Secondary Structural Model of MALAT1 Becomes Unstructured in Chronic Myeloid Leukemia and Undergoes Structural Rearrangement in Cervical Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
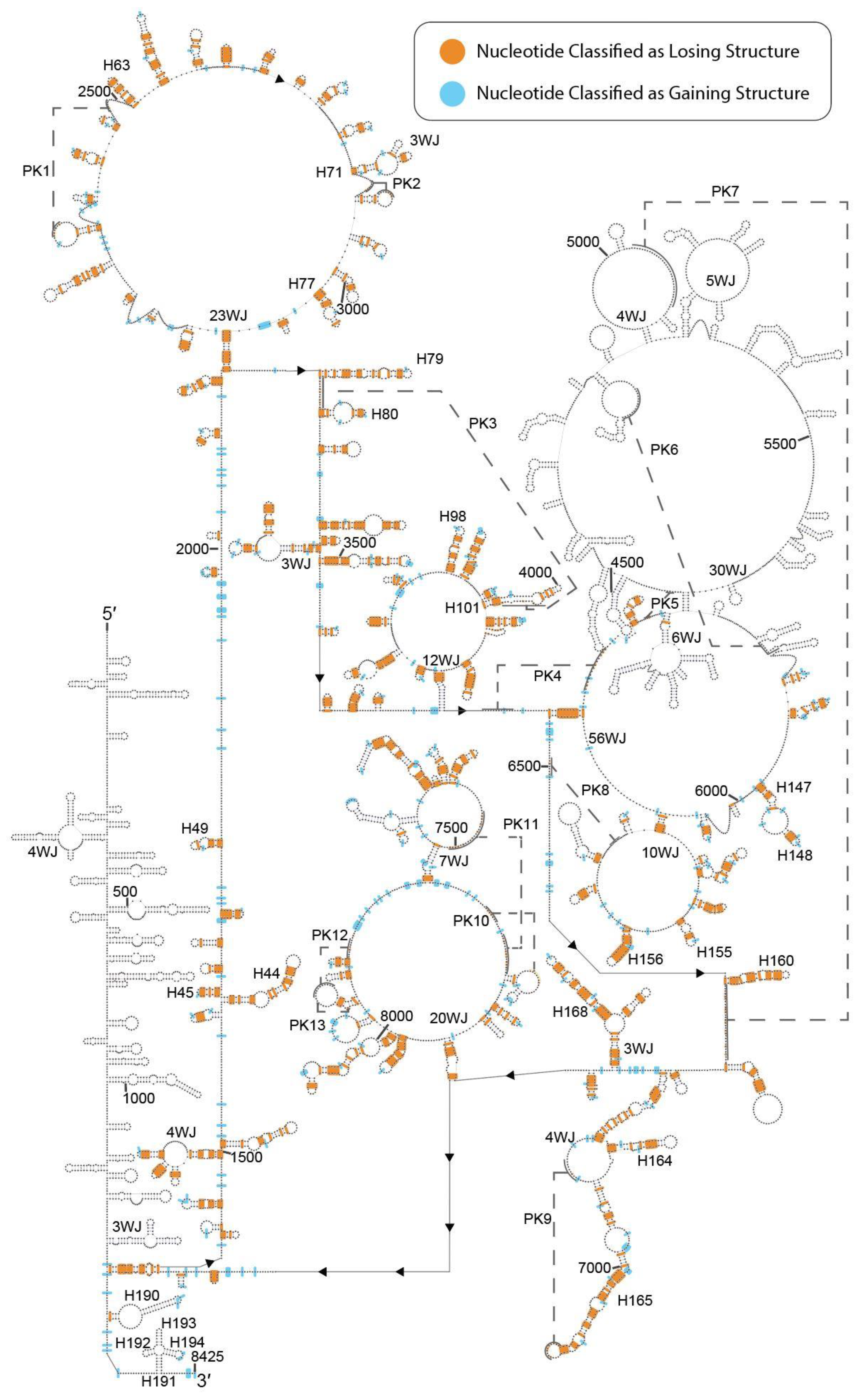
2.1. DMS-Seq Data Suggest Unfolding of MALAT1 Structure in K562 Cells
2.2. Predicted Secondary Structural Changes in K562-MALAT1 Would Impact Multiple RNA- and Protein-Binding Sites
2.3. PARIS Data Suggest Maintenance of Overall Structure of MALAT1 with Rearrangements of Select Long-Range Interactions
2.4. Predicted Structural Changes in HeLa-MALAT1 Would Impact RNA-Binding Sites and Modifications
3. Materials and Methods
3.1. Dataset Acquisition
3.2. Dataset Analysis and Comparison to MALAT1 Secondary Structural Model
3.3. Data and Software Availability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016, 17, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Chillón, I.; Marcia, M. The molecular structure of long non-coding RNAs: Emerging patterns and functional implications. Crit. Rev. Biochem. Mol. Biol. 2020, 55, 662–690. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 2010, 39, 925–938. [Google Scholar] [CrossRef] [PubMed]
- Jeon, Y.; Lee, J.T. YY1 Tethers Xist RNA to the inactive X nucleation center. Cell 2011, 146, 119–133. [Google Scholar] [CrossRef]
- Vance, K.W.; Ponting, C. Transcriptional regulatory functions of nuclear long noncoding RNAs. Trends Genet. 2014, 30, 348–355. [Google Scholar] [CrossRef]
- Ballantyne, M.D.; McDonald, R.A.; Baker, A.H. LncRNA/MicroRNA interactions in the vasculature. Clin. Pharmacol. Ther. 2016, 99, 494–501. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef]
- Paraskevopoulou, M.D.; Hatzigeorgiou, A.G. Analyzing miRNA-lncRNA interactions. In Long Non-Coding RNAs: Methods and Protocols; Feng, Y., Zhang, L., Eds.; Humana Press: New York, NY, USA, 2016; Volume 1402, pp. 271–286. [Google Scholar]
- Yoon, J.H.; Abdelmohsen, K.; Gorospe, M. Functional interactions among microRNAs and long noncoding RNAs. Semin. Cell Dev. Biol. 2014, 34, 9–14. [Google Scholar] [CrossRef]
- Ergun, S.; Oztuzcu, S. Oncocers: ceRNA-mediated cross-talk by sponging miRNAs in oncogenic pathways. Tumor Biol. 2015, 36, 3129–3136. [Google Scholar] [CrossRef]
- Bak, R.O.; Mikkelsen, J.G. miRNA sponges: Soaking up miRNAs for regulation of gene expression. Wiley Interdiscip. Rev. RNA 2013, 5, 317–333. [Google Scholar] [CrossRef]
- Bhan, A.; Soleimani, M.; Mandal, S.S. Long noncoding RNA and cancer: A new paradigm. Cancer Res. 2017, 77, 3965–3981. [Google Scholar] [CrossRef] [PubMed]
- Gutschner, T.; Hämmerle, M.; Diederichs, S. MALAT1—A paradigm for long noncoding RNA function in cancer. J. Mol. Med. 2013, 91, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Bulkley, D.; Wang, J.; Valenstein, M.L.; Yario, T.A.; Steitz, T.A.; Steitz, J.A. Structural insights into the stabilization of MALAT1 noncoding RNA by a bipartite triple helix. Nat. Struct. Mol. Biol. 2014, 21, 633–640. [Google Scholar] [CrossRef] [PubMed]
- McCown, P.J.; Wang, M.C.; Jaeger, L.; Brown, J.A. Secondary structural model of human MALAT1 reveals multiple structure–function relationships. Int. J. Mol. Sci. 2019, 20, 5610. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Zhou, K.I.; Parisien, M.; Dai, Q.; Diatchenko, L.; Pan, T. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. 2017, 45, 6051–6063. [Google Scholar] [CrossRef]
- Liu, N.; Dai, Q.; Zheng, G.; He, C.; Parisien, M.; Pan, T. N6-methyladenosine-dependent RNA structural switches regulate RNA–protein interactions. Nat. Cell Biol. 2015, 518, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Arun, G.; Aggarwal, D.; Spector, D.L. MALAT1 long non-coding RNA: Functional implications. Non Coding RNA 2020, 6, 22. [Google Scholar] [CrossRef]
- Pan, B.; Yang, J.; Wang, X.; Xu, K.; Ikezoe, T. miR-217 sensitizes chronic myelogenous leukemia cells to tyrosine kinase inhibitors by targeting pro-oncogenic anterior gradient 2. Exp. Hematol. 2018, 68, 80–88. [Google Scholar] [CrossRef]
- Sachdeva, M.; Zhu, S.; Wu, F.; Wu, H.; Walia, V.; Kumar, S.; Elble, R.; Watabe, K.; Mo, Y.Y. p53 represses c-Myc through induction of the tumor suppressor miR-145. Proc. Natl. Acad. Sci. USA 2009, 106, 3207–3212. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, C.; Wang, M.; Su, L.; Qu, Y.; Li, J.; Yu, B.; Yan, M.; Yu, Y.; Liu, B.; et al. Decrease of miR-202-3p expression, a novel tumor suppressor, in gastric cancer. PLoS ONE 2013, 8, e69756. [Google Scholar] [CrossRef]
- He, J.; Huang, B.; Zhang, K.; Liu, M.; Xu, T. Long non-coding RNA in cervical cancer: From biology to therapeutic opportunity. Biomed. Pharmacother. 2020, 127, 110209. [Google Scholar] [CrossRef]
- Pan, J.; Bian, Y.; Cao, Z.; Lei, L.; Pan, J.; Huang, J.; Cai, X.; Lan, X.; Zheng, H. Long noncoding RNA MALAT1 as a candidate serological biomarker for the diagnosis of non-small cell lung cancer: A meta-analysis. Thorac. Cancer 2019, 11, 329–335. [Google Scholar] [CrossRef]
- Linder, B.; Grozhik, A.V.; Olarerin-George, A.O.; Meydan, C.; Mason, C.E.; Jaffrey, S.R. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 2015, 12, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Parisien, M.; Dai, Q.; Zheng, G.; He, C.; Pan, T. Probing N6-methyladenosine RNA modification status at single nucleotide resolution in mRNA and long noncoding RNA. RNA 2013, 19, 1848–1856. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Nie, P.; Peng, N.; He, Z.; Liu, M.; Xie, Y.; Miao, Y.; Zuo, Z.; Ren, J. m6AVar: A database of functional variants involved in m6A modification. Nucleic Acids Res. 2018, 46, D139–D145. [Google Scholar] [CrossRef] [PubMed]
- Rouskin, S.; Zubradt, M.; Washietl, S.; Kellis, M.; Weissman, J.S. Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nat. Cell Biol. 2014, 505, 701–705. [Google Scholar] [CrossRef]
- Lu, Z.; Gong, J.; Zhang, Q.C. PARIS: Psoralen analysis of RNA interactions and structures with high throughput and resolution. Comput. Biol. 2018, 1649, 59–84. [Google Scholar] [CrossRef]
- Lu, Z.; Zhang, Q.C.; Lee, B.; Flynn, R.A.; Smith, M.A.; Robinson, J.T.; Davidovich, C.; Gooding, A.R.; Goodrich, K.J.; Mattick, J.S.; et al. RNA duplex map in living cells reveals higher-order transcriptome structure. Cell 2016, 165, 1267–1279. [Google Scholar] [CrossRef]
- Zhao, W.G.; Yu, S.N.; Lu, Z.H.; Ma, Y.H.; Gu, Y.M.; Chen, J. The miR-217 microRNA functions as a potential tumor suppressor in pancreatic ductal adenocarcinoma by targeting KRAS. Carcinogenesis 2010, 31, 1726–1733. [Google Scholar] [CrossRef]
- Polakova, K.M.; Lopotová, T.; Klamova, H.; Burda, P.; Trněný, M.; Stopka, T.; Moravcová, J. Expression patterns of microRNAs associated with CML phases and their disease related targets. Mol. Cancer 2011, 10, 41. [Google Scholar] [CrossRef]
- Gao, X.; Wan, Z.; Wei, M.; Dong, Y.; Zhao, Y.; Chen, X.; Li, Z.; Qin, W.; Yang, G.; Liu, L. Chronic myelogenous leukemia cells remodel the bone marrow niche via exosome-mediated transfer of miR-320. Theranostics 2019, 9, 5642–5656. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lu, W. LncRNA SNHG12 regulates gastric cancer progression by acting as a molecular sponge of miR-320. Mol. Med. Rep. 2017, 17, 2743–2749. [Google Scholar] [CrossRef]
- Nie, Z.Y.; Liu, X.J.; Zhan, Y.; Liu, M.H.; Zhang, X.Y.; Li, Z.Y.; Lu, Y.Q.; Luo, J.M.; Yang, L. miR-140-5p induces cell apoptosis and decreases Warburg effect in chronic myeloid leukemia by targeting SIX1. Biosci. Rep. 2019, 39, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Shuai, S.; Suzuki, H.; Diaz-Navarro, A.; Nadeu, F.; Kumar, S.A.; Gutierrez-Fernandez, A.; Delgado, J.; Pinyol, M.; López-Otín, C.; Puente, X.S.; et al. The U1 spliceosomal RNA is recurrently mutated in multiple cancers. Nat. Cell Biol. 2019, 574, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Latorre, E.; Carelli, S.; Raimondi, I.; D’Agostino, V.; Castiglioni, I.; Zucal, C.; Moro, G.; Luciani, A.; Ghiraldi, G.; Monti, E.; et al. The ribonucleic complex HuR-MALAT1 represses CD133 expression and suppresses epithelial-mesenchymal transition in breast cancer. Cancer Res. 2016, 76, 2626–2636. [Google Scholar] [CrossRef]
- Srikantan, S.; Gorospe, M. HuR function in disease. Front. Biosci. 2012, 17, 189–205. [Google Scholar] [CrossRef]
- Dai, Q.; Moshitch-Moshkovitz, S.; Han, D.; Kol, N.; Amariglio, N.; Rechavi, G.; Dominissini, D.; He, C. Nm-seq maps 2′-O-methylation sites in human mRNA with base precision. Nat. Methods 2017, 14, 695–698. [Google Scholar] [CrossRef]
- Zhang, L.S.; Liu, C.; Ma, H.; Dai, Q.; Sun, H.L.; Luo, G.; Zhang, Z.; Zhang, L.; Hu, L.; Dong, X.; et al. Transcriptome-wide mapping of internal N7-methylguanosine methylome in mammalian mRNA. Mol. Cell 2019, 74, 1304–1316.e8. [Google Scholar] [CrossRef]
- Jacob, R.; Zander, S.; Gutschner, T. The dark side of the epitranscriptome: Chemical modifications in long non-coding RNAss. Int. J. Mol. Sci. 2017, 18, 2387. [Google Scholar] [CrossRef]
- Squires, J.E.; Patel, H.R.; Nousch, M.; Sibbritt, T.; Humphreys, D.T.; Parker, B.J.; Suter, C.M.; Preiss, T. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 2012, 40, 5023–5033. [Google Scholar] [CrossRef]
- Liu, J.; Yue, Y.; Han, D.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G.; Yu, M.; Lu, Z.; Deng, X.; et al. A METTL3–METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014, 10, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, J.; Zhu, J.S. The role of m6A RNA methylation in human cancer. Mol. Cancer 2019, 18, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Lu, Y. Dexmedetomidine suppressed the biological behavior of HK-2 cells treated with LPS by down-regulating ALKBH5. Inflammation 2020, 43, 2256–2263. [Google Scholar] [CrossRef]
- Berulava, T.; Ziehe, M.; Klein-Hitpass, L.; Mladenov, E.; Thomale, J.; Ruther, U.; Horsthemke, B. FTO levels affect RNA modification and the transcriptome. Eur. J. Hum. Genet. 2012, 21, 317–323. [Google Scholar] [CrossRef]
- Xu, C.; Liu, K.; Tempel, W.; Demetriades, M.; Aik, W.; Schofield, C.J.; Min, J. Structures of human ALKBH5 demethylase reveal a unique binding mode for specific single-stranded N6-methyladenosine RNA demethylation. J. Biol. Chem. 2014, 289, 17299–17311. [Google Scholar] [CrossRef] [PubMed]
- Yeo, G. The role of the FTO (Fat Mass and Obesity Related) locus in regulating body size and composition. Mol. Cell. Endocrinol. 2014, 397, 34–41. [Google Scholar] [CrossRef]
- Moursy, A.; Allain, F.H.T.; Cléry, A. Characterization of the RNA recognition mode of hnRNPG extends its role in SMN2 splicing regulation. Nucleic Acids Res. 2014, 42, 6659–6672. [Google Scholar] [CrossRef]
- Pal, A.; Levy, Y. Structure, stability and specificity of the binding of ssDNA and ssRNA with proteins. PLoS Comput. Biol. 2019, 15, e1006768. [Google Scholar] [CrossRef]
- Shin, K.H.; Kim, R.H.; Kim, R.H.; Kang, M.K.; Park, N.H. hnRNPG elicits tumor-suppressive activity in part by upregulating the expression of Txnip. Biochem. Biophys. Res. Commun. 2008, 372, 880–885. [Google Scholar] [CrossRef]
- Shin, K.H.; Kang, M.K.; Kim, R.H.; Christensen, R.; Park, N.H. Heterogeneous nuclear ribonucleoprotein G shows tumor suppressive effect against oral squamous cell carcinoma cells. Clin. Cancer Res. 2006, 12, 3222–3228. [Google Scholar] [CrossRef]
- Shin, K.; Kim, R.H.; Yu, B.; Kang, M.K.; Elashoff, D.; Christensen, R.; Pucar, A.; Park, N.H. Expression and mutation analysis of heterogeneous nuclear ribonucleoprotein G in human oral cancer. Oral Oncol. 2011, 47, 1011–1016. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dai, D.; Wang, H.; Zhu, L.; Jin, H.; Wang, X. N6-methyladenosine links RNA metabolism to cancer progression. Cell Death Dis. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Z.; Xiang, J.J.; Wu, L.G.; Bai, Y.S.; Chen, Z.W.; Yin, X.Q.; Wang, Q.; Guo, W.H.; Peng, Y.; Guo, H.; et al. A genetic variant in long non-coding RNA MALAT1 associated with survival outcome among patients with advanced lung adenocarcinoma: A survival cohort analysis. BMC Cancer 2017, 17, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ritz, J.; Martin, J.S.; Laederach, A. Evaluating our ability to predict the structural disruption of RNA by SNPs. BMC Genom. 2012, 13 (Suppl. 4), 1–11. [Google Scholar] [CrossRef]
- Sabarinathan, R.; Tafer, H.; Seemann, S.E.; Hofacker, I.L.; Stadler, P.F.; Gorodkin, J. RNAsnp: Efficient detection of local RNA secondary structure changes induced by SNPs. Hum. Mutat. 2013, 34, 925. [Google Scholar] [CrossRef]
- Wu, S.; Sun, H.; Wang, Y.; Yang, X.; Meng, Q.; Yang, H.; Zhu, H.; Tang, W.; Li, X.; Aschner, M.; et al. MALAT1 rs664589 polymorphism inhibits binding to miR-194-5p, contributing to colorectal cancer risk, growth, and metastasis. Cancer Res. 2019, 79, 5432–5441. [Google Scholar] [CrossRef]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a shared vision for cancer genomic data. N. Engl. J. Med. 2016, 375, 1109–1112. [Google Scholar] [CrossRef]
- Song, B.; Yan, J.; Liu, C.; Zhou, H.; Zheng, Y. Tumor suppressor role of miR-363-3p in gastric cancer. Med. Sci. Monit. 2015, 21, 4074–4080. [Google Scholar] [CrossRef]
- Casadei, L.; Calore, F.; Creighton, C.J.; Guescini, M.; Batte, K.; Iwenofu, O.H.; Zewdu, A.; Braggio, D.A.; Bill, K.L.; Fadda, P.; et al. Exosome-derived miR-25-3p and miR-92a-3p stimulate liposarcoma progression. Cancer Res. 2017, 77, 3846–3856. [Google Scholar] [CrossRef]
- Wan, Z.; Chen, X.; Gao, X.; Dong, Y.; Zhao, Y.; Wei, M.; Fan, W.; Yang, G.; Liu, L. Chronic myeloid leukemia-derived exosomes attenuate adipogenesis of adipose derived mesenchymal stem cells via transporting miR-92a-3p. J. Cell. Physiol. 2019, 234, 21274–21283. [Google Scholar] [CrossRef]
- Willmann, M.R.; Berkowitz, N.D.; Gregory, B.D. Improved genome-wide mapping of uncapped and cleaved transcripts in eukaryotes—GMUCT 2.0. Methods 2014, 67, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Li, L.L. miR-145 Contributes to the progression of cervical carcinoma by directly regulating FSCN1. Cell Transplant. 2019, 28, 1299–1305. [Google Scholar] [CrossRef]
- Lu, H.; He, Y.; Lin, L.; Qi, Z.; Ma, L.; Li, L.; Su, Y. Long non-coding RNA MALAT1 modulates radiosensitivity of HR-HPV+ cervical cancer via sponging miR-145. Tumor Biol. 2015, 37, 1683–1691. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tang, S.; Le, S.Y.; Lu, R.; Rader, J.S.; Meyers, C.; Zheng, Z.M. Aberrant expression of oncogenic and tumor-suppressive microRNAs in cervical cancer is required for cancer cell growth. PLoS ONE 2008, 3, e2557. [Google Scholar] [CrossRef]
- Zeng, F.; Xue, M.; Xiao, T.; Li, Y.; Xiao, S.; Jiang, B.; Ren, C. MiR-200b promotes the cell proliferation and metastasis of cervical cancer by inhibiting FOXG1. Biomed. Pharmacother. 2016, 79, 294–301. [Google Scholar] [CrossRef]
- Kang, H.W.; Wang, F.; Wei, Q.; Zhao, Y.F.; Liu, M.; Li, X.; Tang, H. miR-20a promotes migration and invasion by regulating TNKS2 in human cervical cancer cells. FEBS Lett. 2012, 586, 897–904. [Google Scholar] [CrossRef]
- Piao, J.; You, K.; Guo, Y.; Zhang, Y.Y.; Li, Z.; Geng, L. Substrate stiffness affects epithelial-mesenchymal transition of cervical cancer cells through miR-106b and its target protein DAB2. Int. J. Oncol. 2017, 50, 2033–2042. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Sheng, W.; Meng, Y.; Cao, Y.; Li, R. LncRNA PTENP1 inhibits cervical cancer progression by suppressing miR-106b. Artif. Cells Nanomed. Biotechnol. 2020, 48, 393–407. [Google Scholar] [CrossRef] [PubMed]
- Engreitz, J.M.; Sirokman, K.; McDonel, P.; Shishkin, A.A.; Surka, C.; Russell, P.; Grossman, S.R.; Chow, A.Y.; Guttman, M.; Lander, E.S. RNA-RNA interactions enable specific targeting of noncoding RNAs to nascent pre-mRNAs and chromatin sites. Cell 2014, 159, 188–199. [Google Scholar] [CrossRef]
- Aw, J.G.A.; Shen, Y.; Wilm, A.; Sun, M.; Ni Lim, X.; Boon, K.L.; Tapsin, S.; Chan, Y.S.; Tan, C.P.; Sim, A.Y.; et al. In vivo mapping of eukaryotic RNA interactomes reveals principles of higher-order organization and regulation. Mol. Cell 2016, 62, 603–617. [Google Scholar] [CrossRef]
- Woodcock, C.B.; Yu, D.; Hajian, T.; Li, J.; Huang, Y.; Dai, N.; Correa, I.R., Jr.; Wu, T.; Vedadi, M.; Zhang, X.; et al. Human MettL3–MettL14 complex is a sequence-specific DNA adenine methyltransferase active on single-strand and unpaired DNA in vitro. Cell Discov. 2019, 5, 1–4. [Google Scholar]
- Amort, T.; Soulière, M.F.; Wille, A.; Jia, X.Y.; Fiegl, H.; Wörle, H.; Micura, R.; Lusser, A. Long non-coding RNAs as targets for cytosine methylation. RNA Biol. 2013, 10, 1002–1008. [Google Scholar] [CrossRef] [PubMed]
- West, J.A.; Davis, C.P.; Sunwoo, H.; Simon, M.D.; Sadreyev, R.I.; Wang, P.I.; Tolstorukov, M.Y.; Kingston, R.E. The long noncoding RNAs NEAT1 and MALAT1 bind active chromatin sites. Mol. Cell 2014, 55, 791–802. [Google Scholar] [CrossRef]
- McCown, P.J.; Ruszkowska, A.; Kunkler, C.N.; Breger, K.; Hulewicz, J.P.; Wang, M.C.; Springer, N.A.; Brown, J.A. Naturally occurring modified ribonucleosides. Wiley Interdiscip. Rev. RNA 2020, 11, e1595. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Dou, R.; Yin, T.L.; Ding, J. MiRNA-106b-5p in human cancers: Diverse functions and promising biomarker. Biomed. Pharmacother. 2020, 127, 110211. [Google Scholar] [CrossRef]
- Cai, N.; Hu, L.; Xie, Y.; Gao, J.H.; Zhai, W.; Wang, L.; Jin, Q.J.; Qin, C.Y.; Qiang, R. MiR-17-5p promotes cervical cancer cell proliferation and metastasis by targeting transforming growth factor-β receptor 2. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 1899–1906. [Google Scholar]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Gruning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, A.D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef]
- Darty, K.; Denise, A.; Ponty, Y. VARNA: Interactive drawing and editing of the RNA secondary structure. Bioinformatics 2009, 25, 1974–1975. [Google Scholar] [CrossRef]
- Li, J.H.; Liu, S.; Zhou, H.; Qu, L.H.; Yang, J.H. StarBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef]
- Cai, Z.; Cao, C.; Ji, L.; Ye, R.; Wang, D.; Xia, C.; Wang, S.; Du, Z.; Hu, N.; Yu, X.; et al. RIC-seq for global in situ profiling of RNA–RNA spatial interactions. Nat. Cell Biol. 2020, 582, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, J.E.; Freier, S.M.; Spector, D.L. 3′ End processing of a long nuclear-retained noncoding RNA yields a tRNA-like cytoplasmic RNA. Cell 2008, 135, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Lv, D.; Xu, K.; Jin, X.; Li, J.; Shi, Y.; Zhang, M.; Jin, X.; Li, Y.; Xu, J.; Li, X. LncSpA: LncRNA spatial atlas of expression across normal and cancer tissues. Cancer Res. 2020, 80, 2067–2071. [Google Scholar] [CrossRef] [PubMed]
- Safra, M.; Sas-Chen, A.; Nir, R.; Winkler, R.; Nachshon, A.; Bar-Yaacov, D.; Erlacher, M.; Rossmanith, W.; Stern-Ginossar, N.; Schwartz, S. The m1A landscape on cytosolic and mitochondrial mRNA at single-base resolution. Nat. Cell Biol. 2017, 551, 251–255. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.C.; McCown, P.J.; Schiefelbein, G.E.; Brown, J.A. Secondary Structural Model of MALAT1 Becomes Unstructured in Chronic Myeloid Leukemia and Undergoes Structural Rearrangement in Cervical Cancer. Non-Coding RNA 2021, 7, 6. https://doi.org/10.3390/ncrna7010006
Wang MC, McCown PJ, Schiefelbein GE, Brown JA. Secondary Structural Model of MALAT1 Becomes Unstructured in Chronic Myeloid Leukemia and Undergoes Structural Rearrangement in Cervical Cancer. Non-Coding RNA. 2021; 7(1):6. https://doi.org/10.3390/ncrna7010006
Chicago/Turabian StyleWang, Matthew C., Phillip J. McCown, Grace E. Schiefelbein, and Jessica A. Brown. 2021. "Secondary Structural Model of MALAT1 Becomes Unstructured in Chronic Myeloid Leukemia and Undergoes Structural Rearrangement in Cervical Cancer" Non-Coding RNA 7, no. 1: 6. https://doi.org/10.3390/ncrna7010006
APA StyleWang, M. C., McCown, P. J., Schiefelbein, G. E., & Brown, J. A. (2021). Secondary Structural Model of MALAT1 Becomes Unstructured in Chronic Myeloid Leukemia and Undergoes Structural Rearrangement in Cervical Cancer. Non-Coding RNA, 7(1), 6. https://doi.org/10.3390/ncrna7010006