
Unveiling the Secretome of the Fungal Plant Pathogen Neofusicoccum parvum Induced by In Vitro Host Mimicry

 , , , ,
, , , ,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Fungal Strains, Plant Material, and Culture Conditions
2.2. RNA Extraction and cDNA Synthesis
2.3. Quantitative PCR
2.4. Extracellular Protein Extraction
2.5. Protein Sample Cleaning
2.6. Protein Quantification
2.7. Protein Quality Evaluation by Electrophoresis
2.8. Tryptic Digestion, Mass Spectrometry Analysis, and Protein Identification
2.9. Bioinformatic Analysis
2.10. Interactomics Analysis
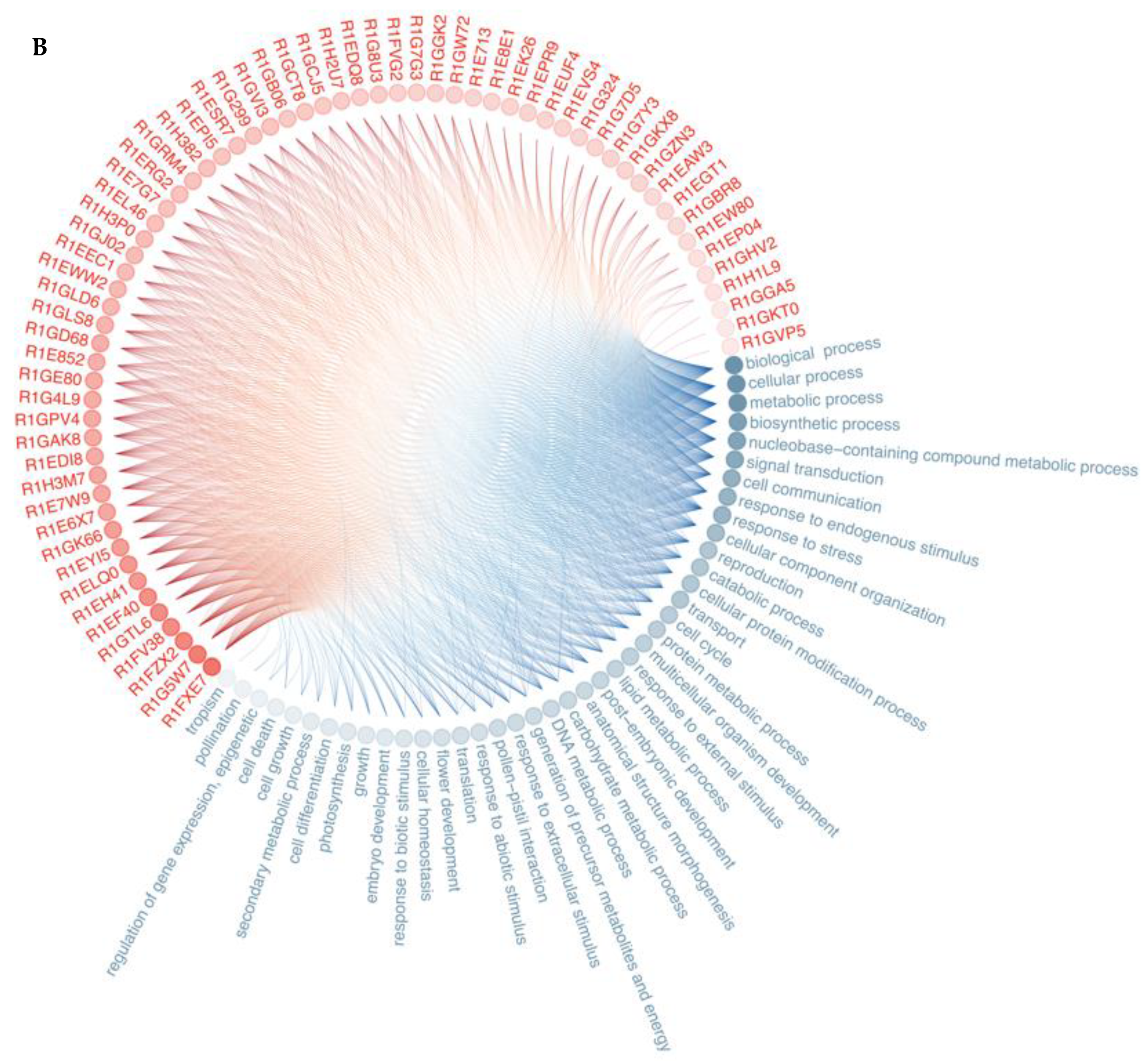
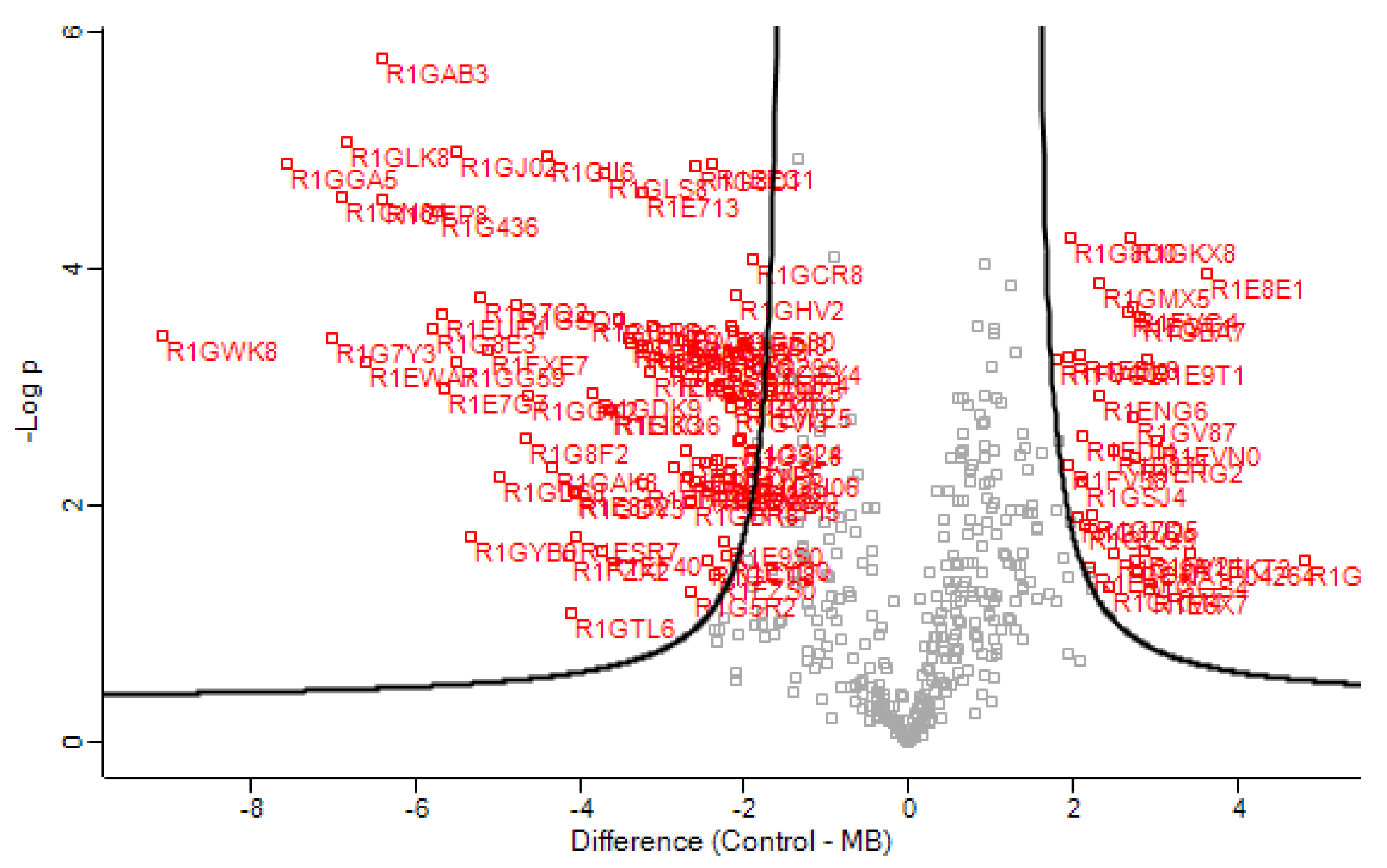
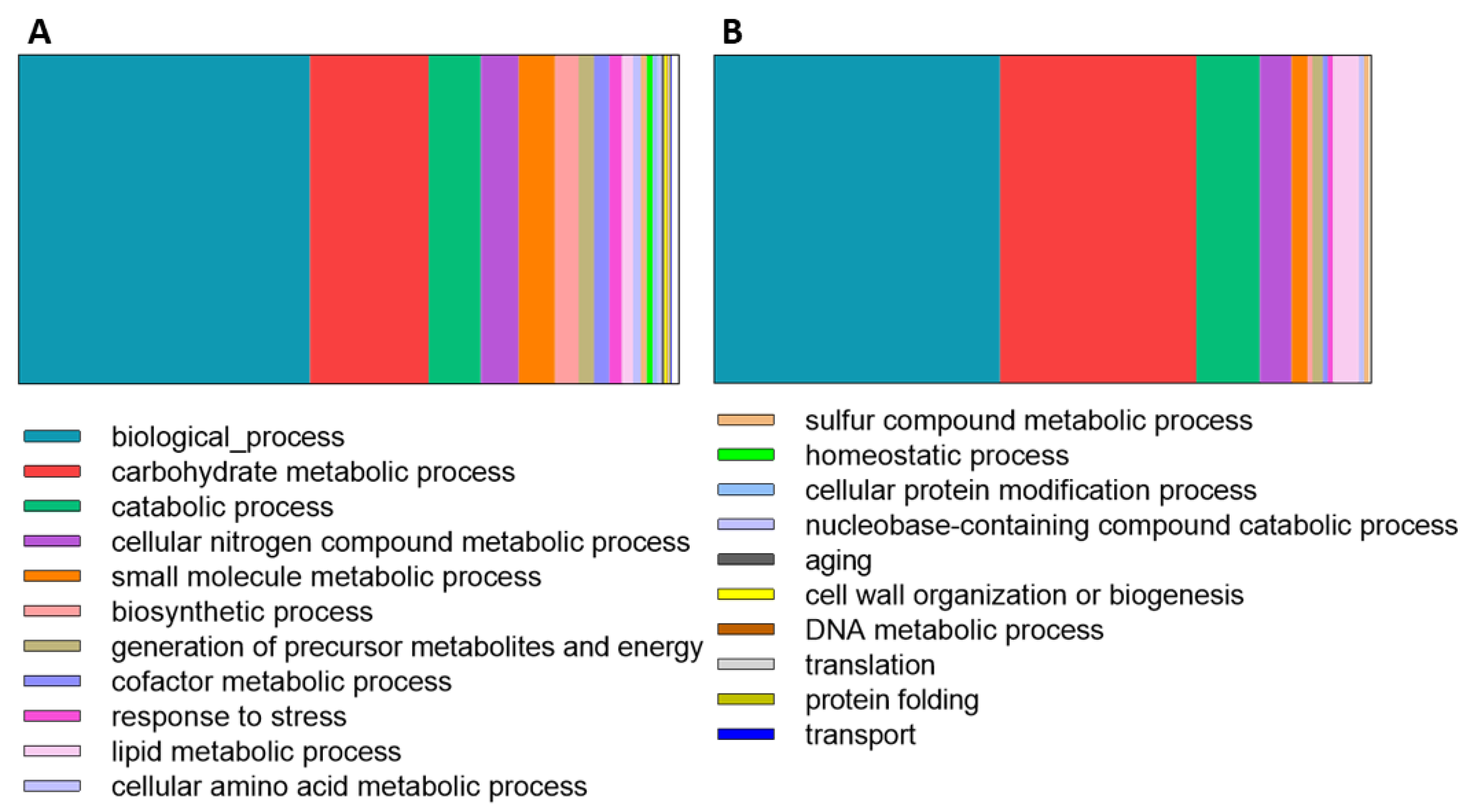
3. Results
3.1. Secretome Analysis
3.2. Protein—Protein Interaction
4. Discussion
4.1. Proteins Involved in Carbohydrate Metabolic Processes
4.2. Defense from Host
4.3. Virulence
4.4. Protein–Protein Interactions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Deus, E.; Silva, J.S.; Castro-Díez, P.; Lomba, A.; Ortiz, M.L.; Vicente, J. Current and future conflicts between eucalypt plantations and high biodiversity areas in the Iberian Peninsula. J. Nat. Conserv. 2018, 45, 107–117. [Google Scholar] [CrossRef]
- ICNF. 6 Inventário Florestal Nacional; Instituto da Conservação da Natureza e das Florestas: Lisboa, Portugal, 2019; p. 284.
- Li, G.; Arnold, R.J.; Liu, F.; Li, J.; Chen, S. Identification and pathogenicity of Lasiodiplodia species from Eucalyptus urophylla × grandis, Polyscias balfouriana and Bougainvillea spectabilis in Southern China. J. Phytopathol. 2015, 163, 956–967. [Google Scholar] [CrossRef]
- Slippers, B.; Roux, J.; Wingfield, M.J.; van der Walt, F.J.J.; Jami, F.; Mehl, J.W.M.; Marais, G.J. Confronting the constraints of morphological taxonomy in the Botryosphaeriales. Persoonia 2014, 33, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Slippers, B.; Wingfield, M.J. Botryosphaeriaceae as endophytes and latent pathogens of woody plants: Diversity, ecology and impact. Fungal Biol. Rev. 2007, 21, 90–106. [Google Scholar] [CrossRef]
- Pérez, C.A.; Wingfield, M.J.; Slippers, B.; Altier, N.A.; Blanchette, R.A. Endophytic and canker-associated Botryosphaeriaceae occurring on non-native Eucalyptus and native Myrtaceae trees in Uruguay. Fungal Divers. 2010, 41, 53–69. [Google Scholar] [CrossRef]
- Phillips, A.J.; Alves, A.; Abdollahzadeh, J.; Slippers, B.; Wingfield, M.J.; Groenewald, J.Z.; Crous, P.W. The Botryosphaeriaceae: Genera and species known from culture. Stud. Mycol. 2013, 76, 51–167. [Google Scholar] [CrossRef]
- Smith, H.; Wingfield, M.J.; Petrini, O. Botryosphaeria dothidea endophytic in Eucalyptus grandis and Eucalyptus nitens in South Africa. Forest Ecol. Manag. 1996, 89, 189–195. [Google Scholar] [CrossRef]
- Barradas, C.; Phillips, A.J.L.; Correia, A.; Diogo, E.; Bragança, H.; Alves, A. Diversity and potential impact of Botryosphaeriaceae species associated with Eucalyptus globulus plantations in Portugal. Eur. J. Plant Pathol. 2016, 146, 245–257. [Google Scholar] [CrossRef]
- Batista, E.; Lopes, A.; Alves, A. Botryosphaeriaceae species on forest trees in Portugal: Diversity, distribution and pathogenicity. Eur. J. Plant Pathol. 2020, 158, 693–720. [Google Scholar] [CrossRef]
- Slippers, B.; Burgess, T.; Pavlic, D.; Ahumada, R.; Maleme, H.; Mohali, S.; Rodas, C.; Wingfield, M.J. A diverse assemblage of Botryosphaeriaceae infect Eucalyptus in native and non-native environments. South For.-J. For. Sci. 2009, 71, 101–110. [Google Scholar] [CrossRef]
- Mohali, S.; Slippers, B.; Wingfield, M.J. Identification of Botryosphaeriaceae from Eucalyptus, Acacia and Pinus in Venezuela. Fungal Divers. 2007, 25, 103–125. [Google Scholar]
- Blanco-Ulate, B.; Rolshausen, P.; Cantu, D. Draft genome sequence of Neofusicoccum parvum isolate UCR-NP2, a fungal vascular pathogen associated with grapevine cankers. Genome Announc 2013, 1, e00339-13. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.F.; Pavlic, D.; Roux, J.; Slippers, B.; Xie, Y.J.; Wingfield, M.J.; Zhou, X.D. Characterization of Botryosphaeriaceae from plantation-grown Eucalyptus species in South China. Plant Pathol. 2011, 60, 739–751. [Google Scholar] [CrossRef]
- Pavlic, D.; Slippers, B.; Coutinho, T.A.; Wingfield, M.J. Botryosphaeriaceae occurring on native Syzygium cordatum in South Africa and their potential threat to Eucalyptus. Plant Pathol. 2007, 56, 624–636. [Google Scholar] [CrossRef]
- Abou-Mansour, E.; Débieux, J.-L.; Ramírez-Suero, M.; Bénard-Gellon, M.; Magnin-Robert, M.; Spagnolo, A.; Chong, J.; Farine, S.; Bertsch, C.; L’Haridon, F.; et al. Phytotoxic metabolites from Neofusicoccum parvum, a pathogen of Botryosphaeria dieback of grapevine. Phytochemistry 2015, 115, 207–215. [Google Scholar] [CrossRef]
- Andolfi, A.; Mugnai, L.; Luque, J.; Surico, G.; Cimmino, A.; Evidente, A. Phytotoxins produced by fungi associated with grapevine trunk diseases. Toxins 2011, 3, 1569–1605. [Google Scholar] [CrossRef]
- Masi, M.; Cimmino, A.; Reveglia, P.; Mugnai, L.; Surico, G.; Evidente, A. Advances on Fungal Phytotoxins and Their Role in Grapevine Trunk Diseases. J. Agric. Food Chem. 2018, 66, 5948–5958. [Google Scholar] [CrossRef]
- Salvatore, M.M.; Alves, A.; Andolfi, A. Secondary metabolites produced by Neofusicoccum species associated with plants: A review. Agriculture 2021, 11, 149. [Google Scholar] [CrossRef]
- Bénard-Gellon, M.; Farine, S.; Goddard, M.L.; Schmitt, M.; Stempien, E.; Pensec, F.; Laloue, H.; Mazet-Kieffer, F.; Fontaine, F.; Larignon, P.; et al. Toxicity of extracellular proteins from Diplodia seriata and Neofusicoccum parvum involved in grapevine Botryosphaeria dieback. Protoplasma 2015, 252, 679–687. [Google Scholar] [CrossRef]
- Nazar Pour, F.; Cobos, R.; Rubio Coque, J.J.; Serôdio, J.; Alves, A.; Félix, C.; Ferreira, V.; Esteves, A.C.; Duarte, A.S. Toxicity of recombinant Necrosis and Ethylene-Inducing Proteins (NLPs) from Neofusicoccum parvum. Toxins 2020, 12, 235. [Google Scholar] [CrossRef]
- Massonnet, M.; Figueroa-Balderas, R.; Galarneau, E.R.A.; Miki, S.; Lawrence, D.P.; Sun, Q.; Wallis, C.M.; Baumgartner, K.; Cantu, D. Neofusicoccum parvum colonization of the grapevine woody stem triggers asynchronous host responses at the site of infection and in the leaves. Front. Plant Sci. 2017, 8, 1117. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.Y.; Zhao, W.S.; Chen, Z.; Xing, Q.K.; Zhang, W.; Chethana, K.W.T.; Xue, M.F.; Xu, J.P.; Phillips, A.J.L.; Wang, Y.; et al. Comparative genome and transcriptome analyses reveal adaptations to opportunistic infections in woody plant degrading pathogens of Botryosphaeriaceae. DNA Res. 2018, 25, 87–102. [Google Scholar] [CrossRef] [PubMed]
- Massonnet, M.; Morales-Cruz, A.; Figueroa-Balderas, R.; Lawrence, D.P.; Baumgartner, K.; Cantu, D. Condition-dependent co-regulation of genomic clusters of virulence factors in the grapevine trunk pathogen Neofusicoccum parvum. Mol. Plant Pathol. 2018, 19, 21–34. [Google Scholar] [CrossRef] [PubMed]
- González-Fernández, R.; Valero-Galván, J.; Gómez-Gálvez, F.J.; Jorrín-Novo, J.V. Unraveling the in vitro secretome of the phytopathogen Botrytis cinerea to understand the interaction with its hosts. Front. Plant Sci. 2015, 6, 839. [Google Scholar] [CrossRef] [PubMed]
- Jami, M.S.; Barreiro, C.; García-Estrada, C.; Martín, J.F. Proteome analysis of the penicillin producer Penicillium chrysogenum: Characterization of protein changes during the industrial strain improvement. Mol. Cell Proteom. 2010, 9, 1182–1198. [Google Scholar] [CrossRef]
- Lyu, X.; Shen, C.; Fu, Y.; Xie, J.; Jiang, D.; Li, G.; Cheng, J. Comparative genomic and transcriptional analyses of the carbohydrate-active enzymes and secretomes of phytopathogenic fungi reveal their significant roles during infection and development. Sci. Rep. 2015, 5, 15565. [Google Scholar] [CrossRef]
- Mandelc, S.; Javornik, B. The secretome of vascular wilt pathogen Verticillium albo-atrum in simulated xylem fluid. Proteomics 2015, 15, 787–797. [Google Scholar] [CrossRef]
- Shah, P.; Atwood, J.A.; Orlando, R.; El Mubarek, H.; Podila, G.K.; Davis, M.R. Comparative proteomic analysis of Botrytis cinerea secretome. J. Proteome Res. 2009, 8, 1123–1130. [Google Scholar] [CrossRef]
- Cobos, R.; Barreiro, C.; Mateos, R.M.; Coque, J.J. Cytoplasmic- and extracellular-proteome analysis of Diplodia seriata: A phytopathogenic fungus involved in grapevine decline. Proteome Sci. 2010, 8, 46. [Google Scholar] [CrossRef]
- Fernandes, I.; Alves, A.; Correia, A.; Devreese, B.; Esteves, A.C. Secretome analysis identifies potential virulence factors of Diplodia corticola, a fungal pathogen involved in cork oak (Quercus suber) decline. Fungal Biol. 2014, 118, 516–523. [Google Scholar] [CrossRef]
- Félix, C.; Duarte, A.S.; Vitorino, R.; Guerreiro, A.C.; Domingues, P.; Correia, A.C.; Alves, A.; Esteves, A.C. Temperature modulates the secretome of the phytopathogenic fungus Lasiodiplodia theobromae. Front. Plant Sci. 2016, 7, 1096. [Google Scholar] [CrossRef] [PubMed]
- Félix, C.; Meneses, R.; Gonçalves, M.F.M.; Tilleman, L.; Duarte, A.S.; Jorrín-Novo, J.V.; Van de Peer, Y.; Deforce, D.; Van Nieuwerburgh, F.; Esteves, A.C.; et al. A multi-omics analysis of the grapevine pathogen Lasiodiplodia theobromae reveals that temperature affects the expression of virulence- and pathogenicity-related genes. Sci. Rep. 2019, 9, 13144. [Google Scholar] [CrossRef]
- Uranga, C.C.; Ghassemian, M.; Hernández-Martínez, R. Novel proteins from proteomic analysis of the trunk disease fungus Lasiodiplodia theobromae (Botryosphaeriaceae). Biochim. Open. 2017, 4, 88–98. [Google Scholar] [CrossRef]
- Alves, A.; Crous, P.W.; Correia, A.; Phillips, A. Morphological and molecular data reveal cryptic speciation in Lasiodiplodia theobromae. Fungal Divers. 2008, 28, 1–13. [Google Scholar]
- Wessel, D.; Flügge, U.I. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 1984, 138, 141–143. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. Quantitative, high-resolution proteomics for data-driven systems biology. Annu. Rev. Biochem. 2011, 80, 273–299. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef]
- Consortium, T.U. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2020, 49, D480–D489. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Blum, M.; Chang, H.-Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 2020, 49, D344–D354. [Google Scholar] [CrossRef] [PubMed]
- Drula, E.; Garron, M.-L.; Dogan, S.; Lombard, V.; Henrissat, B.; Terrapon, N. The carbohydrate-active enzyme database: Functions and literature. Nucleic Acids Res. 2021, 50, D571–D577. [Google Scholar] [CrossRef] [PubMed]
- Pierleoni, A.; Martelli, P.L.; Fariselli, P.; Casadio, R. BaCelLo: A balanced subcellular localization predictor. Bioinformatics 2006, 22, e408–e416. [Google Scholar] [CrossRef]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef]
- Bendtsen, J.D.; Jensen, L.J.; Blom, N.; Von Heijne, G.; Brunak, S. Feature-based prediction of non-classical and leaderless protein secretion. Protein Eng. Des. Sel. 2004, 17, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Coelho, E.D.; Arrais, J.P.; Matos, S.; Pereira, C.; Rosa, N.; Correia, M.J.; Barros, M.; Oliveira, J.L. Computational prediction of the human-microbial oral interactome. BMC Syst. Biol. 2014, 8, 24. [Google Scholar] [CrossRef]
- Team, R.C. R: A Language and Environment for Statistical Computing. Available online: https://www.R-project.org/ (accessed on 1 April 2022).
- Pedersen, T.L. ggraph: An Implementation of Grammar of Graphics for Graphs and Networks. R package Version 2.0.5. Available online: https://CRAN.R-project.org/package=ggraph (accessed on 1 April 2022).
- Csardi, G.; Nepusz, T.T. The igraph software package for complex network research. InterJournal Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the tidyverse. J. Open. Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Nielsen, H.; Tsirigos, K.D.; Brunak, S.; von Heijne, G. A brief history of protein sorting prediction. Protein J. 2019, 38, 200–216. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2020, 49, D412–D419. [Google Scholar] [CrossRef]
- Esteves, E.; Rosa, N.; Correia, M.J.; Arrais, J.P.; Barros, M. New targets for Zika Virus determined by human-viral interactomic: A bioinformatics approach. Biomed Res. Int. 2017, 2017, 1734151. [Google Scholar] [CrossRef] [PubMed]
- Rosa, N.; Campos, B.; Esteves, A.C.; Duarte, A.S.; Correia, M.J.; Silva, R.M.; Barros, M. Tracking the functional meaning of the human oral-microbiome protein-protein interactions. Adv. Protein Chem. Struct. Biol. 2020, 121, 199–235. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, I.O. Infection Mechanism of Diplodia corticola. Ph.D. Thesis, Universidade de Aveiro, Aveiro, Portugal, 2015. Available online: http://hdl.handle.net/10773/15487 (accessed on 21 July 2022).
- Salvatore, M.M.; Félix, C.; Lima, F.; Ferreira, V.; Naviglio, D.; Salvatore, F.; Duarte, A.S.; Alves, A.; Andolfi, A.; Esteves, A.C. Secondary Metabolites Produced by Macrophomina phaseolina Isolated from Eucalyptus globulus. Agriculture 2020, 10, 72. [Google Scholar] [CrossRef]
- Jung, Y.H.; Jeong, S.H.; Kim, S.H.; Singh, R.; Lee, J.E.; Cho, Y.S.; Agrawal, G.K.; Rakwal, R.; Jwa, N.S. Secretome analysis of Magnaporthe oryzae using in vitro systems. Proteomics 2012, 12, 878–900. [Google Scholar] [CrossRef]
- Sibbald, M.J.; Ziebandt, A.K.; Engelmann, S.; Hecker, M.; de Jong, A.; Harmsen, H.J.; Raangs, G.C.; Stokroos, I.; Arends, J.P.; Dubois, J.Y.; et al. Mapping the pathways to staphylococcal pathogenesis by comparative secretomics. Microbiol. Mol. Biol. Rev. 2006, 70, 755–788. [Google Scholar] [CrossRef]
- Nagel, J.H.; Wingfield, M.J.; Slippers, B. Increased abundance of secreted hydrolytic enzymes and secondary metabolite gene clusters define the genomes of latent plant pathogens in the Botryosphaeriaceae. BMC Genom. 2021, 22, 589. [Google Scholar] [CrossRef]
- Sinha, N.; Patra, S.K.; Ghosh, S. Secretome analysis of Macrophomina phaseolina identifies an array of putative virulence factors responsible for charcoal rot disease in plants. Front. Microbiol. 2022, 13, 847832. [Google Scholar] [CrossRef]
- De Silva, N.; Lumyong, S.; Hyde, K.; Bulgakov, T.; Phillips, A.; Yan, J. Mycosphere Essays 9: Defining biotrophs and hemibiotrophs. Mycosphere 2016, 7, 545–559. [Google Scholar] [CrossRef]
- Martínez, M.J.; Alconada, M.T.; Guillén, F.; Vázquez, C.; Reyes, F. Pectic activities from Fusarium oxysporum f. sp. melonis: Purification and characterization of an exopolygalacturonase. FEMS Microbiol. Lett. 1991, 81, 145–149. [Google Scholar]
- Niture, S.K.; Kumar, A.R.; Pant, A. Role of glucose in production and repression of polygalacturonase and pectate lyase from phytopathogenic fungus Fusarium moniliforme NCIM 1276. World J. Microbiol. Biotechnol. 2006, 22, 893–899. [Google Scholar] [CrossRef]
- Panda, T.; Nair, S.R.; Kumar, M.P. Regulation of synthesis of the pectolytic enzymes of Aspergillus niger. Enzym. Microb. Technol. 2004, 34, 466–473. [Google Scholar] [CrossRef]
- Coetzee, B.; Schols, H.A.; Wolfaardt, F. Determination of Pectin Content of Eucalyptus Wood. pp. 327–331. Available online: https://www.degruyter.com/document/doi/10.1515/hf.2011.054/html (accessed on 21 July 2022).
- Kang, Z.; Buchenauer, H. Ultrastructural and cytochemical studies on cellulose, xylan and pectin degradation in wheat spikes infected by Fusarium culmorum. J. Phytopathol. 2000, 148, 263–275. [Google Scholar] [CrossRef]
- Tomassini, A.; Sella, L.; Raiola, A.; D’Ovidio, R.; Favaron, F. Characterization and expression of Fusarium graminearum endo-polygalacturonases in vitro and during wheat infection. Plant Pathol. 2009, 58, 556–564. [Google Scholar] [CrossRef]
- Ma, Z.; Song, T.; Zhu, L.; Ye, W.; Wang, Y.; Shao, Y.; Dong, S.; Zhang, Z.; Dou, D.; Zheng, X.; et al. A Phytophthora sojae glycoside hydrolase 12 protein is a major virulence factor during Soybean infection and is recognized as a PAMP. Plant Cell 2015, 27, 2057–2072. [Google Scholar] [CrossRef] [PubMed]
- Gui, Y.J.; Chen, J.Y.; Zhang, D.D.; Li, N.Y.; Li, T.G.; Zhang, W.Q.; Wang, X.Y.; Short, D.P.G.; Li, L.; Guo, W.; et al. Verticillium dahliae manipulates plant immunity by glycoside hydrolase 12 proteins in conjunction with carbohydrate-binding module 1. Environ. Microbiol. 2017, 19, 1914–1932. [Google Scholar] [CrossRef] [PubMed]
- Van Vu, B.; Itoh, K.; Nguyen, Q.B.; Tosa, Y.; Nakayashiki, H. Cellulases belonging to glycoside hydrolase families 6 and 7 contribute to the virulence of Magnaporthe oryzae. Mol. Plant Microbe Interact. 2012, 25, 1135–1141. [Google Scholar] [CrossRef]
- Nguyen, Q.B.; Itoh, K.; Van Vu, B.; Tosa, Y.; Nakayashiki, H. Simultaneous silencing of endo-β-1,4 xylanase genes reveals their roles in the virulence of Magnaporthe oryzae. Mol. Microbiol. 2011, 81, 1008–1019. [Google Scholar] [CrossRef]
- Lai, M.W.; Liou, R.F. Two genes encoding GH10 xylanases are essential for the virulence of the oomycete plant pathogen Phytophthora parasitica. Curr. Genet. 2018, 64, 931–943. [Google Scholar] [CrossRef]
- Brito, N.; Espino, J.J.; González, C. The endo-beta-1,4-xylanase xyn11A is required for virulence in Botrytis cinerea. Mol. Plant Microbe Interact. 2006, 19, 25–32. [Google Scholar] [CrossRef]
- Apel-Birkhold, P.C.; Walton, J.D. Cloning, disruption, and expression of two endo-beta 1, 4-xylanase genes, XYL2 and XYL3, from Cochliobolus carbonum. Appl. Environ. Microbiol. 1996, 62, 4129–4135. [Google Scholar] [CrossRef]
- Calero-Nieto, F.; Di Pietro, A.; Roncero, M.I.; Hera, C. Role of the transcriptional activator xlnR of Fusarium oxysporum in regulation of xylanase genes and virulence. Mol. Plant Microbe Interact. 2007, 20, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Gómez, E.; Ruíz-Roldán, M.C.; Di Pietro, A.; Roncero, M.I.; Hera, C. Role in pathogenesis of two endo-beta-1,4-xylanase genes from the vascular wilt fungus Fusarium oxysporum. Fungal Genet. Biol. 2002, 35, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Stempien, E.; Goddard, M.L.; Wilhelm, K.; Tarnus, C.; Bertsch, C.; Chong, J. Grapevine Botryosphaeria dieback fungi have specific aggressiveness factor repertory involved in wood decay and stilbene metabolization. PLoS ONE 2017, 12, e0188766. [Google Scholar] [CrossRef]
- Cai, Y.; Gong, Y.; Liu, W.; Hu, Y.; Chen, L.; Yan, L.; Zhou, Y.; Bian, Y. Comparative secretomic analysis of lignocellulose degradation by Lentinula edodes grown on microcrystalline cellulose, lignosulfonate and glucose. J. Proteom. 2017, 163, 92–101. [Google Scholar] [CrossRef]
- Raffaele, S.; Win, J.; Cano, L.M.; Kamoun, S. Analyses of genome architecture and gene expression reveal novel candidate virulence factors in the secretome of Phytophthora infestans. BMC Genom. 2010, 11, 637. [Google Scholar] [CrossRef] [PubMed]
- Seidl, M.F.; Van den Ackerveken, G.; Govers, F.; Snel, B. A domain-centric analysis of oomycete plant pathogen genomes reveals unique protein organization. Plant Physiol. 2011, 155, 628–644. [Google Scholar] [CrossRef] [PubMed]
- Leferink, N.G.; Heuts, D.P.; Fraaije, M.W.; van Berkel, W.J. The growing VAO flavoprotein family. Arch. Biochem. Biophys. 2008, 474, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Do Vale, L.H.; Gómez-Mendoza, D.P.; Kim, M.S.; Pandey, A.; Ricart, C.A.; Ximenes, F.F.E.; Sousa, M.V. Secretome analysis of the fungus Trichoderma harzianum grown on cellulose. Proteomics 2012, 12, 2716–2728. [Google Scholar] [CrossRef]
- Kuroki, M.; Okauchi, K.; Yoshida, S.; Ohno, Y.; Murata, S.; Nakajima, Y.; Nozaka, A.; Tanaka, N.; Nakajima, M.; Taguchi, H.; et al. Chitin-deacetylase activity induces appressorium differentiation in the rice blast fungus Magnaporthe oryzae. Sci. Rep. 2017, 7, 9697. [Google Scholar] [CrossRef]
- Rovenich, H.; Zuccaro, A.; Thomma, B.P. Convergent evolution of filamentous microbes towards evasion of glycan-triggered immunity. New Phytol. 2016, 212, 896–901. [Google Scholar] [CrossRef]
- Sharp, R.G. A review of the applications of chitin and its derivatives in agriculture to modify plant-microbial interactions and improve crop yields. Agronomy 2013, 3, 757–793. [Google Scholar] [CrossRef]
- Kouzai, Y.; Mochizuki, S.; Saito, A.; Ando, A.; Minami, E.; Nishizawa, Y. Expression of a bacterial chitosanase in rice plants improves disease resistance to the rice blast fungus Magnaporthe oryzae. Plant Cell Rep. 2012, 31, 629–636. [Google Scholar] [CrossRef]
- Ride, J.P.; Barber, M.S. Purification and characterization of multiple forms of endochitinase from wheat leaves. Plant Sci. 1990, 71, 185–197. [Google Scholar] [CrossRef]
- Hislop, E.C.; Paver, J.L.; Keon, J.P.R. An acid protease produced by Monilinia fructigena in vitro and in infected apple fruits, and its possible role in pathogenesis. Microbiology 1982, 128, 799–807. [Google Scholar] [CrossRef][Green Version]
- Movahedi, S.; Heale, J.B. Purification and characterization of an aspartic proteinase secreted by Botrytis cinerea Pers ex. Pers in culture and in infected carrots. Physiol. Mol. Plant Pathol. 1990, 36, 289–302. [Google Scholar] [CrossRef]
- Poussereau, N.; Gente, S.; Rascle, C.; Billon-Grand, G.; Fèvre, M. aspS encoding an unusual aspartyl protease from Sclerotinia sclerotiorum is expressed during phytopathogenesis. FEMS Microbiol. Lett. 2001, 194, 27–32. [Google Scholar] [CrossRef]
- Urbanek, H.; Yirdaw, G. Hydrolytic ability of acid protease of Fusarium culmorum and its possible role in phytopathogenesis. Acta Microbiol. Pol. 1984, 33, 131–136. [Google Scholar]
- O’Connell, R.J.; Thon, M.R.; Hacquard, S.; Amyotte, S.G.; Kleemann, J.; Torres, M.F.; Damm, U.; Buiate, E.A.; Epstein, L.; Alkan, N.; et al. Lifestyle transitions in plant pathogenic Colletotrichum fungi deciphered by genome and transcriptome analyses. Nat. Genet. 2012, 44, 1060–1065. [Google Scholar] [CrossRef]
- Lakshman, D.K.; Roberts, D.P.; Garrett, W.M.; Natarajan, S.S.; Darwish, O.; Alkharouf, N.; Pain, A.; Khan, F.; Jambhulkar, P.P.; Mitra, A. Proteomic investigation of Rhizoctonia solani AG 4 identifies secretome and mycelial proteins with roles in plant cell wall degradation and virulence. J. Agric. Food Chem. 2016, 64, 3101–3110. [Google Scholar] [CrossRef]
- González-Fernández, R.; Aloria, K.; Valero-Galván, J.; Redondo, I.; Arizmendi, J.M.; Jorrín-Novo, J.V. Proteomic analysis of mycelium and secretome of different Botrytis cinerea wild-type strains. J. Proteom. 2014, 97, 195–221. [Google Scholar] [CrossRef]
- Ismail, I.A.; Able, A.J. Secretome analysis of virulent Pyrenophora teres f. teres isolates. Proteomics 2016, 16, 2625–2636. [Google Scholar] [CrossRef]
- Kubicek, C.P.; Starr, T.L.; Glass, N.L. Plant cell wall-degrading enzymes and their secretion in plant-pathogenic fungi. Annu. Rev. Phytopathol. 2014, 52, 427–451. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Tsurugi, K. RNA N-glycosidase activity of ricin A-chain. Mechanism of action of the toxic lectin ricin on eukaryotic ribosomes. J. Biol. Chem. 1987, 262, 8128–8130. [Google Scholar] [CrossRef]
- Andersson, K.-M.; Kumar, D.; Bentzer, J.; Friman, E.; Ahrén, D.; Tunlid, A. Interspecific and host-related gene expression patterns in nematode-trapping fungi. BMC Genom. 2014, 15, 968. [Google Scholar] [CrossRef] [PubMed]
- Meerupati, T.; Andersson, K.M.; Friman, E.; Kumar, D.; Tunlid, A.; Ahrén, D. Genomic mechanisms accounting for the adaptation to parasitism in nematode-trapping fungi. PLoS Genet. 2013, 9, e1003909. [Google Scholar] [CrossRef] [PubMed]
- Luhtala, N.; Parker, R. T2 family ribonucleases: Ancient enzymes with diverse roles. Trends Biochem. Sci. 2010, 35, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Pennington, H.G.; Jones, R.; Kwon, S.; Bonciani, G.; Thieron, H.; Chandler, T.; Luong, P.; Morgan, S.N.; Przydacz, M.; Bozkurt, T.; et al. The fungal ribonuclease-like effector protein CSEP0064/BEC1054 represses plant immunity and interferes with degradation of host ribosomal RNA. PLoS Pathog. 2019, 15, e1007620. [Google Scholar] [CrossRef] [PubMed]
- Olombrada, M.; Martínez-del-Pozo, A.; Medina, P.; Budia, F.; Gavilanes, J.G.; García-Ortega, L. Fungal ribotoxins: Natural protein-based weapons against insects. Toxicon 2014, 83, 69–74. [Google Scholar] [CrossRef]
- Carr, M.D.; Bloemink, M.J.; Dentten, E.; Whelan, A.O.; Gordon, S.V.; Kelly, G.; Frenkiel, T.A.; Hewinson, R.G.; Williamson, R.A. Solution structure of the Mycobacterium tuberculosis complex protein MPB70: From tuberculosis pathogenesis to inherited human corneal desease. J. Biol. Chem. 2003, 278, 43736–43743. [Google Scholar] [CrossRef]
- Elkins, T.; Zinn, K.; McAllister, L.; Hoffmann, F.M.; Goodman, C.S. Genetic analysis of a Drosophila neural cell adhesion molecule: Interaction of fasciclin I and Abelson tyrosine kinase mutations. Cell 1990, 60, 565–575. [Google Scholar] [CrossRef]
- Gaspar, Y.; Johnson, K.L.; McKenna, J.A.; Bacic, A.; Schultz, C.J. The complex structures of arabinogalactan-proteins and the journey towards understanding function. Plant Mol. Biol. 2001, 47, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, T.; Noshiro, M.; Shen, M.; Nakamasu, K.; Hashimoto, K.; Kawashima-Ohya, Y.; Gotoh, O.; Kato, Y. Structural and phylogenetic analyses of RGD-CAP/beta ig-h3, a fasciclin-like adhesion protein expressed in chick chondrocytes. Biochim. Biophys. Acta 1998, 1395, 288–292. [Google Scholar] [CrossRef]
- Liu, T.B.; Chen, G.Q.; Min, H.; Lin, F.C. MoFLP1, encoding a novel fungal fasciclin-like protein, is involved in conidiation and pathogenicity in Magnaporthe oryzae. J. Zhejiang Univ. Sci. B 2009, 10, 434–444. [Google Scholar] [CrossRef]
- Seifert, G.J. Fascinating fasciclins: A surprisingly widespread family of proteins that mediate interactions between the cell exterior and the cell surface. Int. J. Mol. Sci. 2018, 19, 1628. [Google Scholar] [CrossRef]
- Li, S.-B.; Xie, Z.-Z.; Hu, C.-G.; Zhang, J.-Z. A review of Auxin response factors (ARFs) in plants. Front. Plant Sci. 2016, 7, 47. [Google Scholar] [CrossRef] [PubMed]
- De Vleesschauwer, D.; Xu, J.; Höfte, M. Making sense of hormone-mediated defense networking: From rice to Arabidopsis. Front. Plant Sci. 2014, 5, 611. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Wang, S. Insights into auxin signaling in plant-pathogen interactions. Front. Plant Sci. 2011, 2, 74. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Cao, Y.; Huang, L.; Zhao, J.; Xu, C.; Li, X.; Wang, S. Activation of the indole-3-acetic acid-amido synthetase GH3-8 suppresses expansin expression and promotes salicylate- and jasmonate-independent basal immunity in rice. Plant Cell 2008, 20, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Domingo, C.; Andrés, F.; Tharreau, D.; Iglesias, D.J.; Talón, M. Constitutive expression of OsGH3.1 reduces auxin content and enhances defense response and resistance to a fungal pathogen in rice. Mol. Plant Microbe Interact. 2009, 22, 201–210. [Google Scholar] [CrossRef]
- Fu, J.; Liu, H.; Li, Y.; Yu, H.; Li, X.; Xiao, J.; Wang, S. Manipulating broad-spectrum disease resistance by suppressing pathogen-induced auxin accumulation in rice. Plant Physiol. 2011, 155, 589–602. [Google Scholar] [CrossRef]
- Gan, P.; Ikeda, K.; Irieda, H.; Narusaka, M.; O’Connell, R.J.; Narusaka, Y.; Takano, Y.; Kubo, Y.; Shirasu, K. Comparative genomic and transcriptomic analyses reveal the hemibiotrophic stage shift of Colletotrichum fungi. New Phytol. 2013, 197, 1236–1249. [Google Scholar] [CrossRef] [PubMed]
- Horbach, R.; Graf, A.; Weihmann, F.; Antelo, L.; Mathea, S.; Liermann, J.C.; Opatz, T.; Thines, E.; Aguirre, J.S.; Deising, H.B. Sfp-Type 4′-Phosphopantetheinyl Transferase Is indispensable for fungal pathogenicity. Plant Cell 2009, 21, 3379–3396. [Google Scholar] [CrossRef] [PubMed]
- McDowell, J.M. Genomic and transcriptomic insights into lifestyle transitions of a hemi-biotrophic fungal pathogen. New Phytol. 2013, 197, 1032–1034. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Name | Gene | Expression Condition | Primer Sequence (5′-3′) | Amplicon Length (bp) | Reference |
|---|---|---|---|---|---|
| Elongation factor 1-α | EF1α | Reference gene | FW: CGGTCACTTGATCTACAAGTGC RV: CCTCGAACTCACCAGTACCG | 302 | [35] |
| Putative exo-beta protein (PL3) | UCRNP2_317 | Up-regulated | FW: ATTCAGCACTCCGGTACCAC RV: GCCGTCCACGGACTTGAT | 255 | Present study |
| Putative aspartic endopeptidase PEP1 protein | UCRNP2_6229 | Up-regulated | FW: AGCTCCAGCTATGGTGGCTA RV: GACGATAGAGAAGCCGATGC | 172 | Present study |
| Protein Name | Accession Number a | Fold Change b | p-Value c | Unique Peptides d | PEP e | Intensity f | Localization g,i |
|---|---|---|---|---|---|---|---|
| Cellulose degradation | |||||||
| GH5—Putative glycoside hydrolase family 5 protein | R1GZQ9 | 2.1 | 1.833 | 6 | 1.87 × 109 | 169.3 | Extracellular |
| GH5—Putative endoglucanase II protein | R1GLD6 | 1.9 | 1.554 | 6 | 8.42 × 108 | 97.74 | Extracellular |
| GH5—Putative cellulase family protein | R1G7G3 | −2.7 | 3.403 | 12 | 2.36 × 1010 | 323.3 | Extracellular |
| GH5—Putative endo-beta-protein | R1GDK9 | −3.9 | 2.961 | 18 | 9.7 × 109 | 323.3 | Extracellular |
| GH3—Putative beta-d-glucoside glucohydrolase protein | R1EK26 | −3.5 | 3.584 | 8 | 1.06 × 109 | 98.06 | Extracellular |
| GH3—Putative beta-glucosidase 1 protein | R1G324 | −2 | 2.565 | 11 | 3.43 × 108 | 84.3 | Extracellular |
| GH7—Glucanase | R1GZN3 | −2.5 | 2.374 | 10 | 1.49 × 1010 | 323.3 | Extracellular |
| AA9/GH61/CBM1—Putative fungal cellulose-binding domain protein | R1GHV2 | −2.1 | 3.779 | 7 | 2.05 × 109 | 204.1 | Extracellular |
| GH12—Putative glycoside hydrolase family 12 protein | R1GQP5 | −3.9 | 3.605 | 8 | 1.05 × 1010 | 120.04 | Extracellular |
| Hemicellulose degradation | |||||||
| GH35—Putative beta-galactosidase B protein | R1E7W9 | −2.5 | 3.430 | 21 | 4.23 × 109 | 255.29 | Extracellular |
| GH43—Putative glycosyl family protein | R1EP04 | −2.9 | 2.330 | 6 | 5.29 × 108 | 73.74 | Extracellular |
| GH10—Beta-xylanase | R1FWZ0 | −3.1 | 3.513 | 14 | 2.31 × 1010 | 323.31 | Extracellular |
| GH43—Putative xylosidase: arabinofuranosidase protein | R1G299 | −2.1 | 3.289 | 6 | 3.79 × 108 | 140.27 | Extracellular |
| GH43—Putative xylosidase glycosyl hydrolase protein | R1G5Y4 | −1.9 | 3.227 | 13 | 3.51 × 1010 | 323.31 | Extracellular |
| GH27—Alpha-galactosidase | R1G8C1 | −2.6 | 4.883 | 12 | 1.8 × 1010 | 286.19 | Extracellular NN h (0.861) |
| GH43—Putative galactan-beta-galactosidase protein | R1GG59 | −5.5 | 3.216 | 14 | 3.67 × 109 | 323.31 | Extracellular |
| GH43—Arabinan endo-1,5-alpha-L-arabinosidase | R1GAB3 | −6.4 | 5.780 | 10 | 4.97 × 109 | 117.45 | Extracellular |
| GH51—Putative alpha-l-arabinofuranosidase a protein | R1EVS4 | −3.1 | 3.133 | 10 | 7.22 × 108 | 190.73 | Extracellular |
| CE5—Putative acetylxylan esterase protein | R1EWW2 | −2.3 | 1.059 | 2 | 1.54 × 109 | 323.31 | Extracellular NN h (0.898) |
| GH11—Endo-1,4-beta-xylanase | R1GCT8 | −2.4 | 1.534 | 7 | 3.41 × 108 | 144.76 | Extracellular |
| GH43/CBM6—Putative glycosyl hydrolase family 43 protein | R1GE80 | −2.2 | 3.527 | 16 | 8.42 × 109 | 307.72 | Extracellular |
| Lignin degradation | |||||||
| AA5—Putative glyoxal oxidase protein | R1EDI4 | 2.1 | 2.596 | 10 | 1.32 × 109 | 86.505 | Extracellular |
| AA1—Putative laccase-1 protein | R1G4L9 | 1.9 | 3.262 | 11 | 1.03 × 1010 | 227.45 | Extracellular |
| AA7—Putative FAD-dependent oxidoreductase protein | R1FVT8 | 2.2 | 1.428 | 14 | 9.82 × 108 | 123.3 | Extracellular |
| AA3—Putative alcohol dehydrogenase protein | R1EH41 | −2.7 | 2.253 | 3 | 2.58 × 108 | 35.849 | Extracellular NN h (0.648) |
| Lignin/celulose degradation | |||||||
| AA3/CBM1—Putative cellobiose dehydrogenase protein | R1H3M7 | 2 | 1.856 | 16 | 1.72 × 109 | 157.89 | Extracellular |
| AA3—Putative GMC oxidoreductase protein | R1FVG2 | 1.8 | 3.233 | 23 | 5.79 × 1010 | 323.31 | Extracellular NN h (0.655) |
| Pectin degradation | |||||||
| GH53—Arabinogalactan endo-beta-1,4-galactanase | R1G7Y3 | −7 | 3.424 | 9 | 6.29 × 109 | 161.62 | Extracellular |
| CE12—Putative rhamnogalacturonan acetylesterase protein | R1GFP8 | −6.4 | 4.594 | 9 | 9.04 × 109 | 155.37 | Extracellular |
| GH53—Arabinogalactan endo-beta-1,4-galactanase | R1GVP5 | −2.3 | 2.391 | 5 | 5.89 × 108 | 56.599 | Extracellular |
| PL3—Putative pectate lyase protein | R1EWA7 | −6.6 | 3.224 | 9 | 6.54 × 109 | 297.03 | Extracellular |
| PL3—Putative pectate lyase protein | R1GN84 | −6.2 | 4.605 | 6 | 4.57 × 109 | 103.84 | Extracellular |
| PL1—Putative pectate lyase a protein | R1GII6 | −4.4 | 4.962 | 13 | 1.75 × 1010 | 323.31 | Extracellular |
| PL4—Putative rhamnogalacturonan lyase protein | R1GJ02 | −5.5 | 4.999 | 18 | 3.48 × 109 | 227.89 | Extracellular |
| PL1—Putative pectate protein | R1GSQ1 | −4.8 | 3.712 | 5 | 1.4 × 109 | 91.554 | Extracellular NN h (0.592) |
| PL3—Putative exo-beta-protein | R1H382 | −2.2 | 2.235 | 24 | 1.8 × 1011 | 323.31 | Extracellular NN h (0.798) |
| GH28—Putative extracellular exo-protein | R1GW72 | −3.2 | 3.359 | 5 | 8.03 × 108 | 57.212 | Extracellular |
| PL4—Rhamnogalacturonate lyase | R1EPI5 | −2 | 2.077 | 6 | 4.03 × 108 | 107.86 | Extracellular |
| PL4—Rhamnogalacturonate lyase | R1GGA5 | −7.6 | 4.898 | 23 | 1.38 × 1010 | 323.31 | Extracellular |
| Chitin degradation | |||||||
| CE4—Putative chitin deacetylase protein | R1E7G7 | −5.6 | 2.993 | 6 | 3.73 × 109 | 53.089 | Extracellular |
| GH75—Endo-chitosanase | R1GTL6 | −4.1 | 1.099 | 4 | 4.37 × 109 | 59.996 | Extracellular |
| Other CAZY | |||||||
| GH16—Putative glycoside hydrolase family 16 protein | R1EVI7 | −2.7 | 2.462 | 3 | 2.12 × 109 | 34.095 | Extracellular |
| Esterase | |||||||
| Carboxylic ester hydrolase | R1GKX8 | 2.7 | 4.271 | 7 | 5.44 × 108 | 70.895 | Extracellular |
| Putative GDSL-like lipase acylhydrolase protein | R1E852 | −4.1 | 2.117 | 6 | 3.13 × 109 | 323.31 | Extracellular |
| Carboxylic ester hydrolase | R1E8C5 | −2.8 | 2.209 | 9 | 7.04 × 108 | 79.717 | Extracellular |
| Putative GDSL-like lipase acylhydrolase protein | R1GK66 | −2.6 | 3.165 | 6 | 4.2 × 108 | 40.614 | Extracellular |
| Carboxylic ester hydrolase | R1GSL8 | −2.1 | 2.550 | 6 | 1.91 × 108 | 83.343 | Extracellular |
| Putative carboxylesterase protein | R1EIK3 | −3.7 | 1.530 | 4 | 7.19 × 108 | 37.138 | Extracellular NN h (0.768) |
| Carboxylic ester hydrolase | R1G8E3 | −5.8 | 3.503 | 9 | 2.21 × 109 | 171.03 | Extracellular |
| Putative carboxylesterase family protein | R1G9C5 | −2.1 | 3.136 | 5 | 1.71 × 108 | 39.971 | Extracellular |
| Putative GDSL lipase acylhydrolase family protein | R1EIF4 | −1.8 | 2.818 | 6 | 3.13 × 109 | 134.94 | Extracellular NN h (0.756) |
| Carboxylic ester hydrolase/tannase family | R1GJW0 | −1.9 | 3.095 | 20 | 4.01 × 109 | 323.31 | Extracellular |
| Protease | |||||||
| Peptidase S1 family—putative carboxypeptidase S1 protein | R1FV38 | 1.9 | 2.351 | 7 | 4.06 × 109 | 175.54 | Extracellular |
| Peptidase S8 family—putative peptidase S8 S53 subtilisin kexin sedolisin protein | R1EAW3 | 2.2 | 1.289 | 5 | 1.54 × 109 | 157.52 | Extracellular |
| Peptidase A1 family—Putative aspartic endopeptidase PEP1 protein | R1GM42 | −3.2 | 4.661 | 4 | 4.3 × 109 | 98.628 | Extracellular |
| Peptidase M43—Putative metalloprotease protein | R1FXE7 | −5.1 | 3.311 | 5 | 3.6 × 109 | 134.74 | Extracellular |
| Peptidase M28 family—peptide hydrolase | R1GBR8 | −2.7 | 2.039 | 6 | 1.35 × 109 | 209.59 | Extracellular |
| Peptidase M35 family—neutral protease 2 | R1EL46 | −2.3 | 0.943 | 5 | 1.62 × 109 | 102.51 | Extracellular |
| Oxidoreductase | |||||||
| Putative FMN-dependent dehydrogenase protein | R1E6X7 | 2.9 | 1.290 | 16 | 6.56 × 108 | 127.23 | Extracellular |
| Putative FAD-binding domain-containing protein | R1E8E1 | 3.6 | 3.973 | 11 | 4.38 × 109 | 264.43 | Extracellular |
| Putative cyclohexanone monooxygenase protein | R1EF40 | −3.7 | 1.628 | 2 | 9.32 × 109 | 20.921 | Extracellular |
| Putative tyrosinase central domain protein | R1ERX8 | −2.4 | 2.164 | 9 | 8.84 × 108 | 90.821 | Extracellular NN h (0.817) |
| Putative FAD FMN-containing dehydrogenase protein | R1GB06 | −3.4 | 3.369 | 16 | 9.58 × 108 | 192.5 | Extracellular |
| Putative berberine-like protein | R1GD68 | −5 | 2.241 | 13 | 2.88 × 109 | 323.31 | Extracellular |
| Putative GMC protein | R1ELQ0 | −2.1 | 0.517 | 6 | 5.13 × 109 | 40.919 | Extracellular |
| Lyase | |||||||
| Putative pectate lyase protein | R1H2U7 | −2.3 | 3.013 | 4 | 2.56 × 108 | 28.73 | Extracellular |
| Putative-secreted protein | R1GFS9 | −3 | 3.218 | 20 | 2.59 × 1011 | 323.31 | Extracellular |
| Putative pectate lyase protein | R1G436 | −5.7 | 4.498 | 17 | 3.09 × 109 | 217.8 | Extracellular |
| Uncharacterized protein | R1GU06 | −1.9 | 2.289 | 7 | 2.48 × 1010 | 323.31 | Extracellular |
| Protein–protein interaction | |||||||
| Putative six-bladed beta-propeller-like protein | R1ENG6 | 2.3 | 2.942 | 3 | 4.52 × 108 | 36.528 | Extracellular |
| Putative six-bladed beta-propeller-like protein | R1E9S0 | −2.3 | 1.704 | 2 | 4.15 × 108 | 21.736 | Extracellular |
| Putative SMP-30 gluconolaconase LRE-like region protein | R1GCJ5 | −2.1 | 0.903 | 5 | 1.13 × 109 | 92.83 | Extracellular NN h (0.754) |
| Carbohydrate binding | |||||||
| Putative alpha-mannosidase family protein | R1EYI5 | 1.8 | 1.196 | 2 | 6.81 × 108 | 23.805 | Extracellular |
| Putative ricin B lectin protein | R1GAK8 | −4.3 | 2.336 | 6 | 1.66 × 109 | 97.147 | Extracellular |
| RNA binding | |||||||
| Putative ribonuclease T2 protein | R1ERG2 | 2.8 | 2.404 | 2 | 5.88 × 108 | 62.528 | Extracellular |
| Uncharacterized protein | R1FZX2 | −4.1 | 1.575 | 6 | 1.9 × 109 | 43.942 | Extracellular |
| Putative extracellular guanyl-specific ribonuclease protein | R1H1L9 | −2.1 | 0.586 | 3 | 4.24 × 109 | 48.559 | Extracellular |
| Other function | |||||||
| Putative allergen V5 Tpx-1-related protein | R1EAF3 | 2.2 | 3.639 | 5 | 5.58 × 109 | 181.15 | Extracellular |
| Putative ethanolamine utilization protein | R1G1U2 | 2 | 2.760 | 5 | 4.1 × 108 | 54.96 | Extracellular NN h (0.223) |
| Putative ABC-type Fe3+ transport system protein | R1FV21 | 2.9 | 1.484 | 6 | 4.15 × 108 | 58.409 | Extracellular |
| Putative major royal jelly protein | R1FVG4 | 2.7 | 1.896 | 15 | 5.75 × 1010 | 323.31 | Extracellular |
| Putative ABC-type Fe3+ transport system protein | R1GBA7 | 2.8 | 1.611 | 15 | 7.33 × 1010 | 323.31 | Extracellular |
| Putative alpha beta hydrolase protein | R1EGT1 | 2.7 | 3.692 | 11 | 3.31 × 109 | 118.1 | Extracellular |
| Putative glutaminase protein | R1GV87 | 2.7 | 3.603 | 10 | 1.15 × 109 | 215.65 | Extracellular |
| Putative fasciclin domain family protein | R1EWZ5 | −2 | 2.878 | 12 | 2.37 × 109 | 129.86 | Extracellular |
| Uncharacterized protein | R1GDV3 | −4.1 | 2.108 | 5 | 3.67 × 1010 | 323.31 | Extracellular |
| Putative BNR Asp-box repeat domain protein | R1GKT0 | −2.2 | 2.950 | 11 | 2.45 × 1010 | 323.31 | Extracellular |
| Putative extracellular aldonolactonase protein | R1E681 | −1.8 | 0.892 | 5 | 2.15 × 109 | 183.99 | Extracellular |
| Unknown | |||||||
| Putative extracellular serine-threonine rich protein | R1E9T1 | 2.9 | 3.242 | 3 | 8.48 × 108 | 78.551 | Extracellular |
| Putative membrane-spanning 4-domains subfamily a member 14 protein | R1EE60 | 2.9 | 3.242 | 3 | 8.48 × 108 | 78.551 | Extracellular |
| Uncharacterized protein | R1EBL8 | 2.1 | 3.282 | 11 | 1.26 × 1010 | 323.31 | Extracellular |
| Putative GPI anchored cell wall protein | R1G7D5 | 2.2 | 1.914 | 4 | 1.35 × 109 | 24.812 | Extracellular |
| Uncharacterized protein | R1GMX5 | 2.3 | 3.894 | 6 | 2.11 × 1010 | 185.31 | Extracellular |
| Uncharacterized protein | R1GRM4 | 2.4 | 1.314 | 4 | 7.62 × 108 | 37.391 | Extracellular |
| Uncharacterized protein | R1G5W7 | 2.1 | 0.681 | 2 | 8.78 × 108 | 20.89 | Extracellular |
| Uncharacterized protein | R1ESR7 | −4 | 1.737 | 3 | 1.4 × 1010 | 48.44 | Extracellular |
| Putative-secreted protein | R1G8U3 | −3.4 | 3.421 | 6 | 4.85 × 108 | 49.633 | Extracellular |
| Uncharacterized protein | R1GYB0 | −5.3 | 1.742 | 7 | 6.21 × 109 | 132.23 | Extracellular |
| Putative GPI anchored cell wall protein | R1ENT4 | −2.4 | 0.948 | 4 | 1.24 × 109 | 40.251 | Extracellular |
| Putative 34-dihydroxy-2-butanone 4-phosphate synthase protein | R1EY60 | −2.1 | 1.093 | 2 | 3.23 × 108 | 44.267 | Extracellular |
| Uncharacterized protein | R1GLY2 | −2.1 | 1.562 | 6 | 3.69 × 108 | 67.175 | Extracellular |
| Putative exo-beta-glucanase protein | R1G5R2 | −2.6 | 1.282 | 7 | 1.19 × 1011 | 323.31 | Extracellular |
| Protein Name | Accession Number | Degree | Organism |
|---|---|---|---|
| Putative gmc protein | R1ELQ0 | 419 | Neofusicoccum parvum (strain UCR-NP2) |
| Uncharacterized protein | R1G5W7 | 406 | Neofusicoccum parvum (strain UCR-NP2) |
| Uncharacterized protein | R1FZX2 | 367 | Neofusicoccum parvum (strain UCR-NP2) |
| Putative metalloprotease protein | R1FXE7 | 258 | Neofusicoccum parvum (strain UCR-NP2) |
| Putative alpha-mannosidase family | R1EYI5 | 225 | Neofusicoccum parvum (strain UCR-NP2) |
| Putative cyclohexanone monooxygenase | R1EF40 | 166 | Neofusicoccum parvum (strain UCR-NP2) |
| Putative GDSL-like lipase acylhydrolase | R1GK66 | 154 | Neofusicoccum parvum (strain UCR-NP2) |
| Putative alcohol dehydrogenase protein | R1EH41 | 117 | Neofusicoccum parvum (strain UCR-NP2) |
| Endo-chitosanase | R1GTL6 | 69 | Neofusicoccum parvum (strain UCR-NP2) |
| Uncharacterized protein | R1ESR7 | 69 | Neofusicoccum parvum (strain UCR-NP2) |
| Auxin response factor | A0A059ACB3 | 33 | Eucalyptus grandis |
| Histone H3 | A0A059AF37 | 28 | Eucalyptus grandis |
| Histone H3 | A0A059BQE5 | 20 | Eucalyptus grandis |
| Protein kinase domain-containing protein | A0A059CUY0 | 19 | Eucalyptus grandis |
| HATPase_c domain-containing protein | A0A059DD44 | 17 | Eucalyptus grandis |
| Glyco_transf_20 domain-containing protein | A0A059CZ70 | 17 | Eucalyptus grandis |
| Uncharacterized protein | A0A059CUY2 | 17 | Eucalyptus grandis |
| Protein kinase domain-containing protein | A0A059CBV7 | 16 | Eucalyptus grandis |
| ERCC4 domain-containing protein | A0A059C0I5 | 16 | Eucalyptus grandis |
| Na_H_Exchanger domain-containing protein | A0A059DJ06 | 15 | Eucalyptus grandis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nazar Pour, F.; Pedrosa, B.; Oliveira, M.; Fidalgo, C.; Devreese, B.; Driessche, G.V.; Félix, C.; Rosa, N.; Alves, A.; Duarte, A.S.; et al. Unveiling the Secretome of the Fungal Plant Pathogen Neofusicoccum parvum Induced by In Vitro Host Mimicry. J. Fungi 2022, 8, 971. https://doi.org/10.3390/jof8090971
Nazar Pour F, Pedrosa B, Oliveira M, Fidalgo C, Devreese B, Driessche GV, Félix C, Rosa N, Alves A, Duarte AS, et al. Unveiling the Secretome of the Fungal Plant Pathogen Neofusicoccum parvum Induced by In Vitro Host Mimicry. Journal of Fungi. 2022; 8(9):971. https://doi.org/10.3390/jof8090971
Chicago/Turabian StyleNazar Pour, Forough, Bruna Pedrosa, Micaela Oliveira, Cátia Fidalgo, Bart Devreese, Gonzalez Van Driessche, Carina Félix, Nuno Rosa, Artur Alves, Ana Sofia Duarte, and et al. 2022. "Unveiling the Secretome of the Fungal Plant Pathogen Neofusicoccum parvum Induced by In Vitro Host Mimicry" Journal of Fungi 8, no. 9: 971. https://doi.org/10.3390/jof8090971
APA StyleNazar Pour, F., Pedrosa, B., Oliveira, M., Fidalgo, C., Devreese, B., Driessche, G. V., Félix, C., Rosa, N., Alves, A., Duarte, A. S., & Esteves, A. C. (2022). Unveiling the Secretome of the Fungal Plant Pathogen Neofusicoccum parvum Induced by In Vitro Host Mimicry. Journal of Fungi, 8(9), 971. https://doi.org/10.3390/jof8090971