
Computational Characterization of ncRNA Fragments in Various Tissues of the Brassica rapa Plant

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Sequence Data
2.2. Mapping the Reads to Genomic Regions and Mature ncRNAs
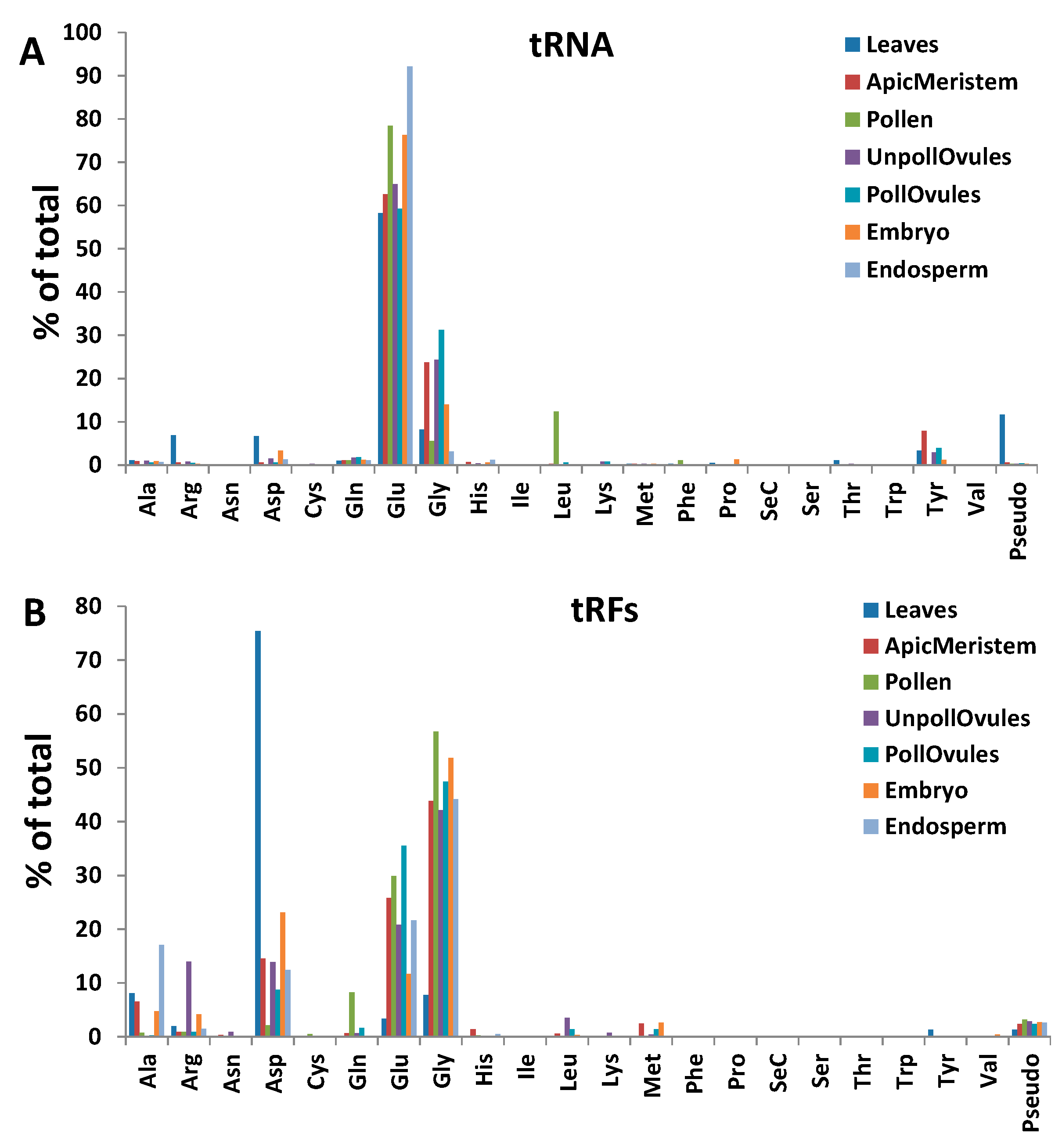
2.3. ncRFs Identification and Description
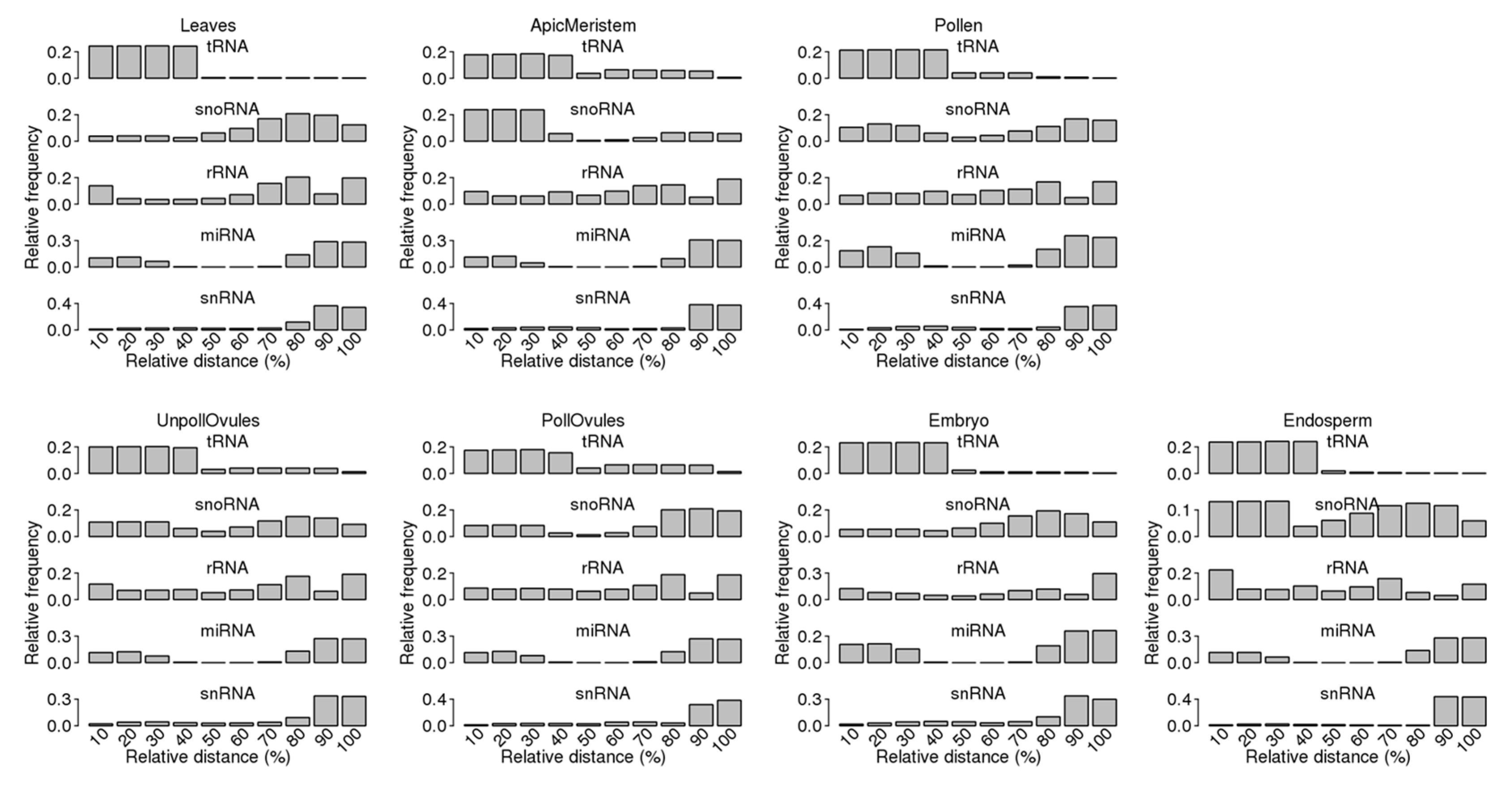
2.4. Analysis of Distribution of ncRNA and ncRF Reads across the Entire Length of Mature ncRNAs
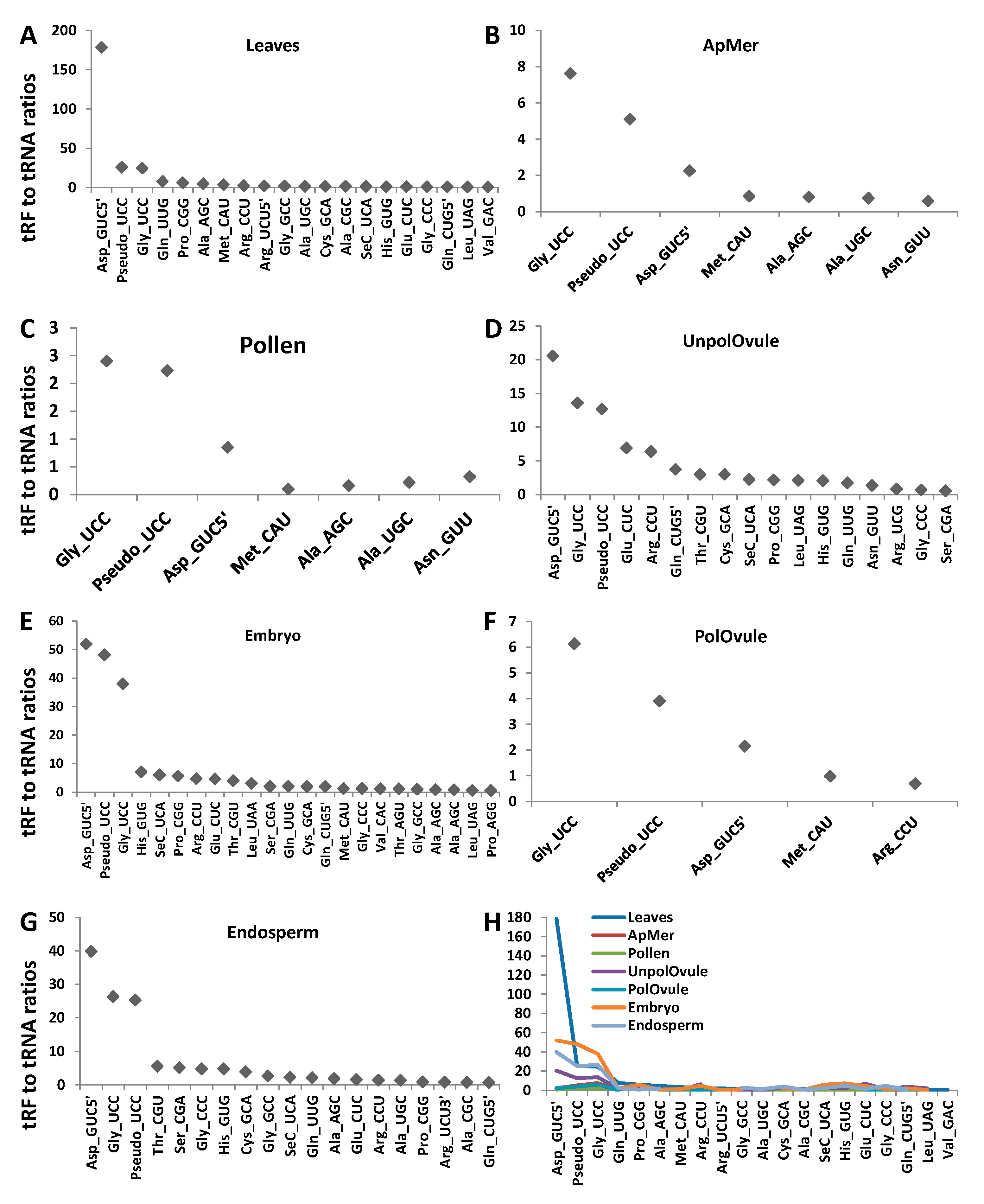
2.5. Analysis of Enrichment of ncRFs Relative to the Number of Mature ncRNAs
2.6. ncRFs Target Prediction
2.7. Classification of snoRNAs
2.8. Gene Ontology Term Analysis of Targets for Each Tissue
2.9. The Identification of Binding Consensus between ncRFs and Their Potential Target
3. Results and Discussion
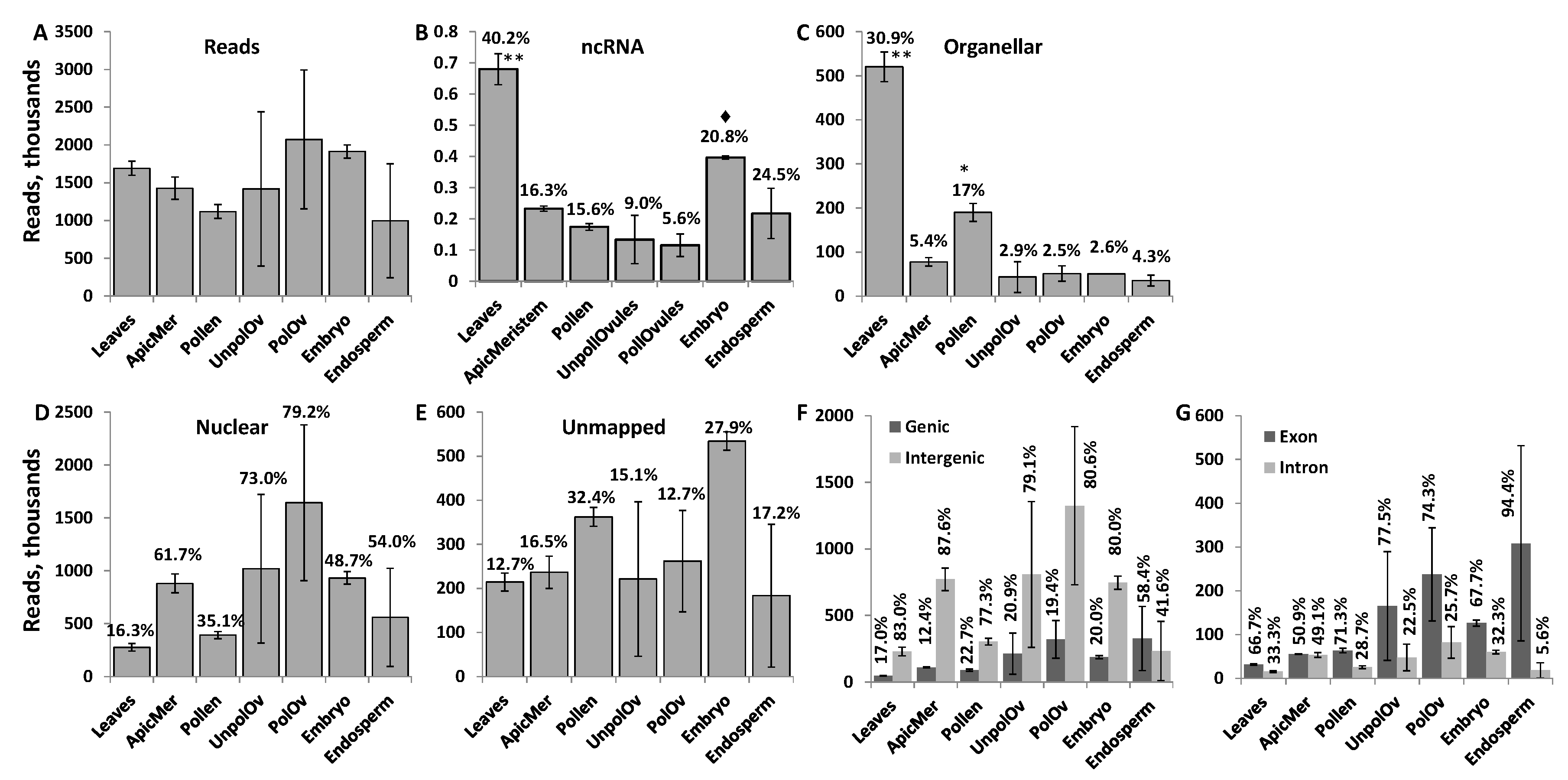
3.1. Characterization of Mapped Reads in Various Tissues
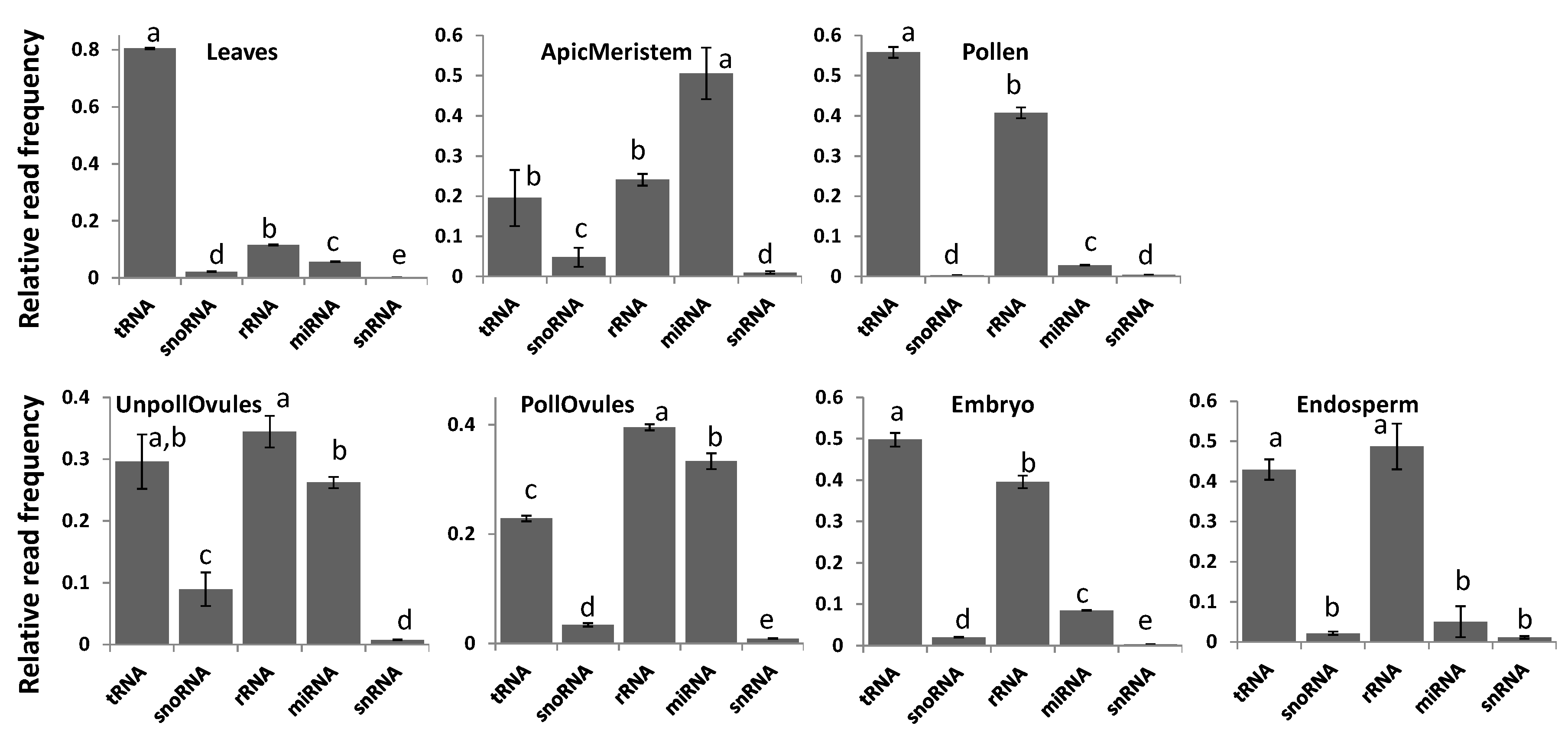
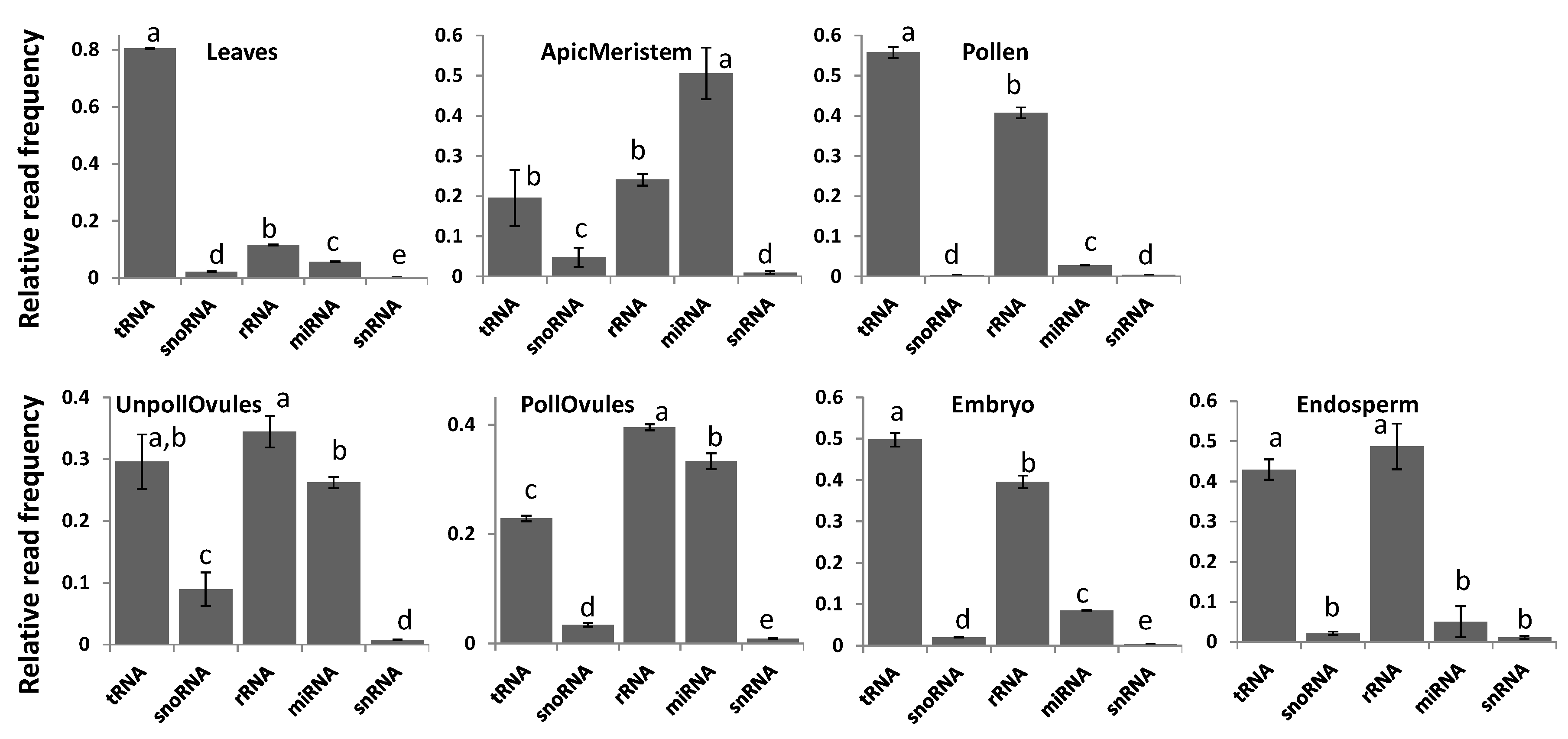
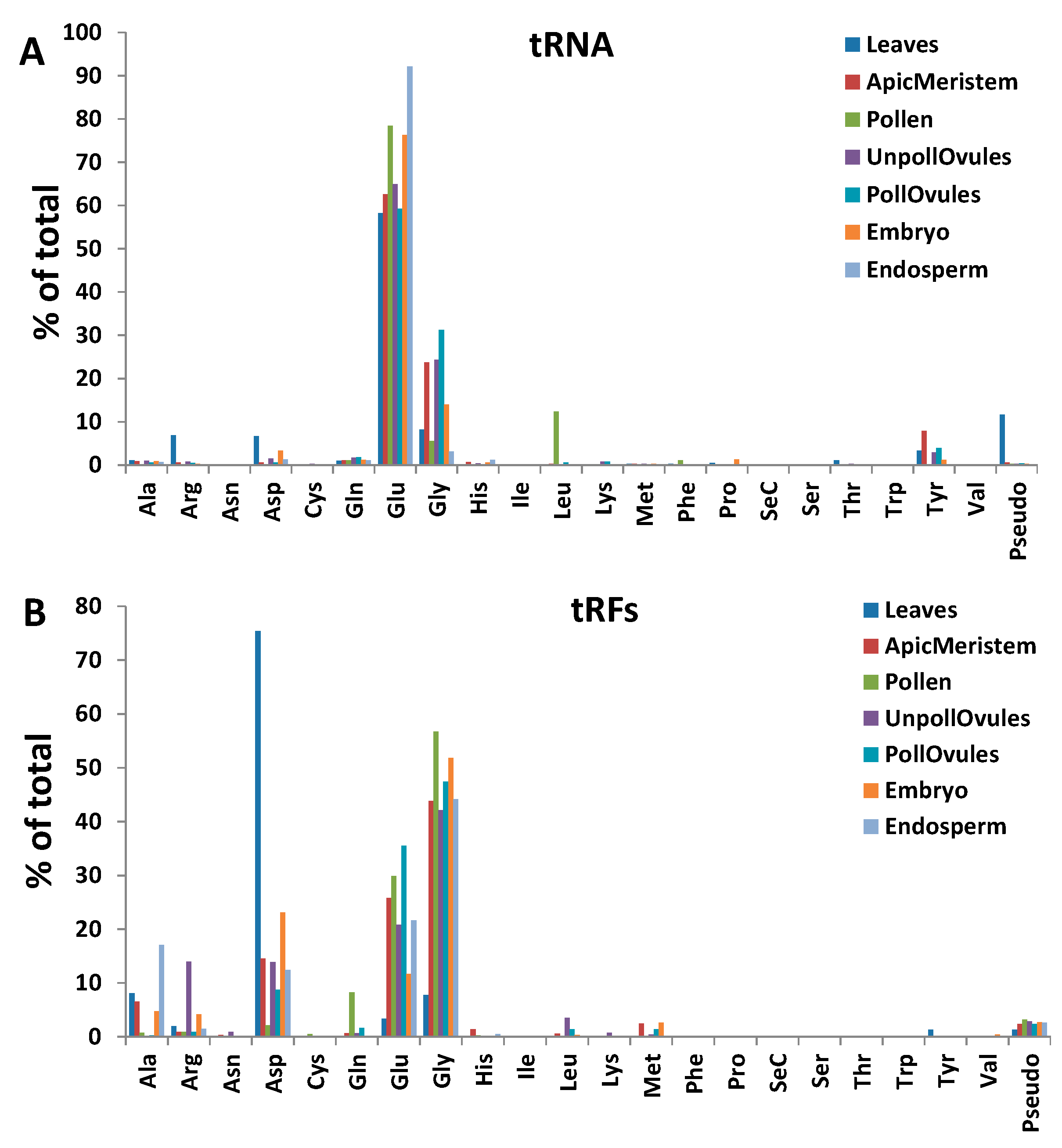
3.2. Characterisation of Types of ncRNA Reads in Various Tissues
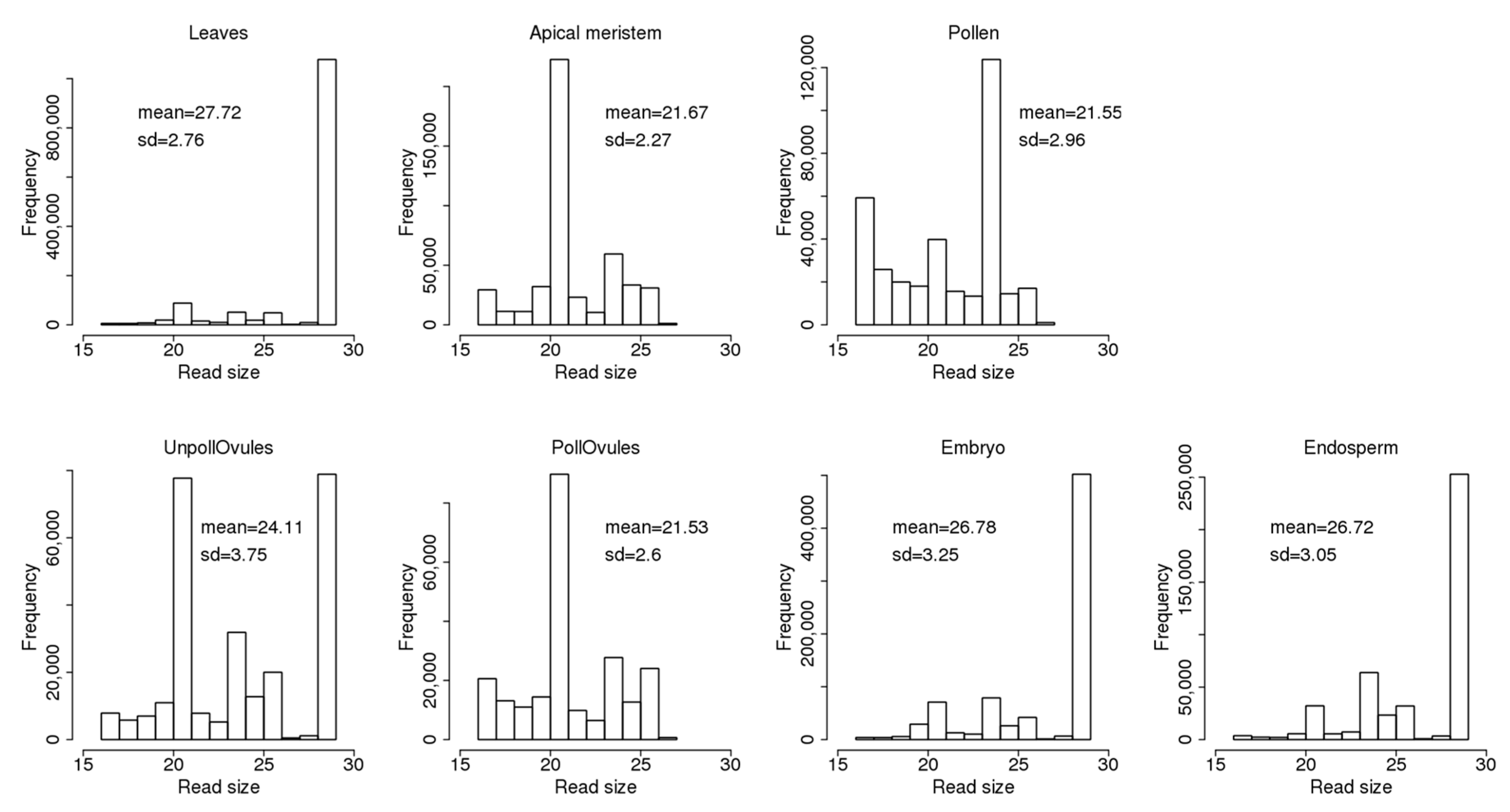
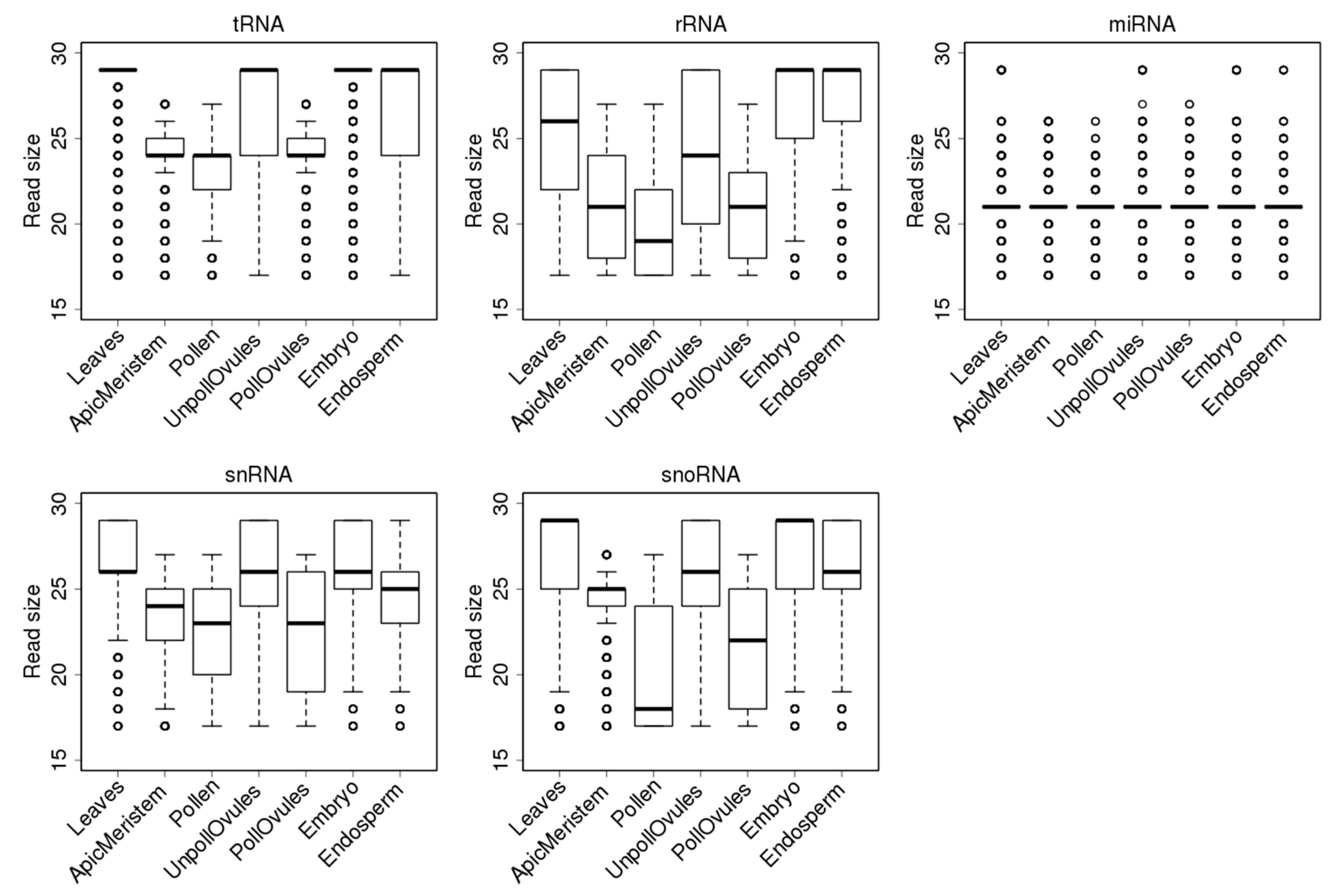
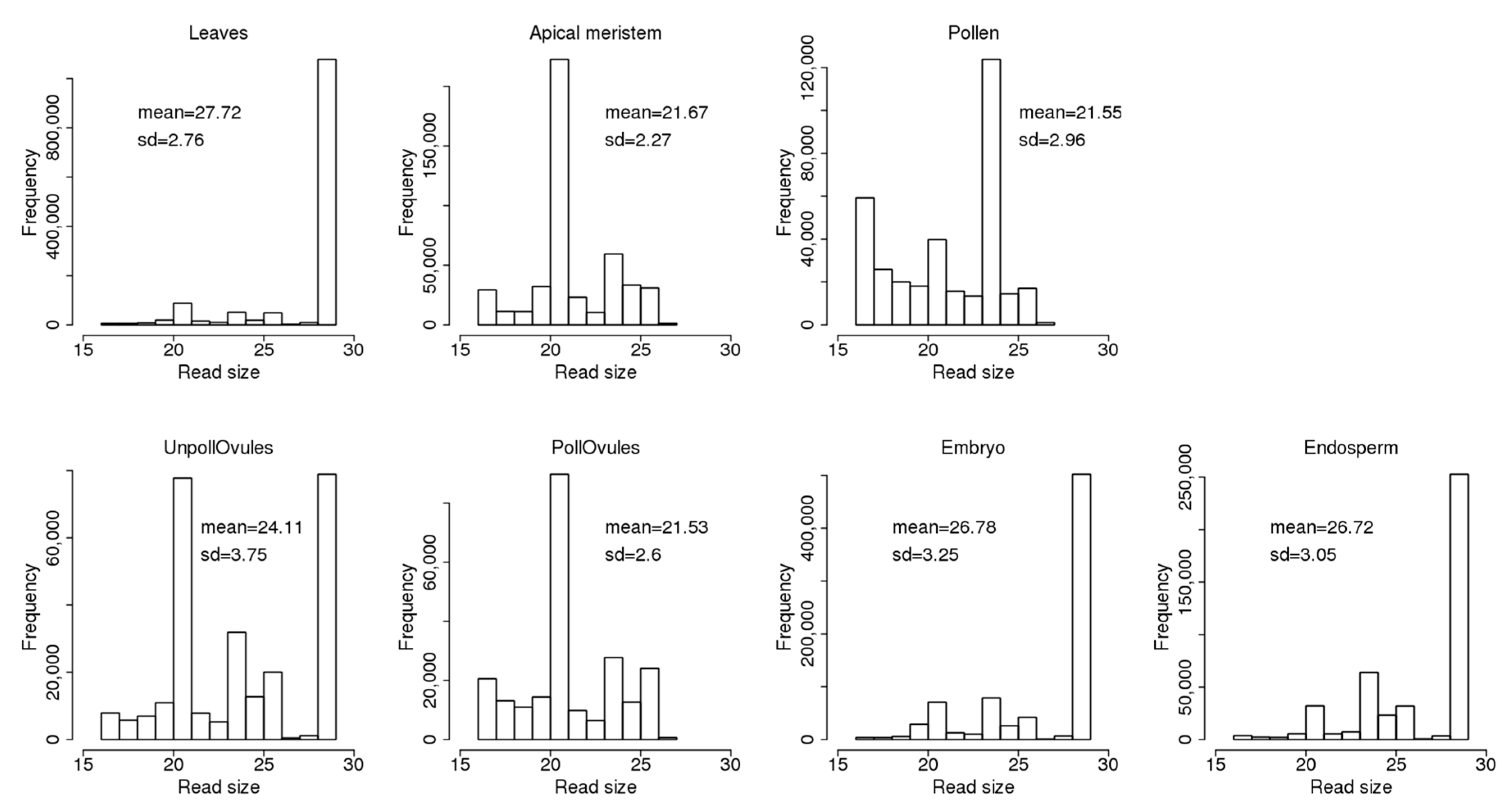
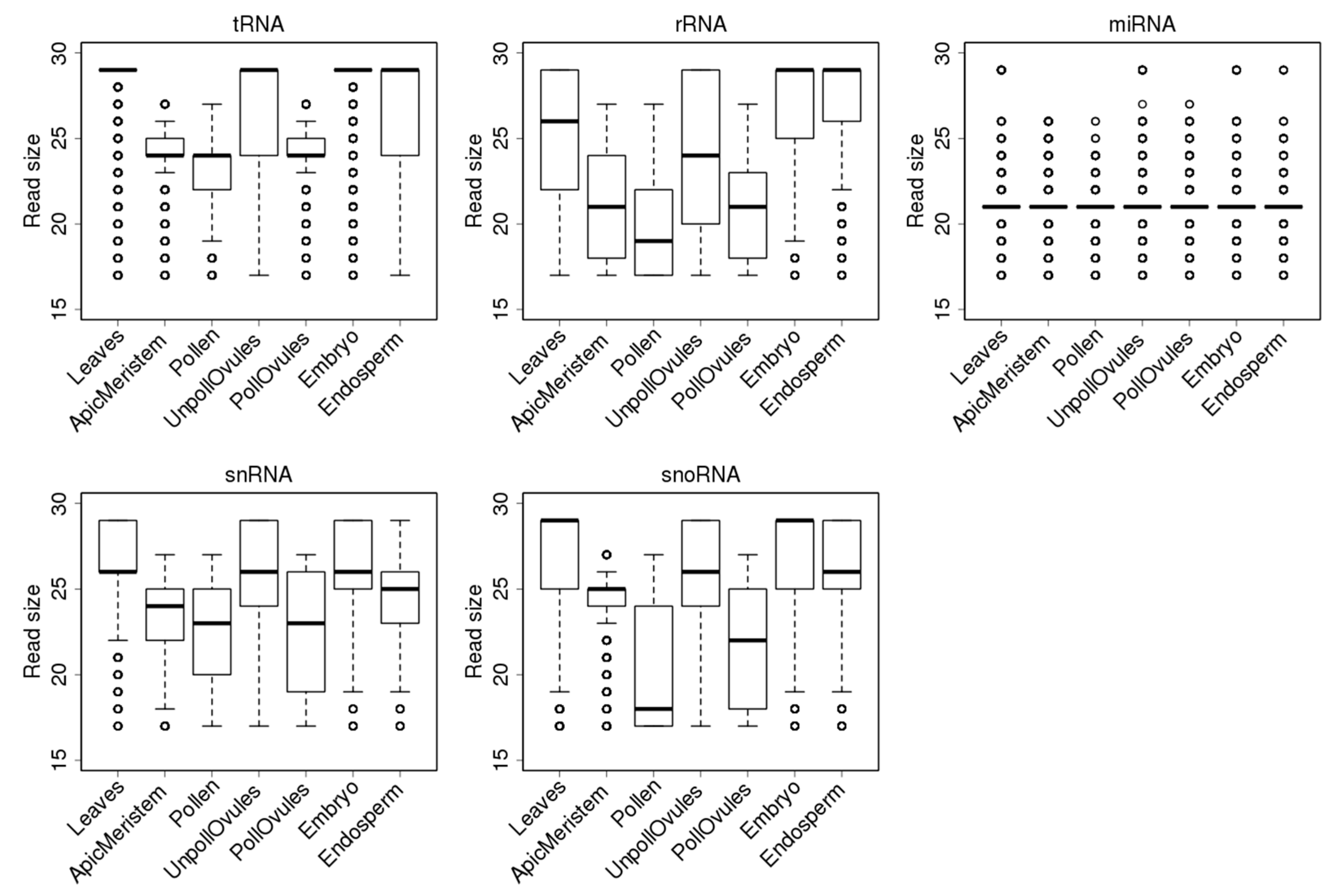
3.3. Characterisation of Size Distribution of ncRNA Fragments
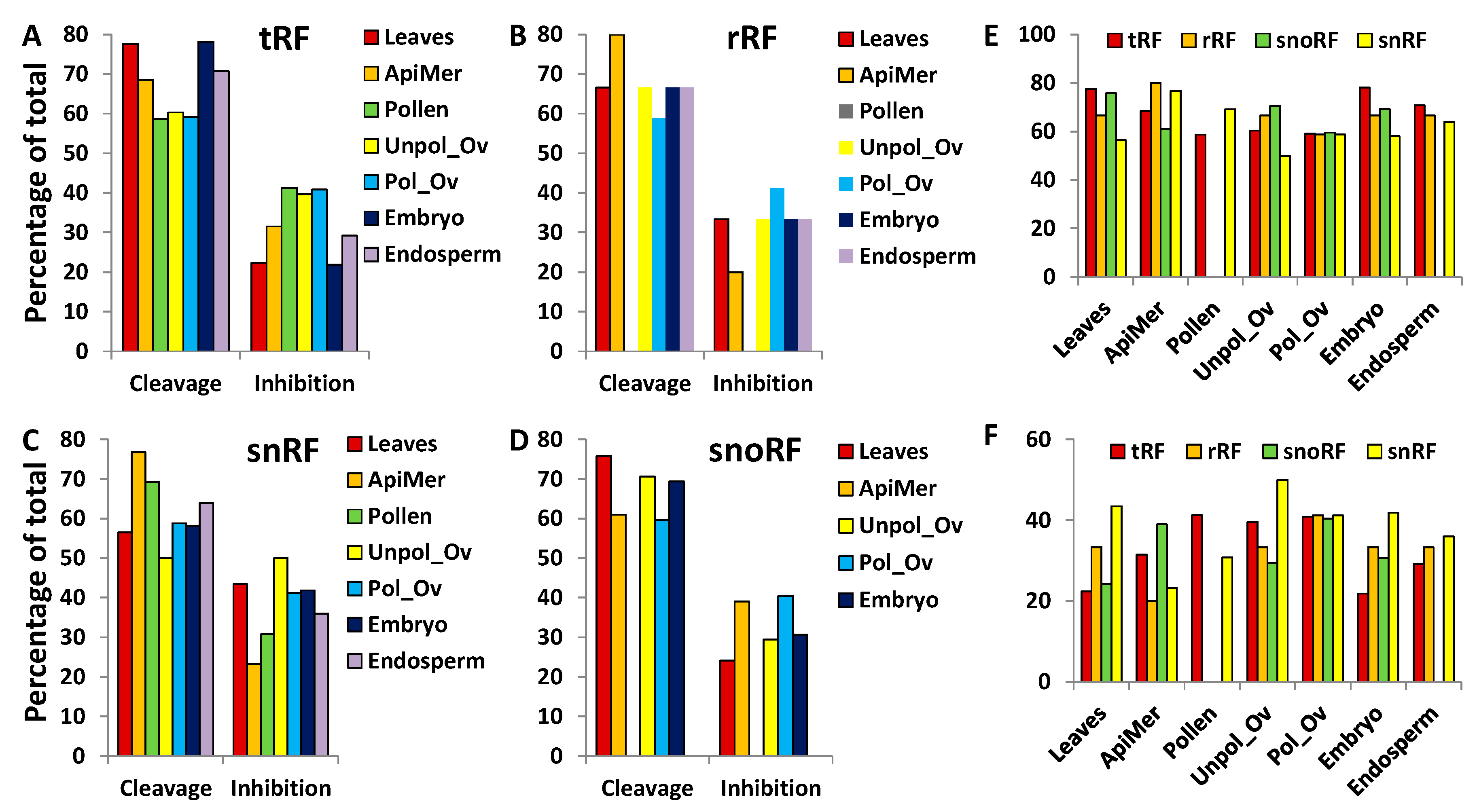
3.4. Characterisation of tRFs, rRFs, snRFs and snoRFs
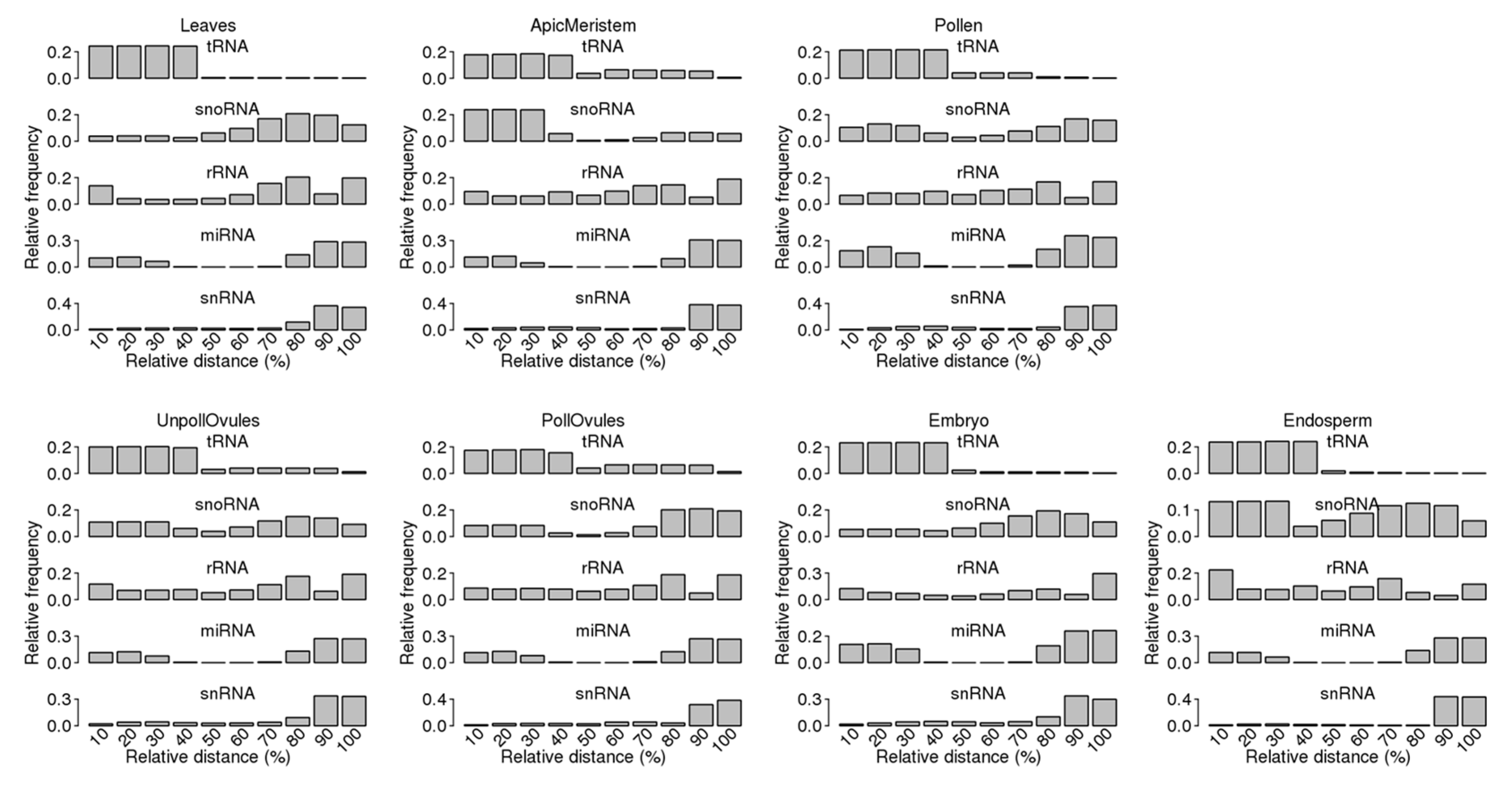
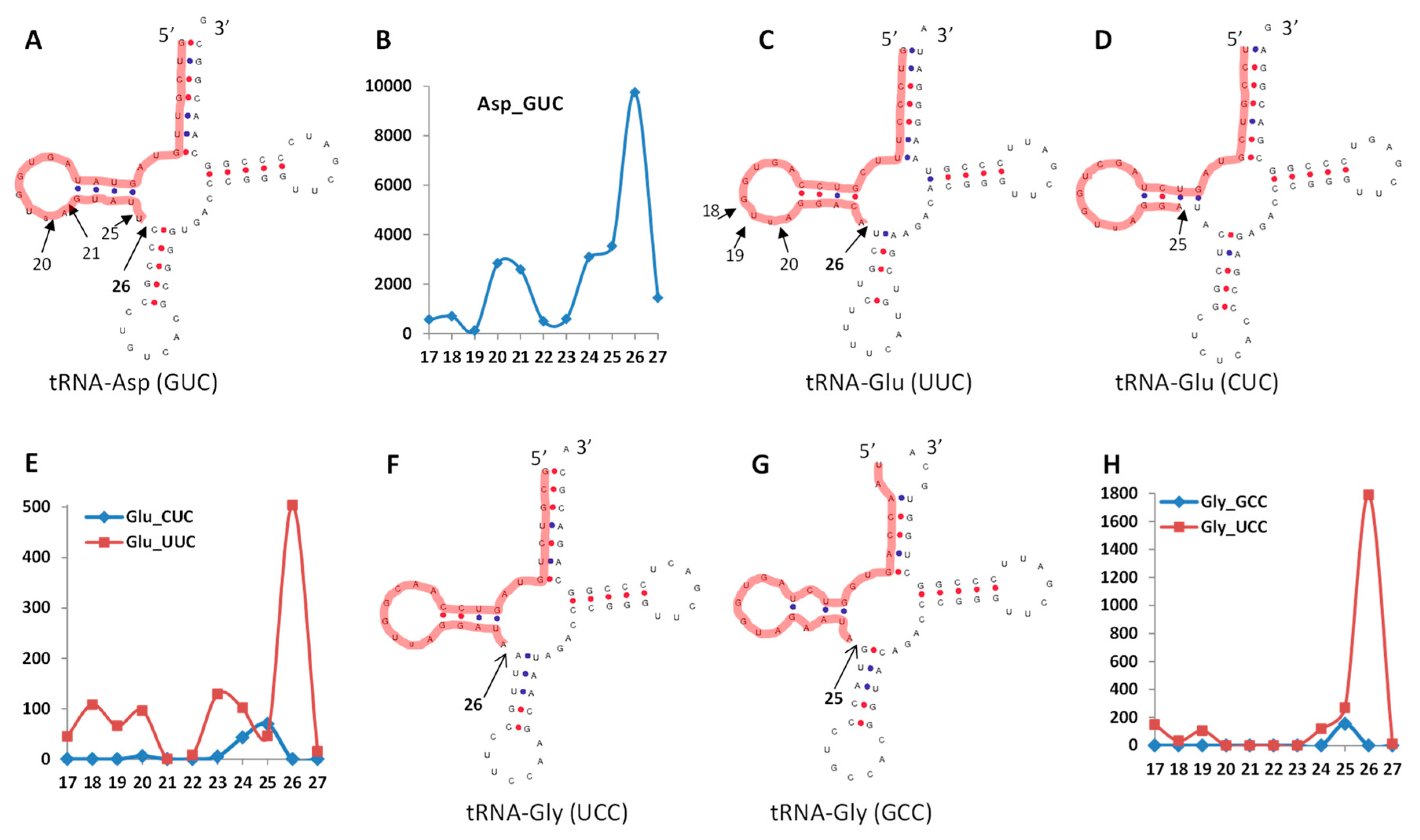
3.4.1. Termini-Specific Processing of ncRNAs into Fragments
3.5. A Prediction of Potential Cleavage Sites in tRFs
3.6. Annotation of Potential mRNA Targets of tRFs, rRFs, snRFs and snoRFs
3.7. SuperViewer Annotation of Biological Processes, Molecular Functions and Cellular Components
3.7.1. The Analysis of tRF Targets
3.7.2. Analysis of snRF and snoRF Targets
3.8. Characterization of tRF Binding Sites
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fu, X.D. Non-coding RNA: A new frontier in regulatory biology. Natl. Sci. Rev. 2014, 1, 190–204. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.X.; Rastetter, R.H.; Wilhelm, D. Non-coding RNAs: An Introduction. Adv. Exp. Med. Biol. 2016, 886, 13–32. [Google Scholar] [PubMed]
- Cole, C.; Sobala, A.; Lu, C.; Thatcher, S.R.; Bowman, A.; Brown, J.W.; Green, P.J.; Barton, G.J.; Hutvagner, G. Filtering of deep sequencing data reveals the existence of abundant Dicer-dependent small RNAs derived from tRNAs. RNA 2009, 15, 2147–2160. [Google Scholar] [CrossRef] [PubMed]
- Haussecker, D.; Huang, Y.; Lau, A.; Parameswaran, P.; Fire, A.Z.; Kay, M.A. Human tRNA-derived small RNAs in the global regulation of RNA silencing. RNA 2010, 16, 673–695. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Silva, M.R.; Frugier, M.; Tosar, J.P.; Correa-Dominguez, A.; Ronalte-Alves, L.; Parodi-Talice, A.; Rovira, C.; Robello, C.; Goldenberg, S.; Cayota, A. A population of tRNA-derived small RNAs is actively produced in Trypanosoma cruzi and recruited to specific cytoplasmic granules. Mol. Biochem. Parasitol. 2010, 171, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ender, C.; Meister, G.; Moore, P.S.; Chang, Y.; John, B. Extensive terminal and asymmetric processing of small RNAs from rRNAs, snoRNAs, snRNAs, and tRNAs. Nucleic Acids Res. 2012, 40, 6787–6799. [Google Scholar] [CrossRef] [PubMed]
- Burroughs, A.M.; Ando, Y.; de Hoon, M.J.; Tomaru, Y.; Suzuki, H.; Hayashizaki, Y.; Daub, C.O. Deep-sequencing of human Argonaute-associated small RNAs provides insight into miRNA sorting and reveals Argonaute association with RNA fragments of diverse origin. RNA Biol. 2011, 8, 158–177. [Google Scholar] [CrossRef] [PubMed]
- Kawaji, H.; Nakamura, M.; Takahashi, Y.; Sandelin, A.; Katayama, S.; Fukuda, S.; Daub, C.O.; Kai, C.; Kawai, J.; Yasuda, J.; et al. Hidden layers of human small RNAs. BMC Genom. 2008, 9, 157. [Google Scholar] [CrossRef] [PubMed]
- Cognat, V.; Morelle, G.; Megel, C.; Lalande, S.; Molinier, J.; Vincent, T.; Small, I.; Duchêne, A.M.; Maréchal-Drouard, L. The nuclear and organellar tRNA-derived RNA fragment population in Arabidopsis thaliana is highly dynamic. Nucleic Acids Res. 2016. [Google Scholar] [CrossRef]
- Hussain, S.; Sajini, A.A.; Blanco, S.; Dietmann, S.; Lombard, P.; Sugimoto, Y.; Paramor, M.; Gleeson, J.G.; Odom, D.T.; Ule, J.; et al. NSun2-mediated cytosine-5 methylation of vault noncoding RNA determines its processing into regulatory small RNAs. Cell Rep. 2013, 4, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, F.E.; Hall, A.E.; Csorba, T.; Turnbull, C.; Dalmay, T. Biogenesis of Y RNA-derived small RNAs is independent of the microRNA pathway. FEBS Lett. 2012, 586, 1226–1230. [Google Scholar] [CrossRef] [PubMed]
- Haiser, H.J.; Karginov, F.V.; Hannon, G.J.; Elliot, M.A. Developmentally regulated cleavage of tRNAs in the bacterium Streptomyces coelicolor. Nucleic Acids Res. 2008, 36, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Tuck, A.C.; Tollervey, D. RNA in pieces. Trends Genet. 2011, 27, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Taft, R.J.; Glazov, E.A.; Lassmann, T.; Hayashizaki, Y.; Carninci, P.; Mattick, J.S. Small RNAs derived from snoRNAs. RNA 2009, 15, 1233–1240. [Google Scholar] [CrossRef] [PubMed]
- Sobala, A.; Hutvagner, G. Transfer RNA-derived fragments: Origins, processing, and functions. Wiley Interdiscip. Rev. RNA 2011, 2, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Rogato, A.; Richard, H.; Sarazin, A.; Voss, B.; Cheminant Navarro, S.; Champeimont, R.; Navarro, L.; Carbone, A.; Hess, W.R.; Falciatore, A. The diversity of small non-coding RNAs in the diatom Phaeodactylum tricornutum. BMC Genom. 2014, 15, 698. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, L.C.; Lin, S.I.; Shih, A.C.; Chen, J.W.; Lin, W.Y.; Tseng, C.Y.; Li, W.H.; Chiou, T.J. Uncovering small RNA-mediated responses to phosphate deficiency in Arabidopsis by deep sequencing. Plant Physiol. 2009, 151, 2120–2132. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; liu, Q.; Zhang, Y.C.; Qu, L.H.; Chen, Y.Q.; Gautheret, D. Genome-wide discovery and analysis of microRNAs and other small RNAs from rice embryogenic callus. RNA Biol. 2011, 8, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Loss-Morais, G.; Waterhouse, P.M.; Margis, R. Description of plant tRNA-derived RNA fragments (tRFs) associated with argonaute and identification of their putative targets. Biol Direct. 2013, 8, 6. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, H.; Sun, Q.; Yao, Y. Characterization of Small RNAs Derived from tRNAs, rRNAs and snoRNAs and Their Response to Heat Stress in Wheat Seedlings. PLoS ONE 2016, 11, e0150933. [Google Scholar] [CrossRef] [PubMed]
- Green, D.; Fraser, W.D.; Dalmay, T. Transfer RNA-derived small RNAs in the cancer transcriptome. Pflugers Arch. 2016, 468, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.Y.; Ma, L.M.; Guo, Y.H.; Zhang, Y.C.; Zhou, H.; Shao, P.; Chen, Y.Q.; Qu, L.H. Deep sequencing of human nuclear and cytoplasmic small RNAs reveals an unexpectedly complex subcellular distribution of miRNAs and tRNA 3′ trailers. PLoS ONE 2010, 5, e10563. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.S.; Ono, M.; Yamada, K.; Endo, A.; Barton, G.J.; Lamond, A.I. Human box C/D snoRNA processing conservation across multiple cell types. Nucleic Acids Res. 2012, 40, 3676–3688. [Google Scholar] [CrossRef] [PubMed]
- Sharp, S.J.; Schaack, J.; Cooley, L.; Burke, D.J.; Soll, D. Structure and transcription of eukaryotic tRNA genes. CRC Crit. Rev. Biochem. 1985, 19, 107–144. [Google Scholar] [CrossRef] [PubMed]
- Torres, A.G.; Batlle, E.; Ribas de Pouplana, L. Role of tRNA modifications in human diseases. Trends Mol. Med. 2014, 20, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Phizicky, E.M.; Hopper, A.K. tRNA biology charges to the front. Genes Dev. 2010, 24, 1832–1860. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, P.; Emara, M.M.; Villen, J.; Gygi, S.P.; Anderson, P. Angiogenin-induced tRNA fragments inhibit translation initiation. Mol. Cell 2011, 43, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Lee, I.; Lee, Y.S.; Bao, X. Small Non-coding Transfer RNA-Derived RNA Fragments (tRFs): Their Biogenesis, Function and Implication in Human Diseases. Genom. Inform. 2015, 13, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Evdokimova, V.; Ruzanov, P.; Imataka, H.; Raught, B.; Svitkin, Y.; Ovchinnikov, L.P.; Sonenberg, N. The major mRNA-associated protein YB-1 is a potent 5' cap-dependent mRNA stabilizer. EMBO J. 2001, 20, 5491–5502. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.; Ivanov, P. tRNA fragments in human health and disease. FEBS Lett. 2014, 588, 4297–4304. [Google Scholar] [CrossRef] [PubMed]
- Matera, A.G.; Terns, R.M.; Terns, M.P. Non-coding RNAs: Lessons from the small nuclear and small nucleolar RNAs. Nat. Rev. Mol. Cell Biol. 2007, 8, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Thomson, D.W.; Pillman, K.A.; Anderson, M.L.; Lawrence, D.M.; Toubia, J.; Goodall, G.J.; Bracken, C.P. Assessing the gene regulatory properties of Argonaute-bound small RNAs of diverse genomic origin. Nucleic Acids Res. 2015, 43, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.D.; Gentner, B.; Cantore, A.; Colleoni, S.; Amendola, M.; Zingale, A.; Baccarini, A.; Lazzari, G.; Galli, C.; Naldini, L. Endogenous microRNA can be broadly exploited to regulate transgene expression according to tissue, lineage and differentiation state. Nat. Biotechnol. 2007, 25, 1457–1467. [Google Scholar] [CrossRef] [PubMed]
- Hackenberg, M.; Huang, P.J.; Huang, C.Y.; Shi, B.J.; Gustafson, P.; Langridge, P. A comprehensive expression profile of microRNAs and other classes of non-coding small RNAs in barley under phosphorous-deficient and -sufficient conditions. DNA Res. 2013, 20, 109–125. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Sun, L.; Kragler, F. The phloem-delivered RNA pool contains small noncoding RNAs and interferes with translation. Plant Physiol. 2009, 150, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Nowacka, M.; Jackowiak, P.; Rybarczyk, A.; Magacz, T.; Strozycki, P.M.; Barciszewski, J.; Figlerowicz, M. 2D-PAGE as an effective method of RNA degradome analysis. Mol. Biol. Rep. 2012, 39, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Nowacka, M.; Strozycki, P.M.; Jackowiak, P.; Hojka-Osinska, A.; Szymanski, M.; Figlerowicz, M. Identification of stable, high copy number, medium-sized RNA degradation intermediates that accumulate in plants under non-stress conditions. Plant Mol. Biol. 2013, 83, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Bilichak, A.; Ilnytskyy, Y.; Woycicki, R.; Kepeshchuk, N.; Fogen, D.; Kovalchuk, I. The elucidation of stress memory inheritance in Brassica rapa plants. Front. Plant Sci. 2015, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17. [Google Scholar] [CrossRef]
- Lee, Y.S.; Shibata, Y.; Malhotra, A.; Dutta, A. A novel class of small RNAs: TRNA-derived RNA fragments (tRFs). Genes Dev. 2009, 23, 2639–2649. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server. Nucleic Acids Res. 2011, 39, W155–W159. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. miRU: An automated plant miRNA target prediction server. Nucleic Acids Res. 2005, 33, W701–W704. [Google Scholar] [CrossRef] [PubMed]
- Brodersen, P.; Sakvarelidze-Achard, L.; Bruun-Rasmussen, M.; Dunoyer, P.; Yamamoto, Y.Y.; Sieburth, L.; Voinnet, O. Widespread translational inhibition by plant miRNAs and siRNAs. Science 2008, 320, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.H.; Shao, P.; Zhou, H.; Chen, Y.Q.; Qu, L.H. DeepBase: A database for deeply annotating and mining deep sequencing data. Nucleic Acids Res. 2010, 38, D123–D120. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Wang, T.; Wang, K.; Wang, X.; Fu, Y.; Huang, L.; Kang, Z. Transcriptome Analysis Provides Insights into the Mechanisms Underlying Wheat Plant Resistance to Stripe Rust at the Adult Plant Stage. PLoS ONE 2016, 11, e0150717. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, H.; Wang, J.; Sun, R.; Wu, J.; Liu, S.; Bai, Y.; Mun, J.H.; Bancroft, I.; Cheng, F.; et al. The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 2011, 43, 1035–1039. [Google Scholar] [CrossRef] [PubMed]
- Tong, C.; Wang, X.; Yu, J.; Wu, J.; Li, W.; Huang, J.; Dong, C.; Hua, W.; Liu, S. Comprehensive analysis of RNA-seq data reveals the complexity of the transcriptome in Brassica rapa. BMC Genom. 2013, 14, 689. [Google Scholar] [CrossRef] [PubMed]
- Devisetty, U.K.; Covington, M.F.; Tat, A.V.; Lekkala, S.; Maloof, J.N. Polymorphism identification and improved genome annotation of Brassica rapa through Deep RNA sequencing. Genes Genomes Genet. 2014, 4, 2065–2078. [Google Scholar] [CrossRef] [PubMed]
- Ruwe, H.; Schmitz-Linneweber, C. Short non-coding RNA fragments accumulating in chloroplasts: Footprints of RNA binding proteins? Nucleic Acids Res. 2012, 40, 3106–3116. [Google Scholar] [CrossRef] [PubMed]
- Haruta, M.; Gray, W.M.; Sussman, M.R. Regulation of the plasma membrane proton pump (H(+)-ATPase) by phosphorylation. Curr. Opin. Plant Biol. 2015, 28, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Nourbakhsh, A.; Collakova, E.; Gillaspy, G.E. Characterization of the inositol monophosphatase gene family in Arabidopsis. Front. Plant Sci. 2014, 5, 725. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Lin, T.; Grandellis, C.; Yu, M.; Hannapel, D.J. The BEL1-like family of transcription factors in potato. J. Exp. Bot. 2014, 65, 709–723. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.; Giraud, E.; Duncan, O.; Law, S.R.; Wang, Y.; Xu, L.; Narsai, R.; Carrie, C.; Walker, H.; Day, D.A.; et al. Cyclin-dependent kinase E1 (CDKE1) provides a cellular switch in plants between growth and stress responses. J. Biol. Chem. 2013, 288, 3449–3459. [Google Scholar] [CrossRef] [PubMed]
- Tam, T.H.; Catarino, B.; Dolan, L. Conserved regulatory mechanism controls the development of cells with rooting functions in land plants. Proc. Natl. Acad. Sci. USA 2015, 112, E3959–E3968. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.S.; Xie, Q.; Fei, J.F.; Chua, N.H. MicroRNA directs mRNA cleavage of the transcription factor NAC1 to downregulate auxin signals for arabidopsis lateral root development. Plant Cell 2005, 17, 1376–1386. [Google Scholar] [CrossRef] [PubMed]
- Lanet, E.; Delannoy, E.; Sormani, R.; Floris, M.; Brodersen, P.; Crete, P.; Voinnet, O.; Robaglia, C. Biochemical evidence for translational repression by Arabidopsis microRNAs. Plant Cell 2009, 21, 1762–1768. [Google Scholar] [CrossRef] [PubMed]
- Ameres, S.L.; Zamore, P.D. Diversifying microRNA sequence and function. Nat. Rev. Mol. Cell Biol. 2013, 14, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Iwakawa, H.O.; Tomari, Y. Molecular insights into microRNA-mediated translational repression in plants. Mol. Cell 2013, 52, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Verdel, A.; Jia, S.; Gerber, S.; Sugiyama, T.; Gygi, S.; Grewal, S.I.; Moazed, D. RNAi-mediated targeting of heterochromatin by the RITS complex. Science 2004, 303, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Allo, M.; Buggiano, V.; Fededa, J.P.; Petrillo, E.; Schor, I.; de la Mata, M.; Agirre, E.; Plass, M.; Eyras, E.; Elela, S.A.; et al. Control of alternative splicing through siRNA-mediated transcriptional gene silencing. Nat. Struct. Mol. Biol. 2009, 16, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Iwasaki, S.; Watanabe, T.; Utsumi, M.; Watanabe, Y. The mechanism selecting the guide strand from small RNA duplexes is different among argonaute proteins. Plant Cell Physiol. 2008, 49, 493–500. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Byeon, B.; Bilichak, A.; Kovalchuk, I. Computational Characterization of ncRNA Fragments in Various Tissues of the Brassica rapa Plant. Non-Coding RNA 2017, 3, 17. https://doi.org/10.3390/ncrna3020017
Byeon B, Bilichak A, Kovalchuk I. Computational Characterization of ncRNA Fragments in Various Tissues of the Brassica rapa Plant. Non-Coding RNA. 2017; 3(2):17. https://doi.org/10.3390/ncrna3020017
Chicago/Turabian StyleByeon, Boseon, Andriy Bilichak, and Igor Kovalchuk. 2017. "Computational Characterization of ncRNA Fragments in Various Tissues of the Brassica rapa Plant" Non-Coding RNA 3, no. 2: 17. https://doi.org/10.3390/ncrna3020017
APA StyleByeon, B., Bilichak, A., & Kovalchuk, I. (2017). Computational Characterization of ncRNA Fragments in Various Tissues of the Brassica rapa Plant. Non-Coding RNA, 3(2), 17. https://doi.org/10.3390/ncrna3020017