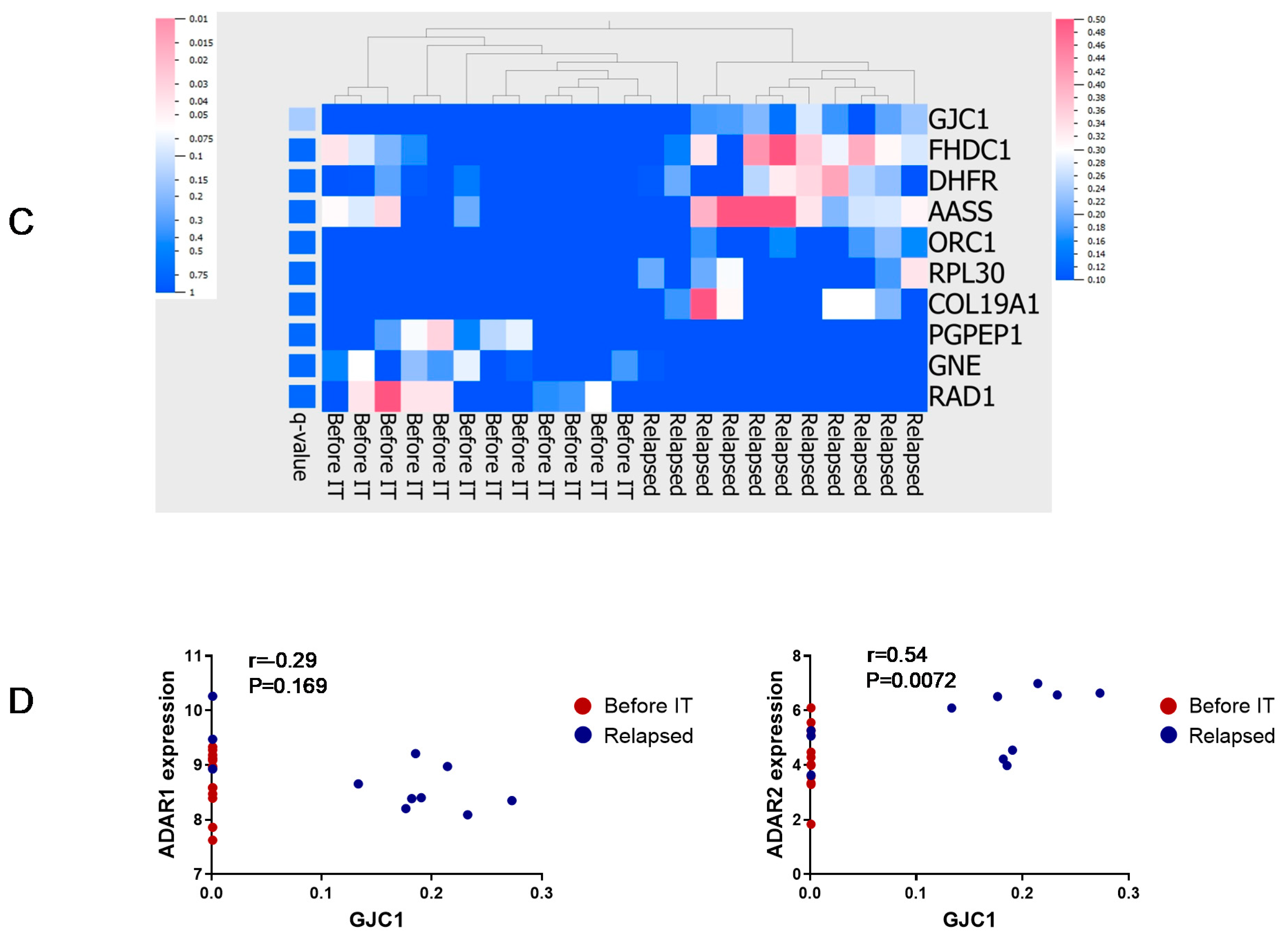
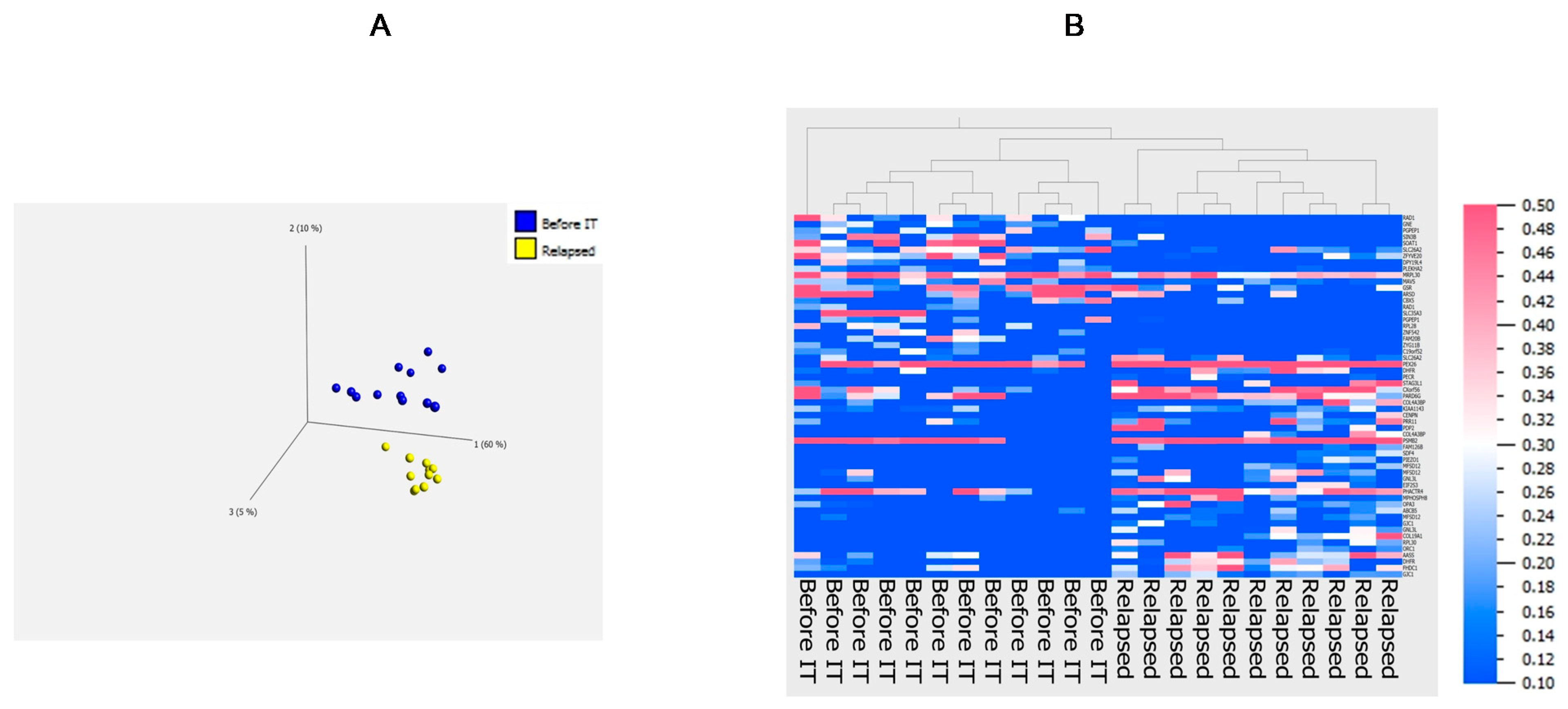
3.4. Principal Component Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADAR | adenosine deaminase acting on RNA |
| ADAT | adenosine deaminase acting on tRNA |
| Alu | Arthrobacter luteus |
| AZIN1 | encoding antizyme inhibitor 1 |
| CCNI | cyclin I |
| COG3 | component of oligomeric Golgi complex 3 |
| COPA | coatomer protein complex subunit α |
| FLNB | filamin B |
| Gabra3 | gamma-aminobutyric acid type A receptor alpha 3 subunit |
| GJC1 | Gap junction gamma-1 protein |
| Glur-B | glutamate receptor subunit B |
| HLA | human leukocyte antigen |
| IGFBP7 | insulin-like growth factor-binding protein 7 |
| LINE | Long interspersed element |
| NEIL1 | DNA base excision repair glycosylase enzyme NEI-like protein 1 |
| NSCLC | non-small cell lung carcinoma |
| PTPN6 | protein tyrosine phosphatase non-receptor type 6 |
| RHOQ | Ras homologue family member Q |
| Tils | tumor infiltrating lymphocytes |
References
- Hsiao, Y.E.; Bahn, J.H.; Yang, Y.; Lin, X.; Tran, S.; Yang, E.W.; Quinones-Valdez, G.; Xiao, X. RNA editing in nascent RNA affects pre-mRNA splicing. Genome Res. 2018, 28, 812–823. [Google Scholar] [CrossRef] [PubMed]
- Bass, B.L. RNA editing by adenosine deaminases that act on RNA. Annu. Rev. Biochem. 2002, 71, 817–846. [Google Scholar] [CrossRef] [PubMed]
- Levanon, E.Y.; Hallegger, M.; Kinar, Y.; Shemesh, R.; Djinovic-Carugo, K.; Rechavi, G.; Jantsch, M.F.; Eisenberg, E. Evolutionarily conserved human targets of adenosine to inosine RNA editing. Nucleic Acids Res. 2005, 33, 1162–1168. [Google Scholar] [CrossRef] [PubMed]
- Gerber, A.P.; Keller, W. RNA editing by base deamination: More enzymes, more targets, new mysteries. Trends Biochem. Sci. 2001, 26, 376–384. [Google Scholar] [CrossRef]
- Blanc, V.; Davidson, N.O. C-to-U RNA editing: Mechanisms leading to genetic diversity. J. Biol. Chem. 2003, 278, 1395–1398. [Google Scholar] [CrossRef]
- Picardi, E.; Manzari, C.; Mastropasqua, F.; Aiello, I.; D’Erchia, A.M.; Pesole, G. Profiling RNA editing in human tissues: Towards the inosinome Atlas. Sci. Rep. 2015, 5, 14941. [Google Scholar] [CrossRef]
- Nishikura, K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol. 2016, 17, 83–96. [Google Scholar] [CrossRef]
- Athanasiadis, A.; Rich, A.; Maas, S. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. Plos Biol. 2004, 2, e391. [Google Scholar] [CrossRef]
- Bazak, L.; Haviv, A.; Barak, M.; Jacob-Hirsch, J.; Deng, P.; Zhang, R.; Isaacs, F.J.; Rechavi, G.; Li, J.B.; Eisenberg, E.; et al. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res. 2014, 24, 365–376. [Google Scholar] [CrossRef]
- Kim, D.D.; Kim, T.T.; Walsh, T.; Kobayashi, Y.; Matise, T.C.; Buyske, S.; Gabriel, A. Widespread RNA editing of embedded alu elements in the human transcriptome. Genome Res. 2004, 14, 1719–1725. [Google Scholar] [CrossRef]
- Nishikura, K. Functions and regulation of RNA editing by ADAR deaminases. Annu. Rev. Biochem. 2010, 79, 321–349. [Google Scholar] [CrossRef] [PubMed]
- Hood, J.L.; Emeson, R.B. Editing of neurotransmitter receptor and ion channel RNAs in the nervous system. Curr. Top. Microbiol. Immunol. 2012, 353, 61–90. [Google Scholar] [PubMed]
- Levanon, E.Y.; Eisenberg, E.; Yelin, R.; Nemzer, S.; Hallegger, M.; Shemesh, R.; Fligelman, Z.Y.; Shoshan, A.; Pollock, S.R.; Sztybel, D.; et al. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat. Biotechnol. 2004, 22, 1001–1005. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Khillan, J.; Gadue, P.; Nishikura, K. Requirement of the RNA editing deaminase ADAR1 gene for embryonic erythropoiesis. Science 2000, 290, 1765–1768. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, M.; Maas, S.; Single, F.N.; Hartner, J.; Rozov, A.; Burnashev, N.; Feldmeyer, D.; Sprengel, R.; Seeburg, P.H. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR. Nature 2000, 406, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Wahlstedt, H.; Daniel, C.; Enstero, M.; Ohman, M. Large-scale mRNA sequencing determines global regulation of RNA editing during brain development. Genome Res. 2009, 19, 978–986. [Google Scholar] [CrossRef]
- Roth, S.H.; Danan-Gotthold, M.; Ben-Izhak, M.; Rechavi, G.; Cohen, C.J.; Louzoun, Y.; Levanon, E.Y. Increased RNA Editing May Provide a Source for Autoantigens in Systemic Lupus Erythematosus. Cell Rep. 2018, 23, 50–57. [Google Scholar] [CrossRef]
- Tusup, M.; Kundig, T.; Pascolo, S. Epitranscriptomics of cancer. World J. Clin. Oncol. 2018, 9, 42–55. [Google Scholar] [CrossRef]
- Maas, S.; Kawahara, Y.; Tamburro, K.M.; Nishikura, K. A-to-I RNA editing and human disease. RNA Biol. 2006, 3, 1–9. [Google Scholar] [CrossRef]
- Keegan, L.P.; Gallo, A.; O’Connell, M.A. The many roles of an RNA editor. Nat. Rev. Genet. 2001, 2, 869–878. [Google Scholar] [CrossRef]
- Han, L.; Diao, L.; Yu, S.; Xu, X.; Li, J.; Zhang, R.; Yang, Y.; Werner, H.M.J.; Eterovic, A.K.; Yuan, Y.; et al. The Genomic Landscape and Clinical Relevance of A-to-I RNA Editing in Human Cancers. Cancer Cell 2015, 28, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Paz-Yaacov, N.; Bazak, L.; Buchumenski, I.; Porath, H.T.; Danan-Gotthold, M.; Knisbacher, B.A.; Eisenberg, E.; Levanon, E.Y. Elevated RNA Editing Activity Is a Major Contributor to Transcriptomic Diversity in Tumors. Cell Rep. 2015, 13, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Mannion, N.M.; Greenwood, S.M.; Young, R.; Cox, S.; Brindle, J.; Read, D.; Nellåker, C.; Vesely, C.; Ponting, C.P.; McLaughlin, P.J.; et al. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep. 2014, 9, 1482–1494. [Google Scholar] [CrossRef] [PubMed]
- Ishizuka, J.J.; Manguso, R.T.; Cheruiyot, C.K.; Bi, K.; Panda, A.; Iracheta-Vellve, A.; Miller, B.C.; Du, P.P.; Yates, K.B.; Dubrot, J.; et al. Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nat. Cell Biol. 2019, 565, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Golji, J.; Brodeur, L.K.; Chung, F.S.; Chen, J.T.; Debeaumont, R.S.; Bullock, C.P.; Jones, M.D.; Kerr, G.; Li, L.; et al. Tumor-derived IFN triggers chronic pathway agonism and sensitivity to ADAR loss. Nat. Med. 2019, 25, 95–102. [Google Scholar] [CrossRef]
- Zhang, M.; Fritsche, J.; Roszik, J.; Williams, L.J.; Peng, X.; Chiu, Y.; Tsou, C.-C.; Hoffgaard, F.; Goldfinger, V.; Schoor, O.; et al. RNA editing derived epitopes function as cancer antigens to elicit immune responses. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef]
- Raaijmakers, M.I.G.; Widmer, D.S.; Maudrich, M.; Koch, T.; Langer, A.; Flace, A.; Schnyder, C.; Dummer, R.; Levesque, M.P. A new live-cell biobank workflow efficiently recovers heterogeneous melanoma cells from native biopsies. Exp. Dermatol. 2015, 24, 377–380. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef]
- Picardi, E.; Pesole, G. REDItools: High-throughput RNA editing detection made easy. Bioinformatics 2013, 29, 1813–1814. [Google Scholar] [CrossRef] [PubMed]
- D’Antonio, M.; Picardi, E.; Castrignanò, T.; D’Erchia, A.M.; Pesole, G. Exploring the RNA Editing Potential of RNA-Seq Data by ExpEdit. Adv. Struct. Saf. Stud. 2014, 1269, 327–338. [Google Scholar]
- Picardi, E.; D’Antonio, M.; Carrabino, D.; Castrignanò, T.; Pesole, G. ExpEdit: A webserver to explore human RNA editing in RNA-Seq experiments. Bioinformatics 2011, 27, 1311–1312. [Google Scholar] [CrossRef] [PubMed]
- Zaranek, A.W.; Levanon, E.Y.; Zecharia, T.; Clegg, T.; Church, G.M. A Survey of Genomic Traces Reveals a Common Sequencing Error, RNA Editing, and DNA Editing. PLoS Genet. 2010, 6, e1000954. [Google Scholar] [CrossRef] [PubMed]
- Shelton, P.M.; Duran, A.; Nakanishi, Y.; Reina-Campos, M.; Kasashima, H.; Llado, V.; Ma, L.; Campos, A.; Garcia-Olmo, D.; Garcia-Arranz, M.; et al. The Secretion of miR-200s by a PKCzeta/ADAR2 Signaling Axis Promotes Liver Metastasis in Colorectal Cancer. Cell Rep. 2018, 23, 1178–1191. [Google Scholar] [CrossRef]
- Galeano, F.; Rossetti, C.; Tomaselli, S.; Cifaldi, L.; Lezzerini, M.; Pezzullo, M.; Boldrini, R.; Massimi, L.; Di Rocco, C.M.; Locatelli, F.; et al. ADAR2-editing activity inhibits glioblastoma growth through the modulation of the CDC14B/Skp2/p21/p27 axis. Oncogene 2013, 32, 998–1009. [Google Scholar] [CrossRef]
- Peng, X.; Xu, X.; Wang, Y.; Hawke, D.; Yu, S.; Han, L.; Zhou, Z.; Mojumdar, K.; Jeong, K.J.; Labrie, M.; et al. A-to-I RNA Editing Contributes to Proteomic Diversity in Cancer. Cancer Cell 2018, 33, 817–828.e7. [Google Scholar] [CrossRef]
- Anantharaman, A.; Tripathi, V.; Khan, A.; Yoon, J.-H.; Singh, D.K.; Gholamalamdari, O.; Guang, S.; Ohlson, J.; Wahlstedt, H.; Öhman, M.; et al. ADAR2 regulates RNA stability by modifying access of decay-promoting RNA-binding proteins. Nucleic Acids Res. 2017, 45, 4189–4201. [Google Scholar] [CrossRef][Green Version]
- Liu, D.; Schilling, B.; Liu, D.; Sucker, A.; Livingstone, E.; Jerby-Arnon, L.; Zimmer, L.; Gutzmer, R.; Satzger, I.; Loquai, C.; et al. Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat. Med. 2019, 25, 1916–1927. [Google Scholar] [CrossRef]






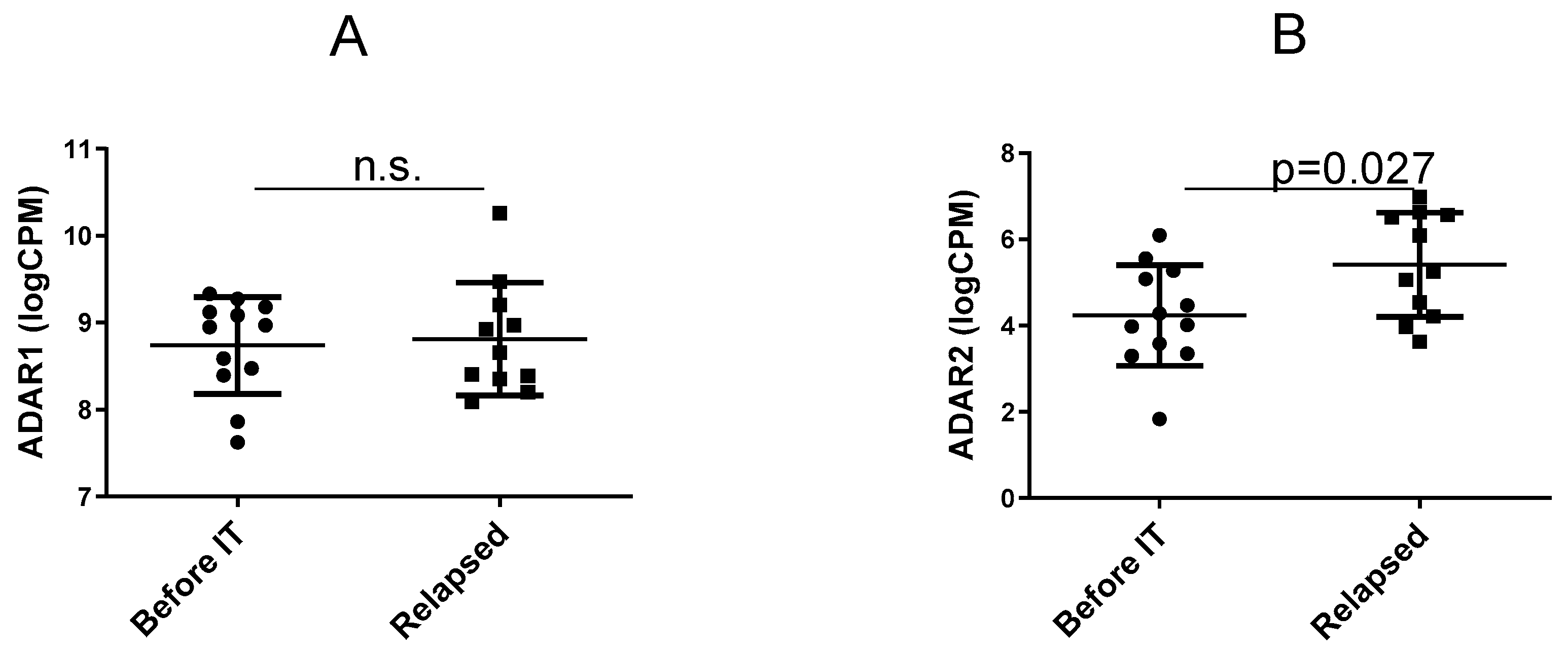{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gender | Tumor | Excision Place | Cell Lines | Treatment | Immunotherapy | Duration | Cell Line Purity | |
|---|---|---|---|---|---|---|---|---|
| Patient 1 | M | Metastasis | Lymph node | MM130820 | Before IT | 87% | ||
| Patient 2 | F | Metastasis | Trunk | MM150325 | Before IT | 98% | ||
| F | Metastasis | Trunk | MM150604 | Relapsed | Ipilimumab | 3 weeks | 74% | |
| Patient 3 | F | Metastasis | Lymph node | MM140905 | Before IT | 99% | ||
| Patient 4 | M | Metastasis | Back | M130107 | Before IT | 59% | ||
| M | Metastasis | Lymph node | M121102 | Before IT | 65% | |||
| Patient 5 | F | Metastasis | Lung | M121008 | Before IT | 20% | ||
| Patient 6 | F | Metastasis | Lymph node | MM130626 | Before IT | 60% | ||
| Patient 7 | F | Metastasis | Leg | MM130106 | Before IT | 57% | ||
| F | Metastasis | Leg | MM131205 | Relapsed | Ipilimumab | 3 weeks | 78% | |
| F | Metastasis | Leg | MM131206 | Relapsed | Ipilimumab | 3 weeks | 65% | |
| Patient 8 | M | Metastasis | Trunk | MM150405 | Before IT | 58% | ||
| M | Metastasis | Lymph node | M150404 | Before IT | ||||
| Patient 9 | F | Metastasis | Skin | MM130926 | Before IT | 50% | ||
| Patient 10 | F | Metastasis | Lymph node | M980513 | Before IT | 83% | ||
| Patient 11 | M | Metastasis | Arm | MM140902 | Relapsed | Ipilimumab | 3 months | 100% |
| M | Metastasis | Arm | M130830 | Relapsed | Ipilimumab | 3 months | 100% | |
| Patient 12 | M | Metastasis | Back | MM130434 | Relapsed | Ipilimumab | 3 months | 77% |
| Patient 13 | M | Metastasis | Trunk | MM121224 | Relapsed | Ipilimumab | 3 months | 75% |
| Patient 14 | M | Metastasis | Skin | M130420 | Relapsed | Ipilimumab | 10 days | 100% |
| M | Metastasis | Liver | M130421 | Relapsed | Ipilimumab | 10 days | 100% | |
| M | Metastasis | Testis | M130425 | Relapsed | Ipilimumab | 10 days | 100% | |
| M | Metastasis | Lung | MM130427 | Relapsed | Ipilimumab | 10 days | 97% |
| Characteristic | Relapsed N = 11 1 | Before IT N = 12 1 |
|---|---|---|
| Age at sampling date | 48 (18–60) | 61 (32–86) |
| Sex | ||
| F | 3 (27%) | 7 (58%) |
| M | 8 (73%) | 5 (42%) |
| Mutation | ||
| BRAF | 2 (18%) | 7 (58%) |
| BRAF and NRAS | 1 (9.1%) | 0 (0%) |
| NRAS | 8 (73%) | 5 (42%) |
| Excision place | ||
| Arm | 2 (18%) | 0 (0%) |
| Back | 1 (9.1%) | 1 (8.3%) |
| Leg | 2 (18%) | 1 (8.3%) |
| Liver | 1 (9.1%) | 0 (0%) |
| Lung | 1 (9.1%) | 1 (8.3%) |
| Lymph node | 0 (0%) | 5 (42%) |
| Skin | 1 (9.1%) | 2 (17%) |
| Testis | 1 (9.1%) | 0 (0%) |
| Trunk | 2 (18%) | 2 (17%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tusup, M.; Cheng, P.F.; Picardi, E.; Raziunaite, A.; Dummer, R.; Levesque, M.P.; French, L.E.; Guenova, E.; Kundig, T.M.; Pascolo, S. Evaluation of the Interplay between the ADAR Editome and Immunotherapy in Melanoma. Non-Coding RNA 2021, 7, 5. https://doi.org/10.3390/ncrna7010005
Tusup M, Cheng PF, Picardi E, Raziunaite A, Dummer R, Levesque MP, French LE, Guenova E, Kundig TM, Pascolo S. Evaluation of the Interplay between the ADAR Editome and Immunotherapy in Melanoma. Non-Coding RNA. 2021; 7(1):5. https://doi.org/10.3390/ncrna7010005
Chicago/Turabian StyleTusup, Marina, Phil F. Cheng, Ernesto Picardi, Austeja Raziunaite, Reinhard Dummer, Mitchell P. Levesque, Lars E. French, Emmanuella Guenova, Thomas M. Kundig, and Steve Pascolo. 2021. "Evaluation of the Interplay between the ADAR Editome and Immunotherapy in Melanoma" Non-Coding RNA 7, no. 1: 5. https://doi.org/10.3390/ncrna7010005
APA StyleTusup, M., Cheng, P. F., Picardi, E., Raziunaite, A., Dummer, R., Levesque, M. P., French, L. E., Guenova, E., Kundig, T. M., & Pascolo, S. (2021). Evaluation of the Interplay between the ADAR Editome and Immunotherapy in Melanoma. Non-Coding RNA, 7(1), 5. https://doi.org/10.3390/ncrna7010005