The Missing “lnc” between Genetics and Cardiac Disease




{kind=link}
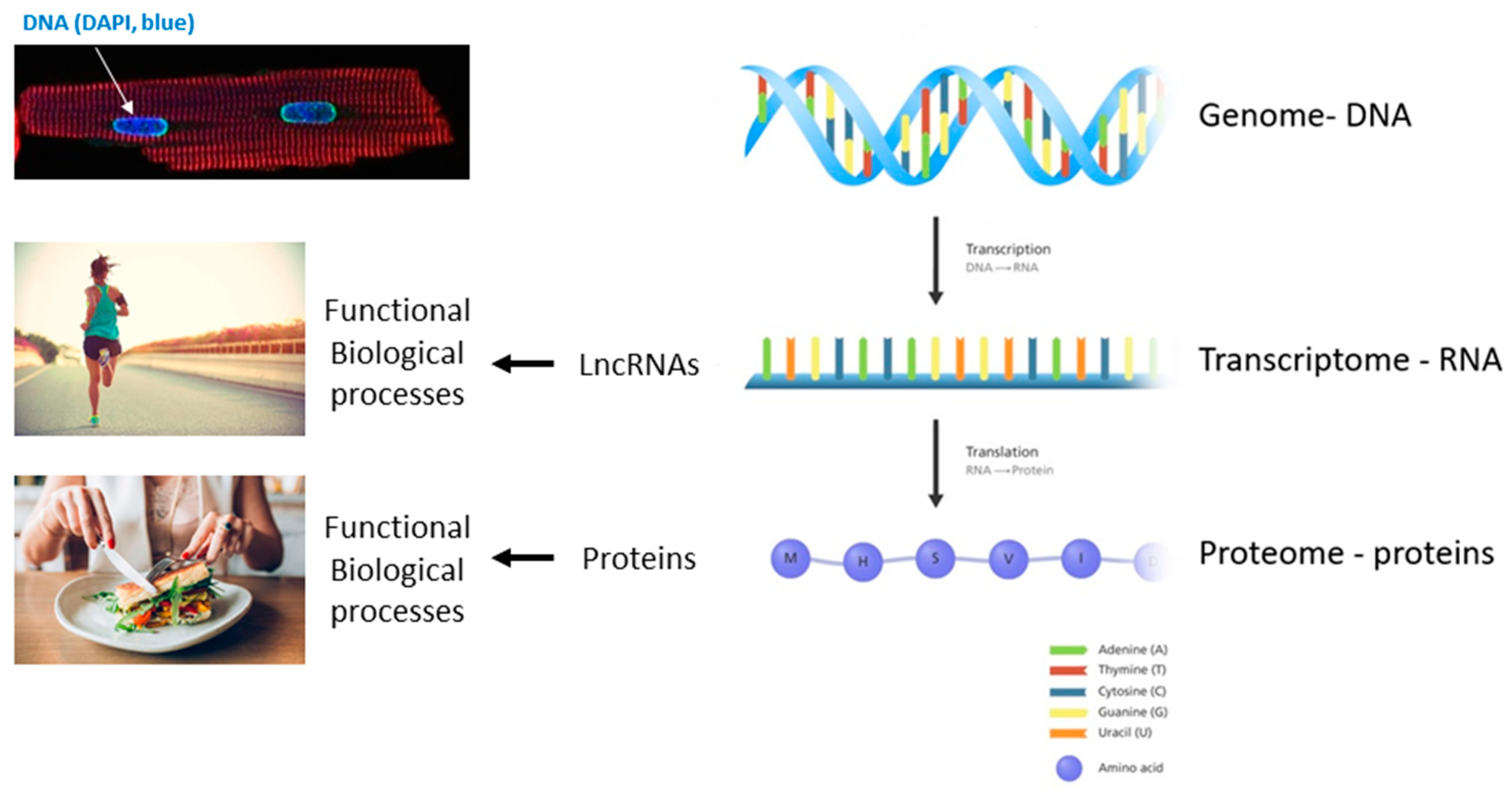
Abstract
Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The Sequence of the Human Genome. Hum. Genome 2001, 291, 51. [Google Scholar] [CrossRef] [PubMed]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef]
- Frankish, A.; Diekhans, M.; Ferreira, A.M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef]
- Ma, L.; Cao, J.; Liu, L.; Du, Q.; Li, Z.; Zou, D.; Bajic, V.B.; Zhang, Z. LncBook: A curated knowledgebase of human long non-coding RNAs. Nucleic Acids Res. 2019, 47, D128–D134. [Google Scholar] [CrossRef]
- Cao, J. The functional role of long non-coding RNAs and epigenetics. Biol. Proced. Online 2014, 16, 11. [Google Scholar] [CrossRef] [PubMed]
- Cobb, M. 60 years ago, Francis Crick changed the logic of biology. PLoS Biol. 2017, 15, e2003243. [Google Scholar] [CrossRef] [PubMed]
- Savarese, G.; Lund, L.H. Global Public Health Burden of Heart Failure. Card. Fail. Rev. 2017, 3, 7–11. [Google Scholar] [CrossRef]
- Cardiovascular Diseases (CVDs). Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 9 January 2020).
- Hobuß, L.; Bär, C.; Thum, T. Long Non-coding RNAs: At the Heart of Cardiac Dysfunction? Front. Physiol. 2019, 10. [Google Scholar] [CrossRef]
- Han, P.; Li, W.; Lin, C.H.; Yang, J.; Shang, C.; Nurnberg, S.T.; Jin, K.K.; Xu, W.; Lin, C.Y.; Lin, C.J.; et al. A long noncoding RNA protects the heart from pathological hypertrophy. Nature 2014, 514, 102–106. [Google Scholar] [CrossRef]
- Harikrishnan, K.N.; Okabe, J.; Mathiyalagan, P.; Khan, A.W.; Jadaan, S.A.; Sarila, G.; Ziemann, M.; Khurana, I.; Maxwell, S.S.; Du, X.J.; et al. Sex-Based Mhrt Methylation Chromatinizes MeCP2 in the Heart. iScience 2019, 17, 288–301. [Google Scholar]
- Wu, Y.; Huang, C.; Meng, X.; Li, J. Long Noncoding RNA MALAT1: Insights into its Biogenesis and Implications in Human Disease. Curr. Pharm. Des. 2015, 21, 5017–5028. [Google Scholar] [CrossRef] [PubMed]
- Gast, M.; Rauch, B.H.; Nakagawa, S.; Haghikia, A.; Jasina, A.; Haas, J.; Nath, N.; Jensen, L.; Stroux, A.; Böhm, A.; et al. Immune system-mediated atherosclerosis caused by deficiency of long non-coding RNA MALAT1 in ApoE−/−mice. Cardiovasc. Res. 2019, 115, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Zhang, L.; Song, J.; Wang, Z.; Huang, X.; Guo, Z.; Chen, F.; Zhao, X. Long noncoding RNA MALAT1 mediates cardiac fibrosis in experimental postinfarct myocardium mice model. J. Cell. Physiol. 2019, 234, 2997–3006. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, X.J.; Ji, Y.X.; Zhang, P.; Deng, K.Q.; Gong, J.; Ren, S.; Wang, X.; Chen, I.; Wang, H.; et al. The long noncoding RNA Chaer defines an epigenetic checkpoint in cardiac hypertrophy. Nat. Med. 2016, 22, 1131–1139. [Google Scholar] [CrossRef]
- Li, X.; Zhou, J.; Huang, K. Inhibition of the lncRNA Mirt1 Attenuates Acute Myocardial Infarction by Suppressing NF-κB Activation. CPB 2017, 42, 1153–1164. [Google Scholar] [CrossRef]
- Zangrando, J.; Zhang, L.; Vausort, M.; Maskali, F.; Marie, P.Y.; Wagner, D.R.; Devaux, Y. Identification of candidate long non-coding RNAs in response to myocardial infarction. BMC Genom. 2015, 15, 460. [Google Scholar] [CrossRef]
- Gomes, C.P.; Spencer, H.; Ford, K.L.; Michel, L.Y.; Baker, A.H.; Emanueli, C.; Balligand, J.L.; Devaux, Y. The Function and Therapeutic Potential of Long Non-coding RNAs in Cardiovascular Development and Disease. Mol. Ther. Nucleic Acids 2017, 8, 494–507. [Google Scholar] [CrossRef]
- Kathiresan, S.; Srivastava, D. Genetics of Human Cardiovascular Disease. Cell 2012, 148, 1242–1257. [Google Scholar] [CrossRef]
- Sabater-Molina, M.; Pérez-Sánchez, I.; Hernández Del Rincón, J.P.; Gimeno, J.R. Genetics of hypertrophic cardiomyopathy: A review of current state. Clin. Genet. 2018, 93, 3–14. [Google Scholar] [CrossRef]
- Garfinkel, A.C.; Seidman, J.G.; Seidman, C.E. Genetic Pathogenesis of Hypertrophic and Dilated Cardiomyopathy. Heart Fail. Clin. 2018, 14, 139–146. [Google Scholar] [CrossRef]
- Roberts, A.M.; Ware, J.S.; Herman, D.S.; Schafer, S.; Baksi, J.; Bick, A.G.; Buchan, R.J.; Walsh, R.; John, S.; Wilkinson, S.; et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci. Transl. Med. 2015, 7, 270ra6. [Google Scholar] [CrossRef]
- Khera, A.V.; Kathiresan, S. Genetics of coronary artery disease: Discovery, biology and clinical translation. Nat. Rev. Genet. 2017, 18, 331–344. [Google Scholar] [CrossRef]
- Paranal, R.M.; Teekakirikul, P.; Ho, C.Y.; Fatkin, D.; Seidman, C.E. 2—Genetic Cardiomyopathies. In Emery and Rimoin’s Principles and Practice of Medical Genetics and Genomics, 7th ed.; Pyeritz, R.E., Korf, B.R., Grody, W.W., Eds.; Academic Press: New York, NY, USA, 2013; pp. 77–114. [Google Scholar] [CrossRef]
- Drummond, M.F.; Sculpher, M.J.; Claxton, K.; Stoddart, G.L.; Torrance, G.W. Methods for the Economic Evaluation of Health Care Programmes; Oxford University Press: Oxford, UK, 2015. [Google Scholar]
- Marziali, A.; Akeson, M. New DNA sequencing methods. Annu. Rev. Biomed. Eng. 2001, 3, 195–223. [Google Scholar] [CrossRef] [PubMed]
- Benn, M.; Nordestgaard, B.G. From genome-wide association studies to Mendelian randomization: Novel opportunities for understanding cardiovascular disease causality, pathogenesis, prevention, and treatment. Cardiovasc. Res. 2018, 114, 1192–1208. [Google Scholar] [CrossRef] [PubMed]
- Brandão, R.D.; van Roozendaal, K.; Tserpelis, D.; Gómez García, E.; Blok, M.J. Characterisation of unclassified variants in the BRCA1/2 genes with a putative effect on splicing. Breast Cancer Res. Treat. 2011, 129, 971–982. [Google Scholar] [CrossRef] [PubMed]
- Cloonan, N.; Forrest, A.R.; Kolle, G.; Gardiner, B.B.; Faulkner, G.J.; Brown, M.K.; Taylor, D.F.; Steptoe, A.L.; Wani, S.; Bethel, G.; et al. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat. Methods 2008, 5, 613–619. [Google Scholar] [CrossRef]
- Sultan, M.; Schulz, M.H.; Richard, H.; Magen, A.; Klingenhoff, A.; Scherf, M.; Seifert, M.; Borodina, T.; Soldatov, A.; Parkhomchuk, D.; et al. A Global View of Gene Activity and Alternative Splicing by Deep Sequencing of the Human Transcriptome. Science 2008, 321, 956–960. [Google Scholar] [CrossRef]
- Liu, X.; Ma, Y.; Yin, K.; Li, W.; Chen, W.; Zhang, Y.; Zhu, C.; Li, T.; Han, B.; Liu, X.; et al. Long non-coding and coding RNA profiling using strand-specific RNA-seq in human hypertrophic cardiomyopathy. Sci. Data 2019, 6, 1–7. [Google Scholar] [CrossRef]
- Cummings, B.B.; Marshall, J.L.; Tukiainen, T.; Lek, M.; Donkervoort, S.; Foley, A.R.; Bolduc, V.; Waddell, L.B.; Sandaradura, S.A.; O’Grady, G.L.; et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci. Transl. Med. 2017, 9, eaal5209. [Google Scholar] [CrossRef]
- Ishii, N.; Ozaki, K.; Sato, H.; Mizuno, H.; Saito, S.; Takahashi, A.; Miyamoto, Y.; Ikegawa, S.; Kamatani, N.; Hori, M.; et al. Identification of a novel non-coding RNA, MIAT, that confers risk of myocardial infarction. J. Hum. Genet. 2006, 51, 1087–1099. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, J.; Sun, L.; Zhu, S. LncRNA myocardial infarction-associated transcript (MIAT) contributed to cardiac hypertrophy by regulating TLR4 via miR-93. Eur. J. Pharmacol. 2018, 818, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Yao, J.; Liu, J.Y.; Li, X.M.; Wang, X.Q.; Li, Y.J.; Tao, Z.F.; Song, Y.C.; Chen, Q.; Jiang, Q. lncRNA-MIAT Regulates Microvascular Dysfunction by Functioning as a Competing Endogenous RNA. Circ. Res. 2015, 116, 1143–1156. [Google Scholar] [CrossRef] [PubMed]
- Check Hayden, E. Technology: The $1,000 genome. Nat. News 2014, 507, 294. [Google Scholar] [CrossRef] [PubMed]
- FDA. FDA Approves First-of-Its Kind Targeted RNA-Based Therapy to Treat a Rare Disease. Available online: http://www.fda.gov/news-events/press-announcements/fda-approves-first-its-kind-targeted-rna-based-therapy-treat-rare-disease (accessed on 1 December 2019).
- Laina, A.; Gatsiou, A.; Georgiopoulos, G.; Stamatelopoulos, K.; Stellos, K. RNA Therapeutics in Cardiovascular Precision Medicine. Front. Physiol. 2018, 9, 953. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azodi, M.; Kamps, R.; Heymans, S.; Robinson, E.L. The Missing “lnc” between Genetics and Cardiac Disease. Non-Coding RNA 2020, 6, 3. https://doi.org/10.3390/ncrna6010003
Azodi M, Kamps R, Heymans S, Robinson EL. The Missing “lnc” between Genetics and Cardiac Disease. Non-Coding RNA. 2020; 6(1):3. https://doi.org/10.3390/ncrna6010003
Chicago/Turabian StyleAzodi, Maral, Rick Kamps, Stephane Heymans, and Emma Louise Robinson. 2020. "The Missing “lnc” between Genetics and Cardiac Disease" Non-Coding RNA 6, no. 1: 3. https://doi.org/10.3390/ncrna6010003
APA StyleAzodi, M., Kamps, R., Heymans, S., & Robinson, E. L. (2020). The Missing “lnc” between Genetics and Cardiac Disease. Non-Coding RNA, 6(1), 3. https://doi.org/10.3390/ncrna6010003