
A Phylogeographic Description of Histoplasma capsulatum in the United States

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Culture and DNA Extraction
2.2. Genomic Sequencing
2.3. Single-Nucleotide Polymorphism (SNP) and Phylogenetic Analysis
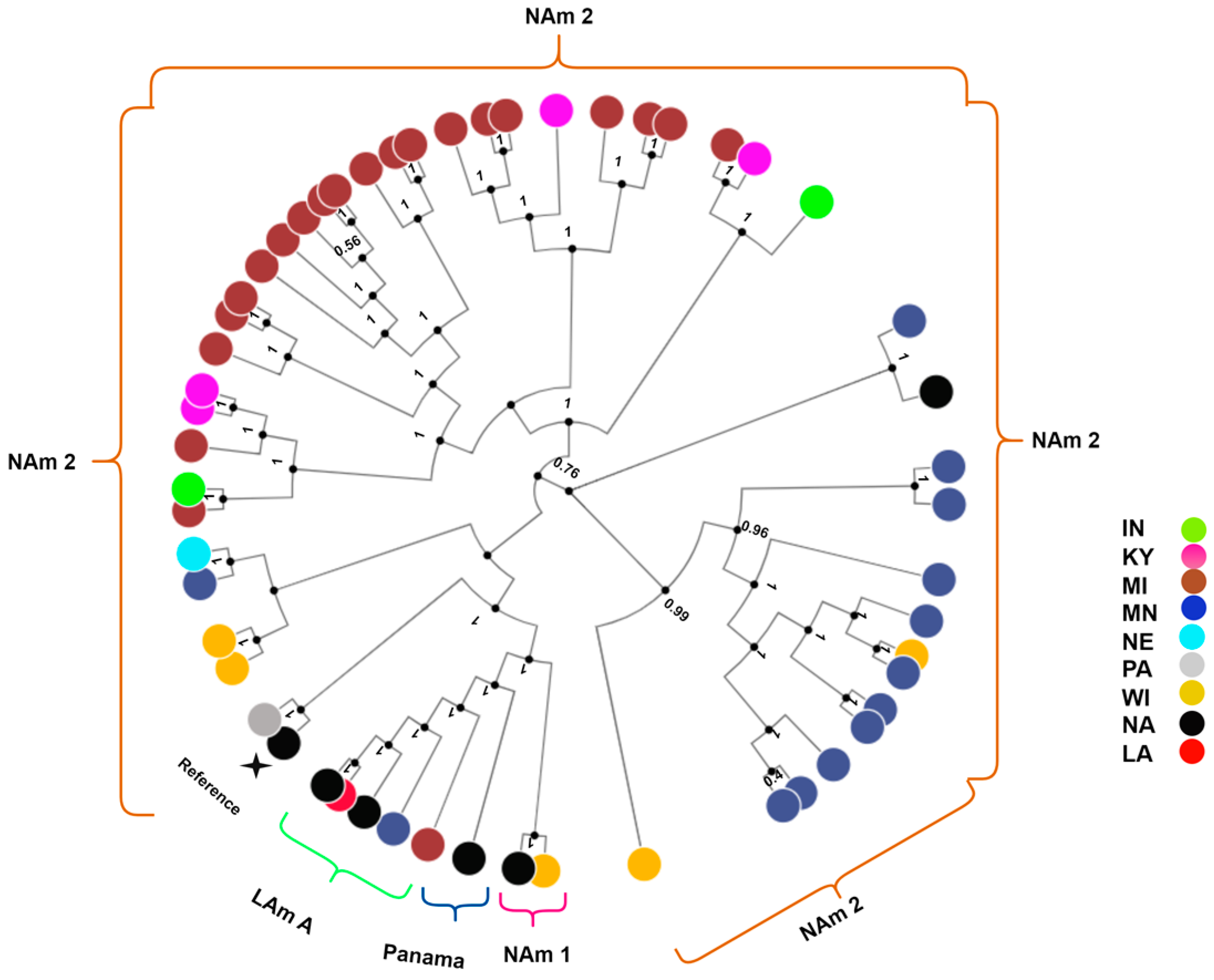
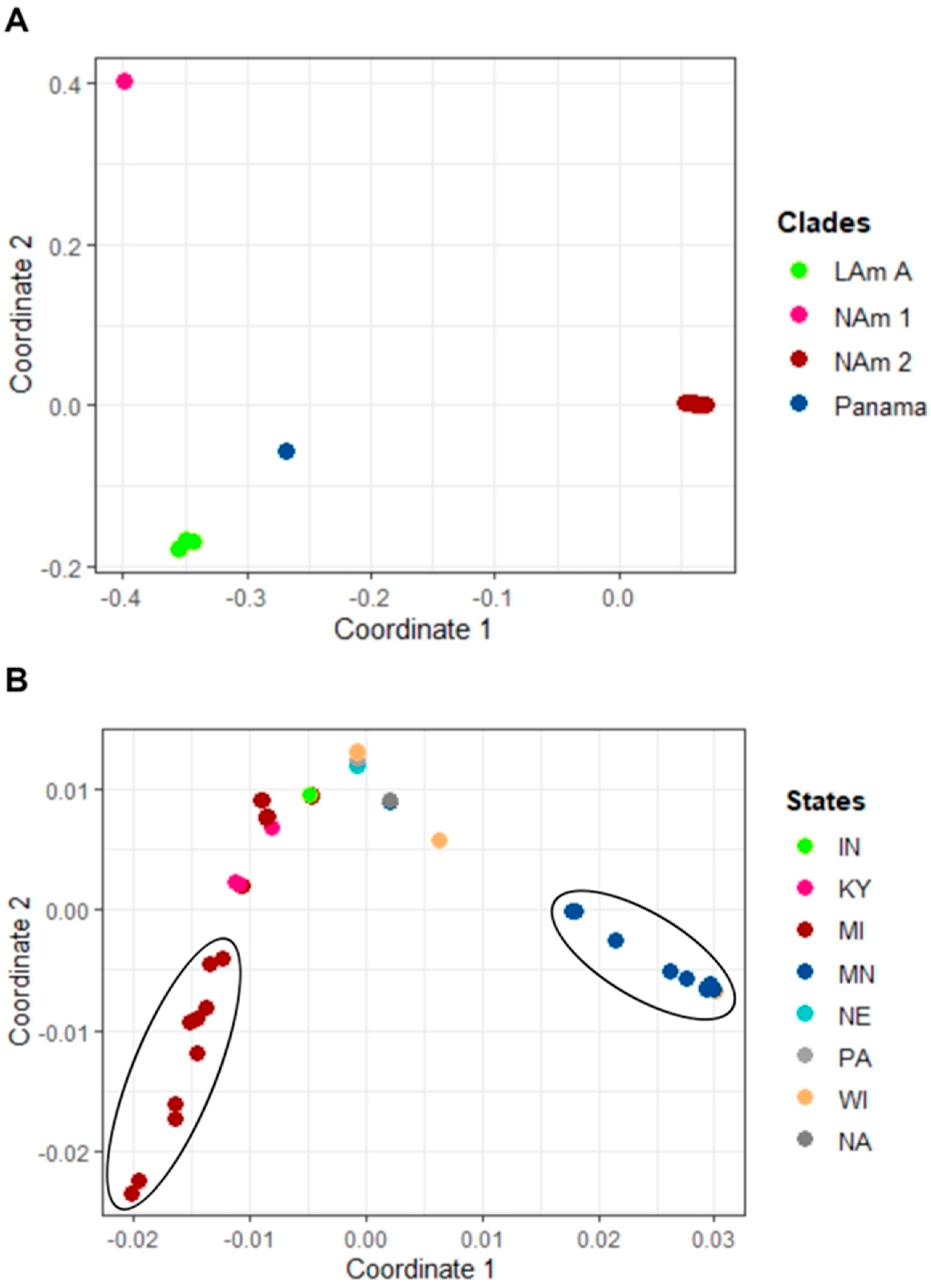
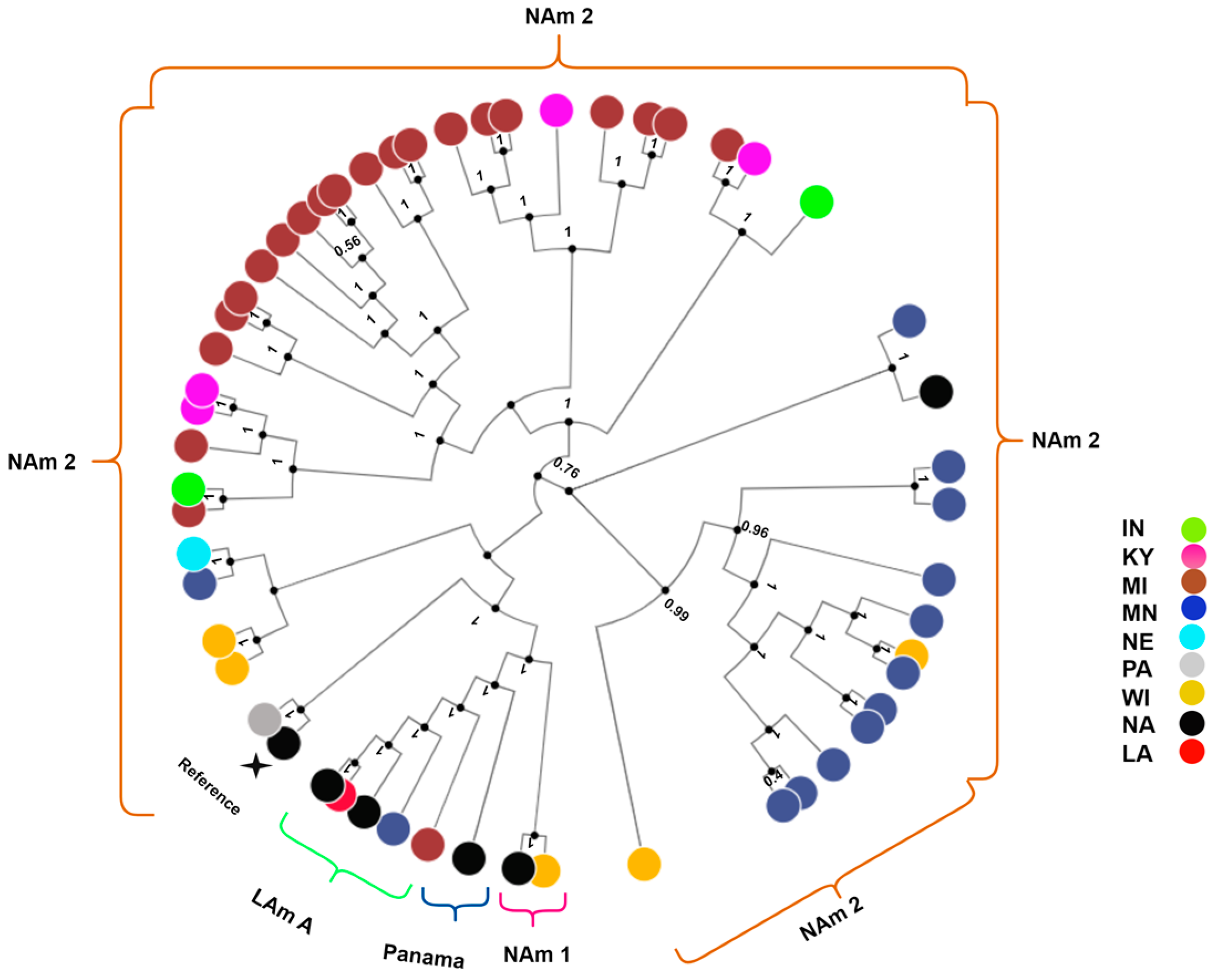
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brömel, C.; Sykes, J.E. Histoplasmosis in Dogs and Cats. Clin. Tech. Small Anim. Pract. 2005, 20, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.L.; Chávez-Tapia, C.B.; Reyes-Montes, M.R. Molecular typing of Histoplasma capsulatum isolated from infected bats, captured in Mexico. Fungal Genet. Biol. 2000, 30, 207–212. [Google Scholar] [CrossRef]
- Benedict, K.; Mody, R.K. Epidemiology of Histoplasmosis Outbreaks, United States, 1938–2013. Emerg. Infect. Dis. 2016, 22, 370–378. [Google Scholar] [CrossRef]
- Cano, M.; Hajjeh, R.A. The epidemiology of histoplasmosis: A review. Semin. Respir. Infect. 2001, 16, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Deepe, G.S., Jr. Outbreaks of histoplasmosis: The spores set sail. PLoS Pathog. 2018, 14, e1007213. [Google Scholar] [CrossRef] [PubMed]
- Gómez, L.F.; Arango, M.; McEwen, J.G.; Gómez, O.M.; Zuluaga, A.; Peláez, C.A.; Acevedo, J.M.; Taylor, M.L.; Jiménez, M.d.P. Molecular epidemiology of Colombian Histoplasma capsulatum isolates obtained from human and chicken manure samples. Heliyon 2019, 5, e02084. [Google Scholar] [CrossRef]
- Rachid, A.; Rezende, L.S.; de Moura, S.F.; Loffy, P.C.; Magalhães, F.L.G.M. A case study of disseminated histoplasmosis linked to common variable immunodeficiency. Braz. J. Infect. Dis. 2003, 7, 268–272. [Google Scholar] [CrossRef]
- Assi, M.A.; Sandid, M.S.; Baddour, L.M.; Roberts, G.D.; Walker, R.C. Systemic Histoplasmosis: A 15-Year Retrospective Institutional Review of 111 Patients. Medicine 2007, 86, 162–169. [Google Scholar] [CrossRef]
- Manos, N.E.; Ferebee, S.H.; Kerschbaum, W.F. Geographic variation in the prevalence of histoplasmin sensitivity. Dis. Chest 1956, 29, 649–668. [Google Scholar] [CrossRef]
- Benedict, K.; McCracken, S.; Signs, K.; Ireland, M.; Amburgey, V.; Serrano, J.A.; Christophe, N.; Gibbons-Burgener, S.; Hallyburton, S.; Warren, K.A.; et al. Enhanced Surveillance for Histoplasmosis—9 States, 2018–2019. Open Forum Infect. Dis. 2020, 7, ofaa343. [Google Scholar] [CrossRef]
- Smith, D.J.; Williams, S.L.; Endemic Mycoses State Partners Group; Benedict, K.M.; Jackson, B.R.; Toda, M. Surveillance for Coccidioidomycosis, Histoplasmosis, and Blastomycosis—United States, 2019. MMWR Morb. Mortal. Wkly. Rep. 2022, 71, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Mazi, P.B.; Sahrmann, J.M.; Olsen, M.A.; Coler-Reilly, A.; Rauseo, A.M.; Pullen, M.; Zuniga-Moya, J.C.; Powderly, W.G.; Spec, A. The Geographic Distribution of Dimorphic Mycoses in the United States for the Modern Era. Clin. Infect. Dis. 2023, 76, 1295–1301. [Google Scholar] [CrossRef]
- Kwon-Chung, K.J.; Bennett, J.E. Medical Mycology. Rev. Inst. Med. Trop. São Paulo 1992, 34, 504. [Google Scholar] [CrossRef]
- Kasuga, T.; White, T.J.; Koenig, G.; McEwen, J.; Restrepo, A.; Castañeda, E.; Da Silva Lacaz, C.; Heins-Vaccari, E.M.; De Freitas, R.S.; Zancopé-Oliveira, R.M.; et al. Phylogeography of the fungal pathogen Histoplasma capsulatum. Mol. Ecol. 2003, 12, 3383–3401. [Google Scholar] [CrossRef]
- Taylor, M.L.; Hernández-García, L.; Estrada-Bárcenas, D.; Salas-Lizana, R.; Zancopé-Oliveira, R.M.; García de la Cruz, S.; Galvão-Dias, M.A.; Curiel-Quesada, E.; Canteros, C.E.; Bojórquez-Torres, G.; et al. Genetic diversity of Histoplasma capsulatum isolated from infected bats randomly captured in Mexico, Brazil, and Argentina, using the polymorphism of (GA)n microsatellite and its flanking regions. Fungal Biol. 2012, 116, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, M.d.M.; Patané, J.S.L.; Taylor, M.L.; Gómez, B.L.; Theodoro, R.C.; de Hoog, S.; Engelthaler, D.M.; Zancopé-Oliveira, R.M.; Felipe, M.S.S.; Barker, B.M. Worldwide Phylogenetic Distributions and Population Dynamics of the Genus Histoplasma. PLoS Neglected Trop. Dis. 2016, 10, e0004732. [Google Scholar] [CrossRef] [PubMed]
- Vite-Garín, T.; Estrada-Bárcenas, D.A.; Gernandt, D.S.; Reyes-Montes, M.d.R.; Sahaza, J.H.; Canteros, C.E.; Ramírez, J.A.; Rodríguez-Arellanes, G.; Serra-Damasceno, L.; Zancopé-Oliveira, R.M.; et al. Histoplasma capsulatum Isolated from Tadarida brasiliensis Bats Captured in Mexico Form a Sister Group to North American Class 2 Clade. J. Fungi 2021, 7, 529. [Google Scholar] [CrossRef] [PubMed]
- Sepúlveda, V.E.; Márquez, R.; Turissini, D.A.; Goldman, W.E.; Matute, D.R. Genome Sequences Reveal Cryptic Speciation in the Human Pathogen Histoplasma capsulatum. mBio 2017, 8, e01339-17. [Google Scholar] [CrossRef]
- Jofre, G.I.; Singh, A.; Mavengere, H.; Sundar, G.; D’Agostino, E.; Chowdhary, A.; Matute, D.R. An Indian lineage of Histoplasma with strong signatures of differentiation and selection. Fungal Genet. Biol. 2022, 158, 103654. [Google Scholar] [CrossRef]
- Robert, V.; Vu, D.; Amor, A.B.; van de Wiele, N.; Brouwer, C.; Jabas, B.; Szoke, S.; Dridi, A.; Triki, M.; Ben Daoud, S.; et al. MycoBank gearing up for new horizons. IMA Fungus 2013, 4, 371–379. [Google Scholar] [CrossRef]
- Köser, C.U.; Ellington, M.J.; Cartwright, E.J.P.; Gillespie, S.H.; Brown, N.M.; Farrington, M.; Holden, M.T.G.; Dougan, G.; Bentley, S.D.; Parkhill, J.; et al. Routine Use of Microbial Whole Genome Sequencing in Diagnostic and Public Health Microbiology. PLoS Pathog. 2012, 8, e1002824. [Google Scholar] [CrossRef] [PubMed]
- Roetzer, A.; Diel, R.; Kohl, T.A.; Rückert, C.; Nübel, U.; Blom, J.; Wirth, T.; Jaenicke, S.; Schuback, S.; Rüsch-Gerdes, S.; et al. Whole Genome Sequencing versus Traditional Genotyping for Investigation of a Mycobacterium tuberculosis Outbreak: A Longitudinal Molecular Epidemiological Study. PLoS Med. 2013, 10, e1001387. [Google Scholar] [CrossRef]
- Bagal, U.R.; Phan, J.; Welsh, R.M.; Misas, E.; Wagner, D.; Gade, L.; Litvintseva, A.P.; Cuomo, C.A.; Chow, N.A. MycoSNP: A portable workflow for performing whole-genome sequencing analysis of Candida auris. In Candida auris: Methods and Protocols, 1st ed.; Lorenz, A., Ed.; Humana: New York, NY, USA, 2022. [Google Scholar]
- Delcher, A.L.; Salzberg, S.L.; Phillippy, A.M. Using MUMmer to identify similar regions in large sequence sets. Curr. Protoc. Bioinform. 2003. Chapter 10, Unit 10.3. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Broad Institute Picard Toolkit: Repository. Available online: http://broadinstitute.github.io/picard/ (accessed on 25 March 2022).
- Lo, C.-C.; Chain, P.S.G. Rapid evaluation and quality control of next generation sequencing data with FaQCs. BMC Bioinform. 2014, 15, 366. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Cheng, A.Y.; Teo, Y.Y.; Ong, R.T. Assessing single nucleotide variant detection and genotype calling on whole-genome sequenced individuals. Bioinformatics 2014, 30, 1707–1713. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Team, R. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Hepler, S.A.; Kaufeld, K.A.; Benedict, K.; Toda, M.; Jackson, B.R.; Liu, X.; Kline, D. Integrating Public Health Surveillance and Environmental Data to Model Presence of Histoplasma in the United States. Epidemiology 2022, 33, 654–659. [Google Scholar] [CrossRef]
- Taylor, M.L.; Reyes-Montes, M.d.R.; Estrada-Bárcenas, D.A.; Zancopé-Oliveira, R.M.; Rodríguez-Arellanes, G.; Ramírez, J.A. Considerations about the Geographic Distribution of Histoplasma Species. Appl. Environ. Microbiol. 2022, 88, e0201021. [Google Scholar] [CrossRef] [PubMed]
- Dingle, T.C.; Croxen, M.A.; Fathima, S.; Shokoples, S.; Sonpar, A.; Saxinger, L.; Schwartz, I.S. Histoplasmosis acquired in Alberta, Canada: An epidemiological and genomic study. Lancet Microbe 2021, 2, e191–e197. [Google Scholar] [CrossRef] [PubMed]
- Almeida-Silva, F.; Teixeira, M.d.M.; Matute, D.R.; de Faria Ferreira, M.; Barker, B.M.; Almeida-Paes, R.; Guimarães, A.J.; Zancopé-Oliveira, R.M. Genomic Diversity Analysis Reveals a Strong Population Structure in Histoplasma capsulatum LAmA (Histoplasma suramericanum). J. Fungi 2021, 7, 865. [Google Scholar] [CrossRef] [PubMed]
- Gusa, A.; Jinks-Robertson, S. Mitotic Recombination and Adaptive Genomic Changes in Human Pathogenic Fungi. Genes 2019, 10, 901. [Google Scholar] [CrossRef] [PubMed]
- Oltean, H.N.; Etienne, K.A.; Roe, C.C.; Gade, L.; McCotter, O.Z.; Engelthaler, D.M.; Litvintseva, A.P. Utility of Whole-Genome Sequencing to Ascertain Locally Acquired Cases of Coccidioidomycosis, Washington, USA. Emerg. Infect. Dis. 2019, 25, 501–506. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Count | Percentage (%) | |
|---|---|---|
| Sex | ||
| Male | 39 | 81 |
| Female | 9 | 19 |
| Total | 48 | 100 |
| Age Group | ||
| <21 | 6 | 13 |
| ≥21 & <65 | 25 | 52 |
| ≥65 | 17 | 35 |
| Total | 48 | 100 |
| Immunosuppressed cases | ||
| Indiana | 1 | 5 |
| Kentucky | 3 | 16 |
| Michigan | 6 | 32 |
| Minnesota | 8 | 42 |
| Wisconsin | 1 | 5 |
| Total | 19 | 100 |
| Specimen Source | ||
| Sputum | 4 | 8 |
| Lymph node | 4 | 8 |
| Bronchial specimen | 17 | 35 |
| Lung tissue | 3 | 6 |
| Blood | 12 | 25 |
| Bone marrow | 3 | 6 |
| Others * | 5 | 10 |
| Total | 48 | 100 |
| States | ||
| Indiana | 2 | 4 |
| Kentucky | 4 | 8 |
| Louisiana | 1 | 2 |
| Michigan | 21 | 44 |
| Minnesota | 13 | 27 |
| Nebraska | 1 | 2 |
| Pennsylvania | 1 | 2 |
| Wisconsin | 5 | 10 |
| Total | 48 | 100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bagal, U.R.; Gade, L.; Benedict, K.; Howell, V.; Christophe, N.; Gibbons-Burgener, S.; Hallyburton, S.; Ireland, M.; McCracken, S.; Metobo, A.K.; et al. A Phylogeographic Description of Histoplasma capsulatum in the United States. J. Fungi 2023, 9, 884. https://doi.org/10.3390/jof9090884
Bagal UR, Gade L, Benedict K, Howell V, Christophe N, Gibbons-Burgener S, Hallyburton S, Ireland M, McCracken S, Metobo AK, et al. A Phylogeographic Description of Histoplasma capsulatum in the United States. Journal of Fungi. 2023; 9(9):884. https://doi.org/10.3390/jof9090884
Chicago/Turabian StyleBagal, Ujwal R., Lalitha Gade, Kaitlin Benedict, Victoria Howell, Natalie Christophe, Suzanne Gibbons-Burgener, Sara Hallyburton, Malia Ireland, Stephanie McCracken, Alison Keyser Metobo, and et al. 2023. "A Phylogeographic Description of Histoplasma capsulatum in the United States" Journal of Fungi 9, no. 9: 884. https://doi.org/10.3390/jof9090884
APA StyleBagal, U. R., Gade, L., Benedict, K., Howell, V., Christophe, N., Gibbons-Burgener, S., Hallyburton, S., Ireland, M., McCracken, S., Metobo, A. K., Signs, K., Warren, K. A., Litvintseva, A. P., & Chow, N. A. (2023). A Phylogeographic Description of Histoplasma capsulatum in the United States. Journal of Fungi, 9(9), 884. https://doi.org/10.3390/jof9090884