The Significance of Lipids to Biofilm Formation in Candida albicans: An Emerging Perspective

Abstract
:1. Introduction
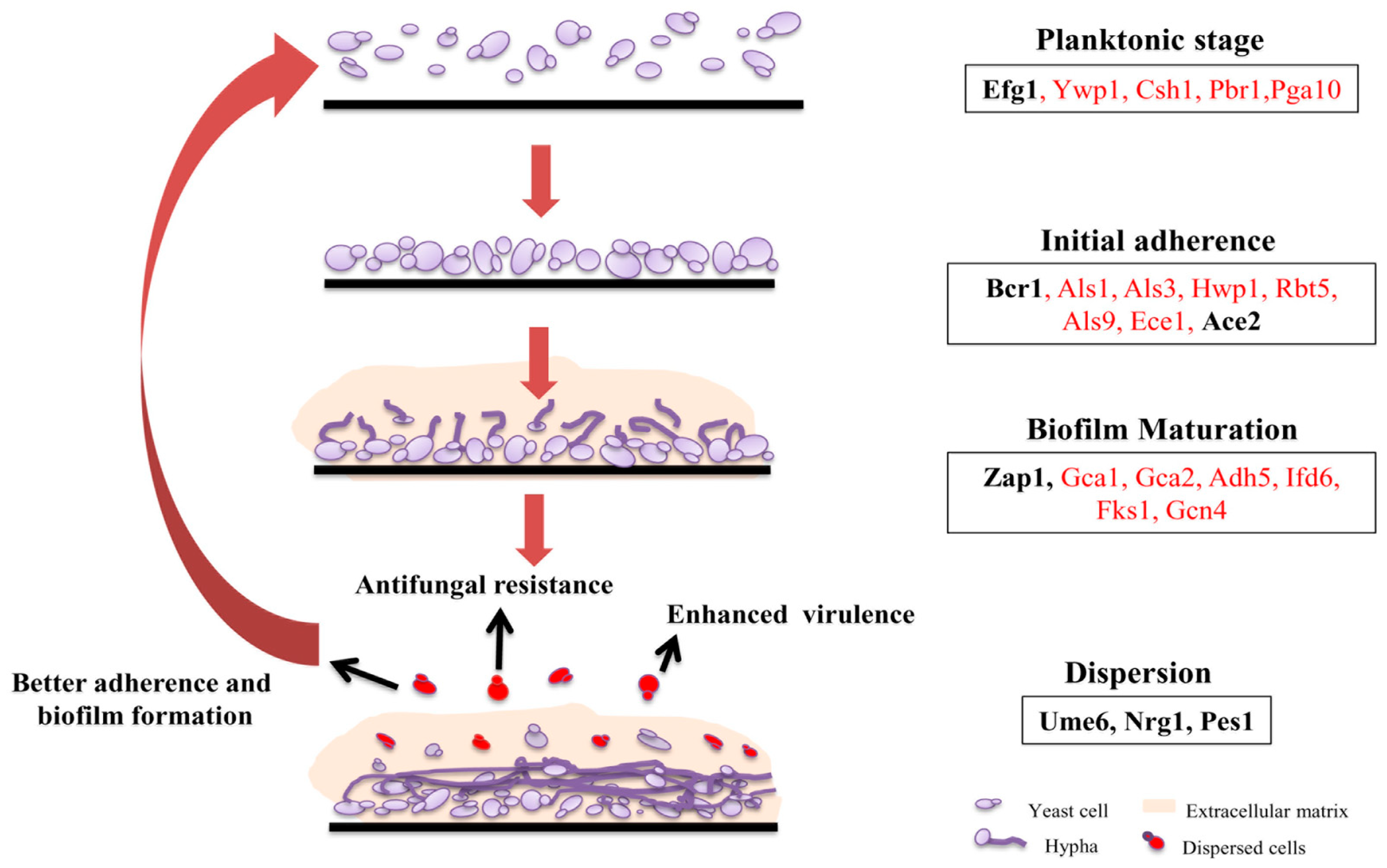
2. Biofilm Architecture, Development, and Regulation in C. albicans
2.1. Adherence
2.2. Formation of Hyphal Cells
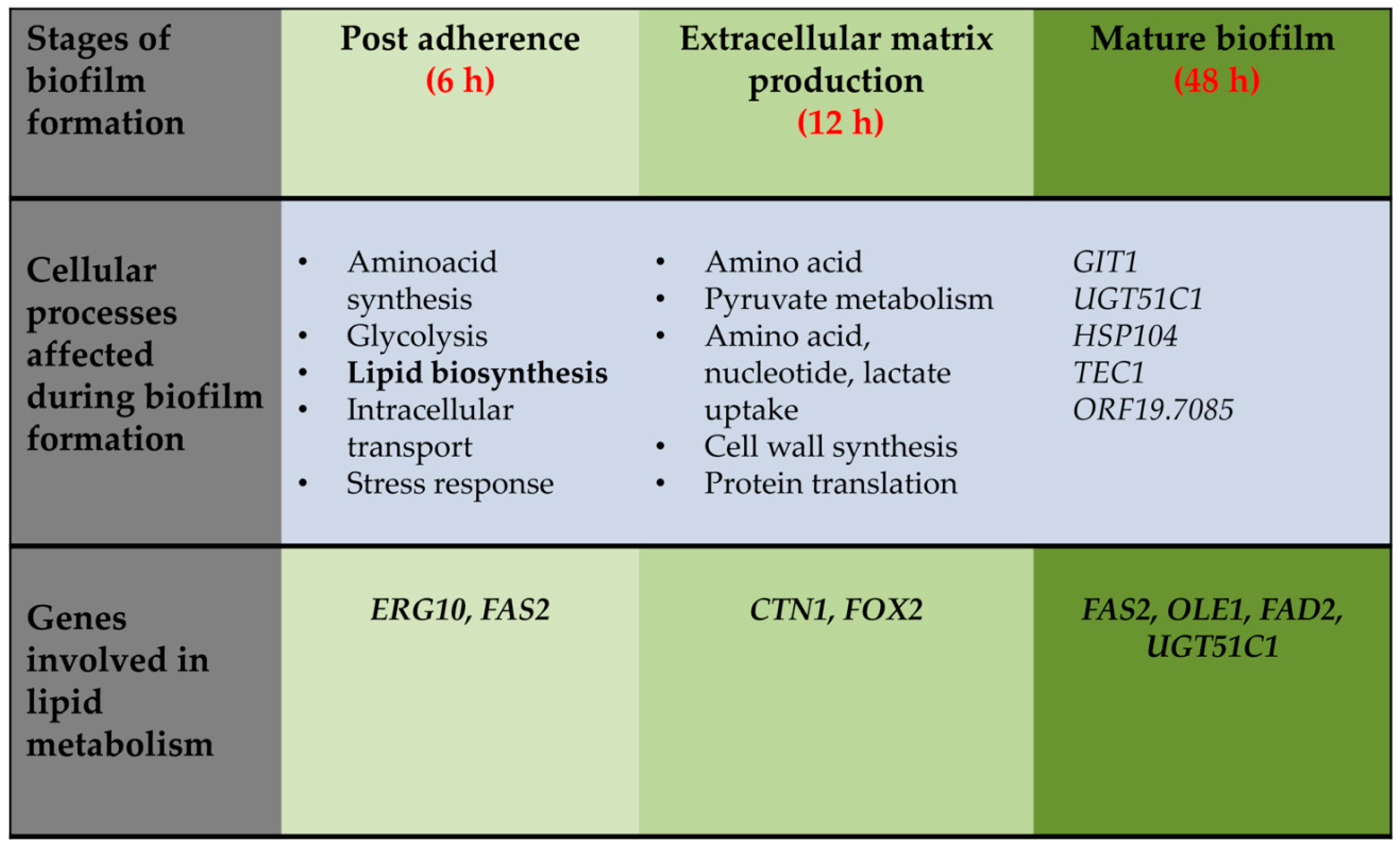
2.3. Extracellular Matrix Production
2.4. Dispersion
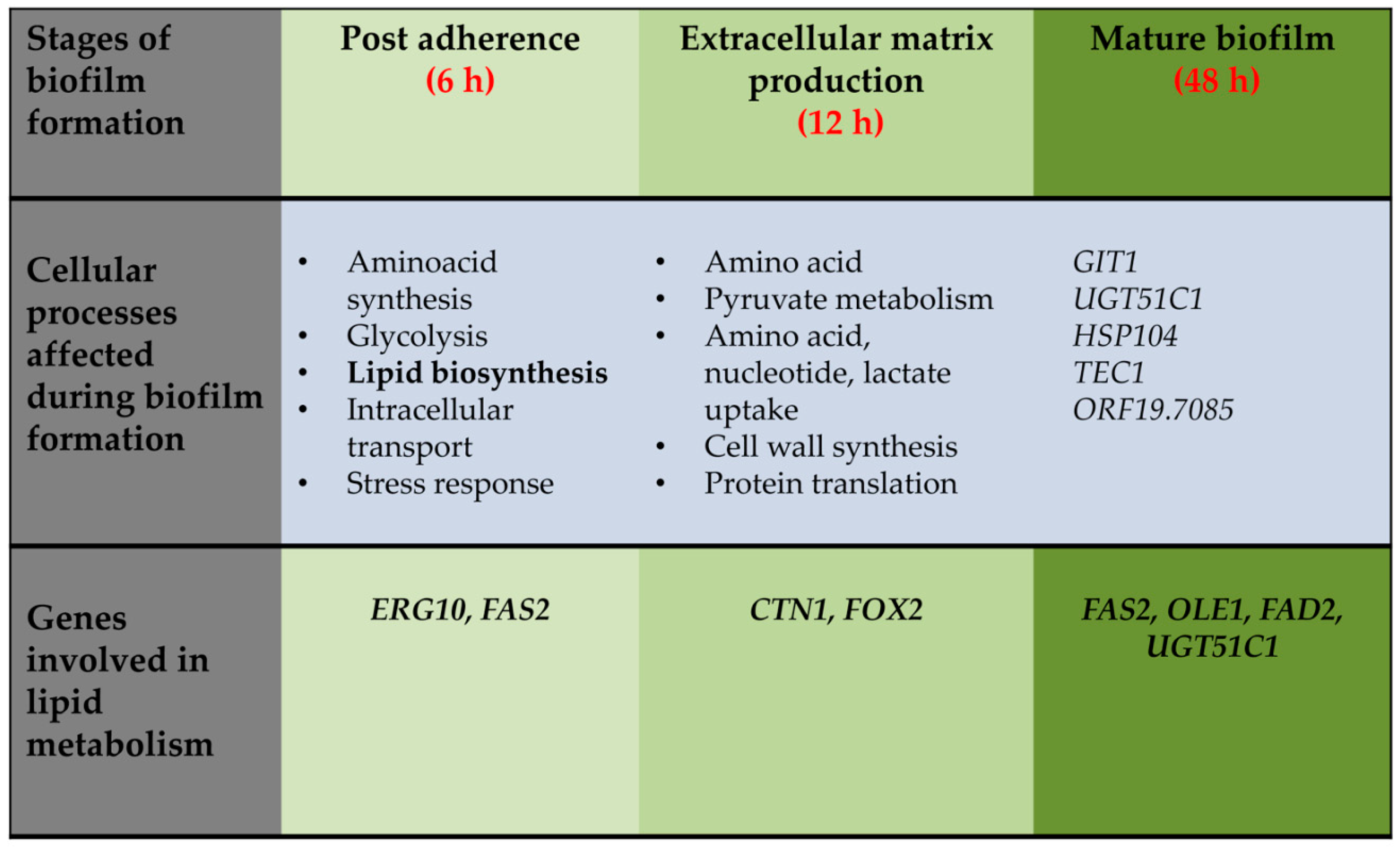
3. Structural and Functional Contribution of Lipids to Biofilm Formation
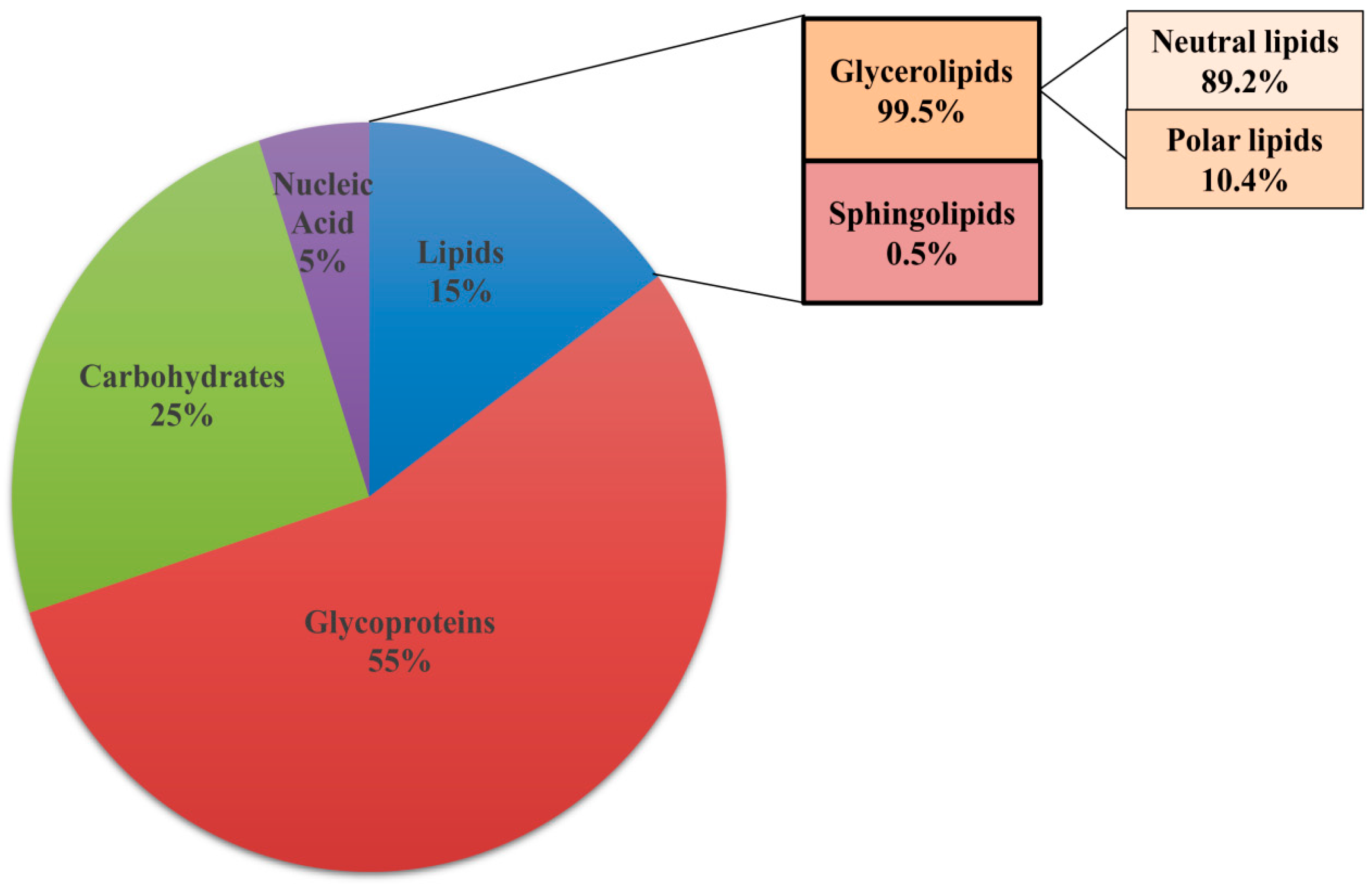
3.1. Lipids are Constituents of Fungal Membrane
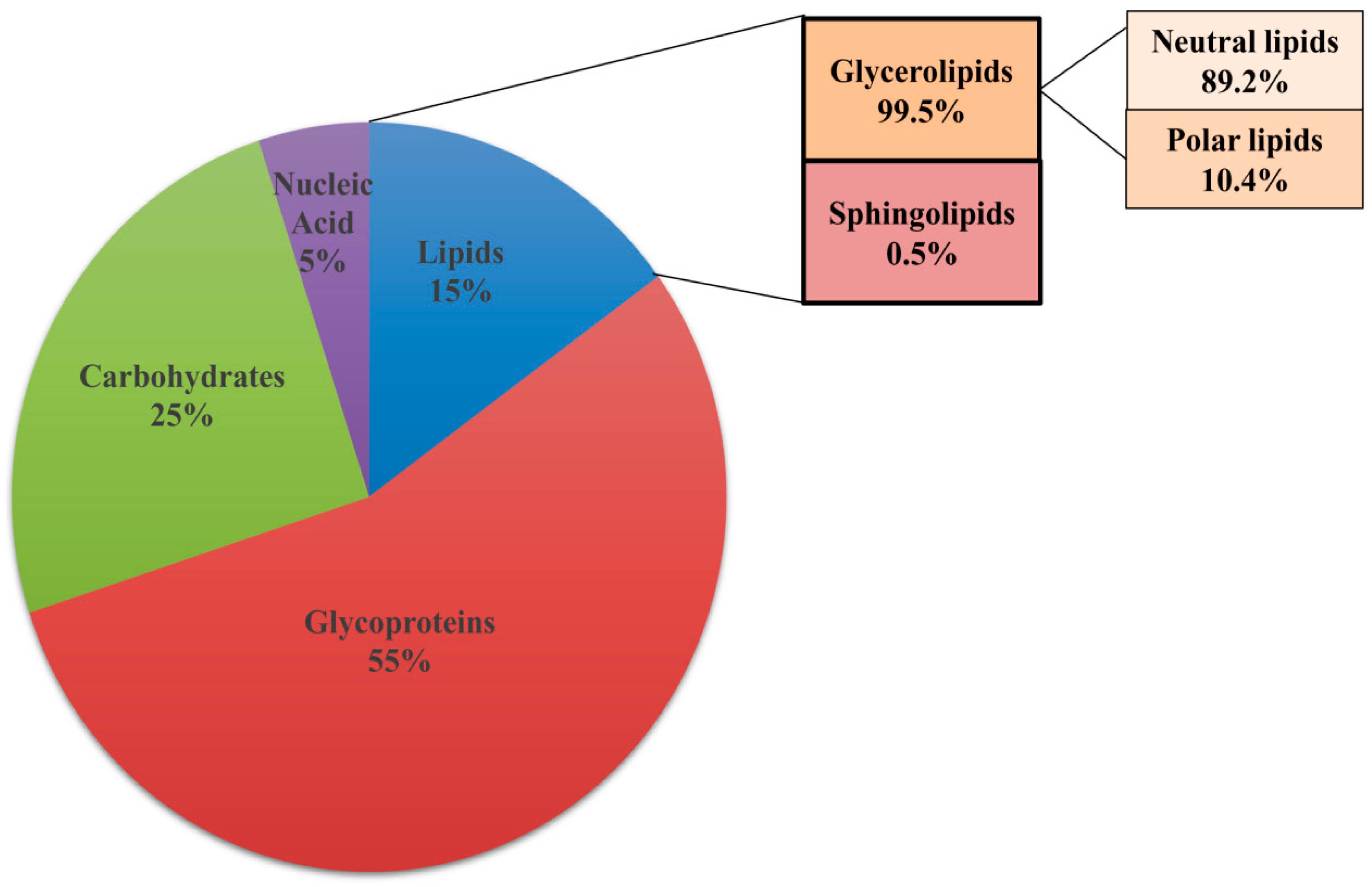
3.2. Lipids Are Constituents of Extracellular Matrix
3.3. Lipids Facilitate Formation of Lipid Rafts
3.4. Lipid Signaling Modulates Biofilm Formation
4. Lipids Influence Clinically-Relevant Traits Associated with Biofilms
4.1. Role of Lipids in Antifungal Drug Resistance
4.2. Role of Lipids in Mixed-Species Biofilm Formation
5. Targeting Lipid Biosynthesis Impedes Biofilm Formation
6. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Percival, S.L.; Malic, S.; Cruz, H.; Williams, D.W. Introduction to biofilms. In Biofilms and Veterinary Medicine; Springer: Berlin/Heidelberg, Germany, 2011; pp. 41–68. [Google Scholar]
- Harty, D.W.; Handley, P.S. Expression of the surface properties of the fibrillar Streptococcus salivarius HB and its adhesion deficient mutants grown in continuous culture under glucose limitation. Microbiology 1989, 135, 2611–2621. [Google Scholar] [CrossRef] [PubMed]
- Allewell, N.M. Introduction to biofilms thematic minireview series. J. Biol. Chem. 2016, 291, 12527–12528. [Google Scholar] [CrossRef] [PubMed]
- Fanning, S.; Mitchell, A.P. Fungal biofilms. PLoS Pathog. 2012, 8, e1002585. [Google Scholar] [CrossRef] [PubMed]
- Kojic, E.M.; Darouiche, R.O. Candida Infections of Medical Devices. Clin. Microbiol. Rev. 2004, 17, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.B.; Gulati, M.; Johnson, A.D.; Nobile, C.J. Development and regulation of single-and multi-species Candida albicans biofilms. Nat. Rev. Microbiol. 2018, 16, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Rella, A.; Farnoud, A.M.; Del Poeta, M. Plasma membrane lipids and their role in fungal virulence. Prog. Lipid Res. 2016, 61, 63–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowlett, V.W.; Mallampalli, V.K.; Karlstaedt, A.; Dowhan, W.; Taegtmeyer, H.; Margolin, W.; Vitrac, H. The impact of membrane phospholipid alterations in Escherichia coli on cellular function and bacterial stress adaptation. J. Bacteriol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Hawser, S.P.; Douglas, L.J. Biofilm formation by Candida species on the surface of catheter materials in vitro. Infect. Immun. 1994, 62, 915–921. [Google Scholar] [PubMed]
- Chandra, J.; Kuhn, D.M.; Mukherjee, P.K.; Hoyer, L.L.; McCormick, T.; Ghannoum, M.A. Biofilm formation by the fungal pathogen Candida albicans: Development, architecture, and drug resistance. J. Bacteriol. 2001, 183, 5385–5394. [Google Scholar] [CrossRef] [PubMed]
- Baillie, G.S.; Douglas, L.J. Role of dimorphism in the development of Candida albicans biofilms. J. Med. Microbiol. 1999, 48, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Ramage, G.; Vandewalle, K.; Wickes, B.L.; López-Ribot, J.L. Characteristics of biofilm formation by Candida albicans. Rev. Iberoam. Micol. 2001, 18, 163–170. [Google Scholar] [PubMed]
- Douglas, L.J. Candida biofilms and their role in infection. Trends Microbiol. 2003, 11, 30–36. [Google Scholar] [CrossRef]
- Nobile, C.J.; Mitchell, A.P. Genetics and genomics of Candida albicans biofilm formation. Cell. Microbiol. 2006, 8, 1382–1391. [Google Scholar] [CrossRef] [PubMed]
- Zarnowski, R.; Westler, W.M.; Lacmbouh, G.A.; Marita, J.M.; Bothe, J.R.; Bernhardt, J.; Sahraoui, A.L.-H.; Fontaine, J.; Sanchez, H.; Hatfield, R.D. Novel entries in a fungal biofilm matrix encyclopedia. MBio 2014, 5, e01333-14. [Google Scholar] [CrossRef] [PubMed]
- Uppuluri, P.; Chaturvedi, A.K.; Srinivasan, A.; Banerjee, M.; Ramasubramaniam, A.K.; Köhler, J.R.; Kadosh, D.; Lopez-Ribot, J.L. Dispersion as an important step in the Candida albicans biofilm developmental cycle. PLoS Pathog. 2010, 6, e1000828. [Google Scholar] [CrossRef] [PubMed]
- Nobile, C.J.; Fox, E.P.; Nett, J.E.; Sorrells, T.R.; Mitrovich, Q.M.; Hernday, A.D.; Tuch, B.B.; Andes, D.R.; Johnson, A.D. A recently evolved transcriptional network controls biofilm development in Candida albicans. Cell 2012, 148, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Andes, D.; Nett, J.; Oschel, P.; Albrecht, R.; Marchillo, K.; Pitula, A. Development and characterization of an in vivo central venous catheter Candida albicans biofilm model. Infect. Immun. 2004, 72, 6023–6031. [Google Scholar] [CrossRef] [PubMed]
- Nett, J.E.; Marchillo, K.; Spiegel, C.A.; Andes, D.R. Development and Validation of an In Vivo Candida albicans Biofilm Denture Model. Infect. Immun. 2010, 78, 3650–3659. [Google Scholar] [CrossRef] [PubMed]
- Nett, J.; Andes, D. Candida albicans biofilm development, modeling a host–pathogen interaction. Curr. Opin. Microbiol. 2006, 9, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Fox, E.P.; Bui, C.K.; Nett, J.E.; Hartooni, N.; Mui, M.C.; Andes, D.R.; Nobile, C.J.; Johnson, A.D. An expanded regulatory network temporally controls C andida albicans biofilm formation: Expanded biofilm regulatory network. Mol. Microbiol. 2015, 96, 1226–1239. [Google Scholar] [CrossRef] [PubMed]
- Frade, J.P.; Arthington-Skaggs, B.A. Effect of serum and surface characteristics on Candida albicans biofilm formation. Mycoses 2011, 54, e154–e162. [Google Scholar] [CrossRef] [PubMed]
- Parsek, M.R.; Greenberg, E.P. Sociomicrobiology: The connections between quorum sensing and biofilms. Trends Microbiol. 2005, 13, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Alves, C.T.; Silva, S.; Pereira, L.; Williams, D.W.; Azeredo, J.; Henriques, M. Effect of progesterone on Candida albicans vaginal pathogenicity. Int. J. Med. Microbiol. 2014, 304, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Oh, S.-H.; Yeater, K.M.; Hoyer, L.L. Analysis of the Candida albicans Als2p and Als4p adhesins suggests the potential for compensatory function within the Als family. Microbiology 2005, 151, 1619–1630. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Svarovsky, M.J.; Karlsson, A.J.; Wagner, J.P.; Marchillo, K.; Oshel, P.; Andes, D.; Palecek, S.P. Eap1p, an adhesin that mediates Candida albicans biofilm formation in vitro and in vivo. Eukaryot. Cell 2007, 6, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Swidergall, M.; Filler, S.G. Oropharyngeal Candidiasis: Fungal Invasion and Epithelial Cell Responses. PLoS Pathog. 2017, 13, e1006056. [Google Scholar] [CrossRef] [PubMed]
- Finkel, J.S.; Xu, W.; Huang, D.; Hill, E.M.; Desai, J.V.; Woolford, C.A.; Nett, J.E.; Taff, H.; Norice, C.T.; Andes, D.R. Portrait of Candida albicans adherence regulators. PLoS Pathog. 2012, 8, e1002525. [Google Scholar] [CrossRef] [PubMed]
- Nobile, C.J.; Mitchell, A.P. Regulation of cell-surface genes and biofilm formation by the C. albicans transcription factor Bcr1p. Curr. Biol. 2005, 15, 1150–1155. [Google Scholar] [CrossRef] [PubMed]
- Sudbery, P.E. Growth of Candida albicans hyphae. Nat. Rev. Microbiol. 2011, 9, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidou, N.; Morrissey, J.P. Co-occurence of filamentation defects and impaired biofilms in Candida albicans protein kinase mutants. FEMS Yeast Res. 2015, 15, fov092. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, R.; May, A.; Sherry, L.; Kean, R.; Williams, C.; Jones, B.L.; Burgess, K.V.; Heringa, J.; Abeln, S.; Brandt, B.W. Integrating Candida albicans metabolism with biofilm heterogeneity by transcriptome mapping. Sci. Rep. 2016, 6, 35436. [Google Scholar] [CrossRef] [PubMed]
- Al-Fattani, M.A.; Douglas, L.J. Biofilm matrix of Candida albicans and Candida tropicalis: Chemical composition and role in drug resistance. J. Med. Microbiol. 2006, 55, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Nett, J.E.; Sanchez, H.; Cain, M.T.; Andes, D.R. Genetic basis of Candida biofilm resistance due to drug-sequestering matrix glucan. J. Infect. Dis. 2010, 202, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Nobile, C.J.; Nett, J.E.; Hernday, A.D.; Homann, O.R.; Deneault, J.-S.; Nantel, A.; Andes, D.R.; Johnson, A.D.; Mitchell, A.P. Biofilm matrix regulation by Candida albicans Zap1. PLoS Biol. 2009, 7, e1000133. [Google Scholar] [CrossRef] [PubMed]
- Nett, J.E.; Sanchez, H.; Cain, M.T.; Ross, K.M.; Andes, D.R. Interface of Candida albicans Biofilm Matrix-Associated Drug Resistance and Cell Wall Integrity Regulation. Eukaryot. Cell 2011. [Google Scholar] [CrossRef] [PubMed]
- Martins, M.; Uppuluri, P.; Thomas, D.P.; Cleary, I.A.; Henriques, M.; Lopez-Ribot, J.L.; Oliveira, R. Presence of extracellular DNA in the Candida albicans biofilm matrix and its contribution to biofilms. Mycopathologia 2010, 169, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Robbins, N.; Uppuluri, P.; Nett, J.; Rajendran, R.; Ramage, G.; Lopez-Ribot, J.L.; Andes, D.; Cowen, L.E. Hsp90 Governs Dispersion and Drug Resistance of Fungal Biofilms. PLoS Pathog. 2011, 7, e1002257. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, R.S.; Uppuluri, P.; Zaas, A.K.; Collins, C.; Senn, H.; Perfect, J.R.; Heitman, J.; Cowen, L.E. Hsp90 orchestrates temperature-dependent Candida albicans morphogenesis via Ras1-PKA signaling. Curr. Biol. 2009, 19, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Granger, B.L. Insight into the anti-adhesive effect of yeast wall protein 1 of Candida albicans. Eukaryot. Cell 2012. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, M.; Thompson, D.S.; Lazzell, A.; Carlisle, P.L.; Pierce, C.; Monteagudo, C.; Lopez-Ribot, J.L.; Kadosh, D. UME6, a novel filament-specific regulator of Candida albicans hyphal extension and virulence. Mol. Biol. Cell 2008, 19, 1354–1365. [Google Scholar] [CrossRef] [PubMed]
- Braun, B.R.; Kadosh, D.; Johnson, A.D. NRG1, a repressor of filamentous growth in C. albicans, is down-regulated during filament induction. EMBO J. 2001, 20, 4753–4761. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Cowen, L.E.; Griffin, A.M.; Chan, L.; Köhler, J.R. The Candida albicans pescadillo homolog is required for normal hypha-to-yeast morphogenesis and yeast proliferation. Proc. Natl. Acad. Sci. USA 2008. [Google Scholar] [CrossRef] [PubMed]
- Pierce, C.G.; Vila, T.; Romo, J.A.; Montelongo-Jauregui, D.; Wall, G.; Ramasubramanian, A.; Lopez-Ribot, J.L. The Candida albicans Biofilm matrix: Composition, structure and function. J. Fungi 2017, 3, 14. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, P.K.; Chandra, J.; Kuhn, D.M.; Ghannoum, M.A. Mechanism of fluconazole resistance in Candida albicans biofilms: Phase-specific role of efflux pumps and membrane sterols. Infect. Immun. 2003, 71, 4333–4340. [Google Scholar] [CrossRef] [PubMed]
- Hallstrom, T.C.; Lambert, L.; Schorling, S.; Balzi, E.; Goffeau, A.; Moye-Rowley, W.S. Coordinate control of sphingolipid biosynthesis and multidrug resistance in Saccharomyces cerevisiae. J. Biol. Chem. 2001, 276, 23674–23680. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.W.; Konopka, J.B. Lipid raft polarization contributes to hyphal growth in Candida albicans. Eukaryot. Cell 2004, 3, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Toulmay, A.; Schneiter, R. Lipid-dependent surface transport of the proton pumping ATPase: A model to study plasma membrane biogenesis in yeast. Biochimie 2007, 89, 249–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lattif, A.A.; Chandra, J.; Chang, J.; Liu, S.; Zhou, G.; Chance, M.R.; Ghannoum, M.A.; Mukherjee, P.K. Proteomics and pathway mapping analyses reveal phase-dependent over-expression of proteins associated with carbohydrate metabolic pathways in Candida albicans biofilms. Open Proteom. J. 2008, 1, 5–26. [Google Scholar] [CrossRef]
- Yeater, K.M.; Chandra, J.; Cheng, G.; Mukherjee, P.K.; Zhao, X.; Rodriguez-Zas, S.L.; Kwast, K.E.; Ghannoum, M.A.; Hoyer, L.L. Temporal analysis of Candida albicans gene expression during biofilm development. Microbiology 2007, 153, 2373–2385. [Google Scholar] [CrossRef] [PubMed]
- García, R.; Bermejo, C.; Grau, C.; Pérez, R.; Rodríguez-Peña, J.M.; Francois, J.; Nombela, C.; Arroyo, J. The global transcriptional response to transient cell wall damage in Saccharomyces cerevisiae and its regulation by the cell integrity signaling pathway. J. Biol. Chem. 2004, 279, 15183–15195. [Google Scholar] [CrossRef] [PubMed]
- Lattif, A.A.; Mukherjee, P.K.; Chandra, J.; Roth, M.R.; Welti, R.; Rouabhia, M.; Ghannoum, M.A. Lipidomics of Candida albicans biofilms reveals phase-dependent production of phospholipid molecular classes and role for lipid rafts in biofilm formation. Microbiology 2011, 157, 3232–3242. [Google Scholar] [CrossRef] [PubMed]
- Hitchcock, C.A.; Barrett-Bee, K.J.; Russell, N.J. The lipid composition and permeability to azole of an azole- and polyene-resistant mutant of Candida albicans. J. Med. Vet. Mycol. 1987, 25, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Vellucci, V.F.; Kyc, S.; Hostetter, M.K. Simvastatin inhibits Candida albicans biofilm in vitro. Pediatr. Res. 2009, 66, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Flemming, H.C.; Wingender, J. The biofilm matrix. Nat. Rev. Microbiol. 2010, 8, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Baillie, G.S.; Douglas, L.J. Matrix polymers of Candida biofilms and their possible role in biofilm resistance to antifungal agents. J. Antimicrob. Chemother. 2000, 46, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Zarnowski, R.; Sanchez, H.; Andes, D.R. Large-scale production and isolation of Candida biofilm extracellular matrix. Nat. Protoc. 2016, 11, 2320–2327. [Google Scholar] [CrossRef] [PubMed]
- Hornby, J.M.; Jensen, E.C.; Lisec, A.D.; Tasto, J.J.; Jahnke, B.; Shoemaker, R.; Dussault, P.; Nickerson, K.W. Quorum sensing in the dimorphic fungus Candida albicans is mediated by farnesol. Appl. Environ. Microbiol. 2001, 67, 2982–2992. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Del Poeta, M. Lipid signalling in pathogenic fungi. Cell. Microbiol. 2011, 13, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Calderone, R.A. Candida and Candidiasis, 2nd ed.; ASM Press: Washington, DC, USA, 2012; ISBN 978-1-55581-717-6. [Google Scholar]
- Ramage, G.; Saville, S.P.; Wickes, B.L.; López-Ribot, J.L. Inhibition of Candida albicans biofilm formation by farnesol, a quorum-sensing molecule. Appl. Environ. Microbiol. 2002, 68, 5459–5463. [Google Scholar] [CrossRef] [PubMed]
- Katragkou, A.; McCarthy, M.; Alexander, E.L.; Antachopoulos, C.; Meletiadis, J.; Jabra-Rizk, M.A.; Petraitis, V.; Roilides, E.; Walsh, T.J. In vitro interactions between farnesol and fluconazole, amphotericin B or micafungin against Candida albicans biofilms. J. Antimicrob. Chemother. 2014, 70, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.G.; Padilla, J.; Koumas, L.; Ray, D.; Phipps, R.P. Prostaglandins as modulators of immunity. Trends Immunol. 2002, 23, 144–150. [Google Scholar] [CrossRef]
- Noverr, M.C.; Toews, G.B.; Huffnagle, G.B. Production of prostaglandins and leukotrienes by pathogenic fungi. Infect. Immun. 2002, 70, 400–402. [Google Scholar] [CrossRef] [PubMed]
- Alem, M.A.S.; Douglas, L.J. Prostaglandin production during growth of Candida albicans biofilms. J. Med. Microbiol. 2005, 54, 1001–1005. [Google Scholar] [CrossRef] [PubMed]
- Krause, J.; Geginat, G.; Tammer, I. Prostaglandin E2 from Candida albicans stimulates the growth of Staphylococcus aureus in mixed biofilms. PLoS ONE 2015, 10, e0135404. [Google Scholar] [CrossRef] [PubMed]
- Alem, M.A.; Douglas, L.J. Effects of aspirin and other nonsteroidal anti-inflammatory drugs on biofilms and planktonic cells of Candida albicans. Antimicrob. Agents Chemother. 2004, 48, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Harriott, M.M.; Noverr, M.C. Importance of Candida–bacterial polymicrobial biofilms in disease. Trends Microbiol. 2011, 19, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Carlson, E. Synergistic effect of Candida albicans and Staphylococcus aureus on mouse mortality. Infect. Immun. 1982, 38, 921–924. [Google Scholar] [PubMed]
- Adam, B.; Baillie, G.S.; Douglas, L.J. Mixed species biofilms of Candida albicans and Staphylococcus epidermidis. J. Med. Microbiol. 2002, 51, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Taff, H.T.; Mitchell, K.F.; Edward, J.A.; Andes, D.R. Mechanisms of Candida biofilm drug resistance. Future Microbiol. 2013, 8, 1325–1337. [Google Scholar] [CrossRef] [PubMed]
- Law, D.; Moore, C.B.; Wardle, H.M.; Ganguli, L.A.; Keaney, M.G.; Denning, D.W. High prevalence of antifungal resistance in Candida spp. from patients with AIDS. J. Antimicrob. Chemother. 1994, 34, 659–668. [Google Scholar] [CrossRef] [PubMed]
- White, T.C.; Marr, K.A.; Bowden, R.A. Clinical, cellular, and molecular factors that contribute to antifungal drug resistance. Clin. Microbiol. Rev. 1998, 11, 382–402. [Google Scholar] [CrossRef] [PubMed]
- White, T.C. Increased mRNA levels of ERG16, CDR, and MDR1 correlate with increases in azole resistance in Candida albicans isolates from a patient infected with human immunodeficiency virus. Antimicrob. Agents Chemother. 1997, 41, 1482–1487. [Google Scholar] [CrossRef] [PubMed]
- Marichal, P.; Koymans, L.; Willemsens, S.; Bellens, D.; Verhasselt, P.; Luyten, W.; Borgers, M.; Ramaekers, F.C.; Odds, F.C.; Bossche, H.V. Contribution of mutations in the cytochrome P450 14α-demethylase (Erg11p, Cyp51p) to azole resistance in Candida albicans. Microbiology 1999, 145, 2701–2713. [Google Scholar] [CrossRef] [PubMed]
- Sanglard, D.; Kuchler, K.; Ischer, F.; Pagani, J.L.; Monod, M.; Bille, J. Mechanisms of resistance to azole antifungal agents in Candida albicans isolates from AIDS patients involve specific multidrug transporters. Antimicrob. Agents Chemother. 1995, 39, 2378–2386. [Google Scholar] [CrossRef] [PubMed]
- Albertson, G.D.; Niimi, M.; Cannon, R.D.; Jenkinson, H.F. Multiple efflux mechanisms are involved in Candida albicans fluconazole resistance. Antimicrob. Agents Chemother. 1996, 40, 2835–2841. [Google Scholar] [CrossRef] [PubMed]
- Sanglard, D.; Ischer, F.; Koymans, L.; Bille, J. Amino acid substitutions in the cytochrome P-450 lanosterol 14α-demethylase (CYP51A1) from azole-resistant Candida albicans clinical isolates contribute to resistance to azole antifungal agents. Antimicrob. Agents Chemother. 1998, 42, 241–253. [Google Scholar] [PubMed]
- Sanglard, D.; Ischer, F.; Monod, M.; Bille, J. Susceptibilities of Candida albicans multidrug transporter mutants to various antifungal agents and other metabolic inhibitors. Antimicrob. Agents Chemother. 1996, 40, 2300–2305. [Google Scholar] [CrossRef] [PubMed]
- Lamb, D.C.; Kelly, D.E.; Schunck, W.-H.; Shyadehi, A.Z.; Akhtar, M.; Lowe, D.J.; Baldwin, B.C.; Kelly, S.L. The mutation T315A in Candida albicans sterol 14α-demethylase causes reduced enzyme activity and fluconazole resistance through reduced affinity. J. Biol. Chem. 1997, 272, 5682–5688. [Google Scholar] [CrossRef] [PubMed]
- Kelly, S.L.; Lamb, D.C.; Corran, A.J.; Baldwin, B.C.; Kelly, D.E. Mode of Action and Resistance to Azole Antifungals Associated with the Formation of 14α-Methylergosta-8,24(28)-dien-3β,6α-diol. Biochem. Biophys. Res. Commun. 1995, 207, 910–915. [Google Scholar] [CrossRef] [PubMed]
- Lupetti, A.; Danesi, R.; Campa, M.; Tacca, M.D.; Kelly, S. Molecular basis of resistance to azole antifungals. Trends Mol. Med. 2002, 8, 76–81. [Google Scholar] [CrossRef]
- Hitchcock, C.A.; Barrett-Bee, K.J.; Russell, N.J. The lipid composition and permeability to the triazole antifungal antibiotic ICI 153066 of serum-grown mycelial cultures of Candida albicans. J. Gen. Microbiol. 1989, 135, 1949–1955. [Google Scholar] [CrossRef] [PubMed]
- García-Sánchez, S.; Aubert, S.; Iraqui, I.; Janbon, G.; Ghigo, J.-M.; d’Enfert, C. Candida albicans biofilms: A developmental state associated with specific and stable gene expression patterns. Eukaryot. Cell 2004, 3, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Nett, J.E.; Lepak, A.J.; Marchillo, K.; Andes, D.R. Time course global gene expression analysis of an in vivo Candida biofilm. J. Infect. Dis. 2009, 200, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Elias, S.; Banin, E. Multi-species biofilms: Living with friendly neighbors. FEMS Microbiol. Rev. 2012, 36, 990–1004. [Google Scholar] [CrossRef] [PubMed]
- Pulimood, S.; Ganesan, L.; Alangaden, G.; Chandrasekar, P. Polymicrobial candidemia. Diagn. Microbiol. Infect. Dis. 2002, 44, 353–357. [Google Scholar] [CrossRef]
- Klotz, S.A.; Chasin, B.S.; Powell, B.; Gaur, N.K.; Lipke, P.N. Polymicrobial bloodstream infections involving Candida species: Analysis of patients and review of the literature. Diagn. Microbiol. Infect. Dis. 2007, 59, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-H.; Yoon, Y.K.; Kim, M.J.; Sohn, J.W. Risk factors for and clinical implications of mixed Candida/bacterial bloodstream infections. Clin. Microbiol. Infect. 2013, 19, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Jack, A.A.; Daniels, D.E.; Jepson, M.A.; Vickerman, M.M.; Lamont, R.J.; Jenkinson, H.F.; Nobbs, A.H. Streptococcus gordonii comCDE (competence) operon modulates biofilm formation with Candida albicans. Microbiology 2015, 161, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Jarosz, L.M.; Deng, D.M.; van der Mei, H.C.; Crielaard, W.; Krom, B.P. Streptococcus mutans competence-stimulating peptide inhibits Candida albicans hypha formation. Eukaryot. Cell 2009, 8, 1658–1664. [Google Scholar] [CrossRef] [PubMed]
- de Repentigny, J.; Lévesque, R.; Mathieu, L.G. Increase in the in vitro susceptibility of Staphylococcus aureus to antimicrobial agents in the presence of Candida albicans. Can. J. Microbiol. 1979, 25, 429–435. [Google Scholar] [CrossRef]
- Harriott, M.M.; Noverr, M.C. Candida albicans and Staphylococcus aureus Form Polymicrobial Biofilms: Effects on Antimicrobial Resistance. Antimicrob. Agents Chemother. 2009, 53, 3914–3922. [Google Scholar] [CrossRef] [PubMed]
- Budtz-Jørgensen, E.; Mojon, P.; Banon-Clément, J.M.; Baehni, P. Oral candidosis in long-term hospital care: Comparison of edentulous and dentate subjects. Oral Dis. 1996, 2, 285–290. [Google Scholar] [CrossRef]
- AL-Dwairi, Z.N. Prevalence and risk factors associated with denture-related stomatitis in healthy subjects attending a dental teaching hospital in North Jordan. J. Ir. Dent. Assoc. 2008, 54, 80–83. [Google Scholar] [PubMed]
- Verran, J.; Motteram, K.L. The effect of adherent oral streptococci on the subsequent adherence of Candida albicans to acrylic in vitro. J. Dent. 1987, 15, 73–76. [Google Scholar] [CrossRef]
- Branting, C.; Sund, M.L.; Linder, L.E. The influence of Streptococcus mutans on adhesion of Candida albicans to acrylic surfaces in vitro. Arch. Oral Biol. 1989, 34, 347–353. [Google Scholar] [CrossRef]
- Inoue, Y.; Shiraishi, A.; Hada, T.; Hirose, K.; Hamashima, H.; Shimada, J. The antibacterial effects of terpene alcohols on Staphylococcus aureus and their mode of action. FEMS Microbiol. Lett. 2004, 237, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Sengupta, A.; Niepa, T.H.R.; Lee, B.-H.; Weljie, A.; Freitas-Blanco, V.S.; Murata, R.M.; Stebe, K.J.; Lee, D.; Koo, H. Candida albicans stimulates Streptococcus mutans microcolony development via cross-kingdom biofilm-derived metabolites. Sci. Rep. 2017, 7, 41332. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, R.A.; Monteiro, D.R.; Arias, L.S.; Fernandes, G.L.; Delbem, A.C.B.; Barbosa, D.B. Biofilm formation by Candida albicans and Streptococcus mutans in the presence of farnesol: A quantitative evaluation. Biofouling 2016, 32, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Rittershaus, P.C.; Kechichian, T.B.; Allegood, J.C.; Merrill, A.H.; Hennig, M.; Luberto, C.; Del Poeta, M. Glucosylceramide synthase is an essential regulator of pathogenicity of Cryptococcus neoformans. J. Clin. Investig. 2006, 116, 1651–1659. [Google Scholar] [CrossRef] [PubMed]
- Dickson, R.C.; Naglee, E.E.; Wells, G.B.; Naglee, M.M.; Lester, R.L. Synthesis of mannose-(inositol-P)2-ceramide, the major sphingolipid in Saccharomyces cerevisiae, requires the IPT1 (YDR072c) gene. J. Biol. Chem. 1997, 272, 29620–29625. [Google Scholar] [CrossRef] [PubMed]
- Alfatah, M.; Bari, V.K.; Nahar, A.S.; Bijlani, S.; Ganesan, K. Critical role for CaFEN1 and CaFEN12 of Candida albicans in cell wall integrity and biofilm formation. Sci. Rep. 2017, 7, 40281. [Google Scholar] [CrossRef] [PubMed]
- Cassilly, C.D.; Farmer, A.T.; Montedonico, A.E.; Smith, T.K.; Campagna, S.R.; Reynolds, T.B. Role of phosphatidylserine synthase in shaping the phospholipidome of Candida albicans. FEMS Yeast Res. 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-L.; Montedonico, A.E.; Kauffman, S.; Dunlap, J.R.; Menn, F.-M.; Reynolds, T.B. Phosphatidylserine synthase and phosphatidylserine decarboxylase are essential for cell wall integrity and virulence in Candida albicans. Mol. Microbiol. 2010, 75, 1112–1132. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, P.K.; Sheehan, D.J.; Hitchcock, C.A.; Ghannoum, M.A. Combination treatment of invasive fungal infections. Clin. Microbiol. Rev. 2005, 18, 163–194. [Google Scholar] [CrossRef] [PubMed]
- Bruzual, I.; Riggle, P.; Hadley, S.; Kumamoto, C.A. Biofilm formation by fluconazole-resistant Candida albicans strains is inhibited by fluconazole. J. Antimicrob. Chemother. 2007, 59, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Bink, A.; Kucharíková, S.; Neirinck, B.; Vleugels, J.; Van Dijck, P.; Cammue, B.P.A.; Thevissen, K. The Nonsteroidal Antiinflammatory Drug Diclofenac Potentiates the In Vivo Activity of Caspofungin Against Candida albicans Biofilms. J. Infect. Dis. 2012, 206, 1790–1797. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.S.A.; Ahmad, I. Antibiofilm activity of certain phytocompounds and their synergy with fluconazole against Candida albicans biofilms. J. Antimicrob. Chemother. 2012, 67, 618–621. [Google Scholar] [CrossRef] [PubMed]
- Seigneuret, M.; Devaux, P.F. ATP-dependent asymmetric distribution of spin-labeled phospholipids in the erythrocyte membrane: Relation to shape changes. Proc. Natl. Acad. Sci. USA 1984, 81, 3751–3755. [Google Scholar] [CrossRef] [PubMed]
- Daleke, D.L.; Huestis, W.H. Erythrocyte morphology reflects the transbilayer distribution of incorporated phospholipids. J. Cell Biol. 1989, 108, 1375–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devaux, P.F. Static and dynamic lipid asymmetry in cell membranes. Biochemistry 1991, 30, 1163–1173. [Google Scholar] [CrossRef] [PubMed]
- Zachowski, A. Phospholipids in animal eukaryotic membranes: Transverse asymmetry and movement. Biochem. J. 1993, 294, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Murate, M.; Abe, M.; Kasahara, K.; Iwabuchi, K.; Umeda, M.; Kobayashi, T. Transbilayer distribution of lipids at nano scale. J. Cell Sci. 2015, 128, 1627–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yabas, M.; Coupland, L.A.; Cromer, D.; Winterberg, M.; Teoh, N.C.; D’Rozario, J.; Kirk, K.; Bröer, S.; Parish, C.R.; Enders, A. Mice deficient in the putative phospholipid flippase ATP11C exhibit altered erythrocyte shape, anemia, and reduced erythrocyte life span. J. Biol. Chem. 2014, 289, 19531–19537. [Google Scholar] [CrossRef] [PubMed]
- Naito, T.; Takatsu, H.; Miyano, R.; Takada, N.; Nakayama, K.; Shin, H.-W. Phospholipid flippase ATP10A translocates phosphatidylcholine and is involved in plasma membrane dynamics. J. Biol. Chem. 2015. [Google Scholar] [CrossRef] [PubMed]
- Takada, N.; Naito, T.; Inoue, T.; Nakayama, K.; Takatsu, H.; Shin, H. Phospholipid-flipping activity of P4-ATPase drives membrane curvature. EMBO J. 2018, 37, e97705. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Sircaik, S.; Husain, F.; Thomas, E.; Ror, S.; Rastogi, S.; Alim, D.; Bapat, P.; Andes, D.R.; Nobile, C.J.; et al. Distinct roles of the 7-transmembrane receptor protein Rta3 in regulating the asymmetric distribution of phosphatidylcholine across the plasma membrane and biofilm formation in Candida albicans. Cell. Microbiol. 2017, 19, e12767. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Lipid Molecular Species | Planktonic Stage | Biofilm Stage | |||
|---|---|---|---|---|---|
| Early Phase | Late Phase | Early Phase | Late Phase | ||
| Phospholipids | Phosphatidylcholine | * | * | ** | ** |
| Phosphatidylethanolamine | * | * | ** | ** | |
| Phosphatidylinositol | * | * | *** | *** | |
| Phosphatidylserine | * | * | ** | ** | |
| Phosphatidic acid | * | * | ** | ** | |
| Phosphatidylglycerol | * | * | ** | ** | |
| Sphingolipids | Inositolphosphorylceramide | * | * | *** | *** |
| Mannosylinositolphosphorylceramide | * | * | *** | *** | |
| Mannosyldiinositolphosphorylceramide | * | *** | *** | nd | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alim, D.; Sircaik, S.; Panwar, S.L. The Significance of Lipids to Biofilm Formation in Candida albicans: An Emerging Perspective. J. Fungi 2018, 4, 140. https://doi.org/10.3390/jof4040140
Alim D, Sircaik S, Panwar SL. The Significance of Lipids to Biofilm Formation in Candida albicans: An Emerging Perspective. Journal of Fungi. 2018; 4(4):140. https://doi.org/10.3390/jof4040140
Chicago/Turabian StyleAlim, Darakshan, Shabnam Sircaik, and Sneh Lata Panwar. 2018. "The Significance of Lipids to Biofilm Formation in Candida albicans: An Emerging Perspective" Journal of Fungi 4, no. 4: 140. https://doi.org/10.3390/jof4040140
APA StyleAlim, D., Sircaik, S., & Panwar, S. L. (2018). The Significance of Lipids to Biofilm Formation in Candida albicans: An Emerging Perspective. Journal of Fungi, 4(4), 140. https://doi.org/10.3390/jof4040140