Tipping the Balance: C. albicans Adaptation in Polymicrobial Environments

{kind=link}
Abstract
1. Introduction
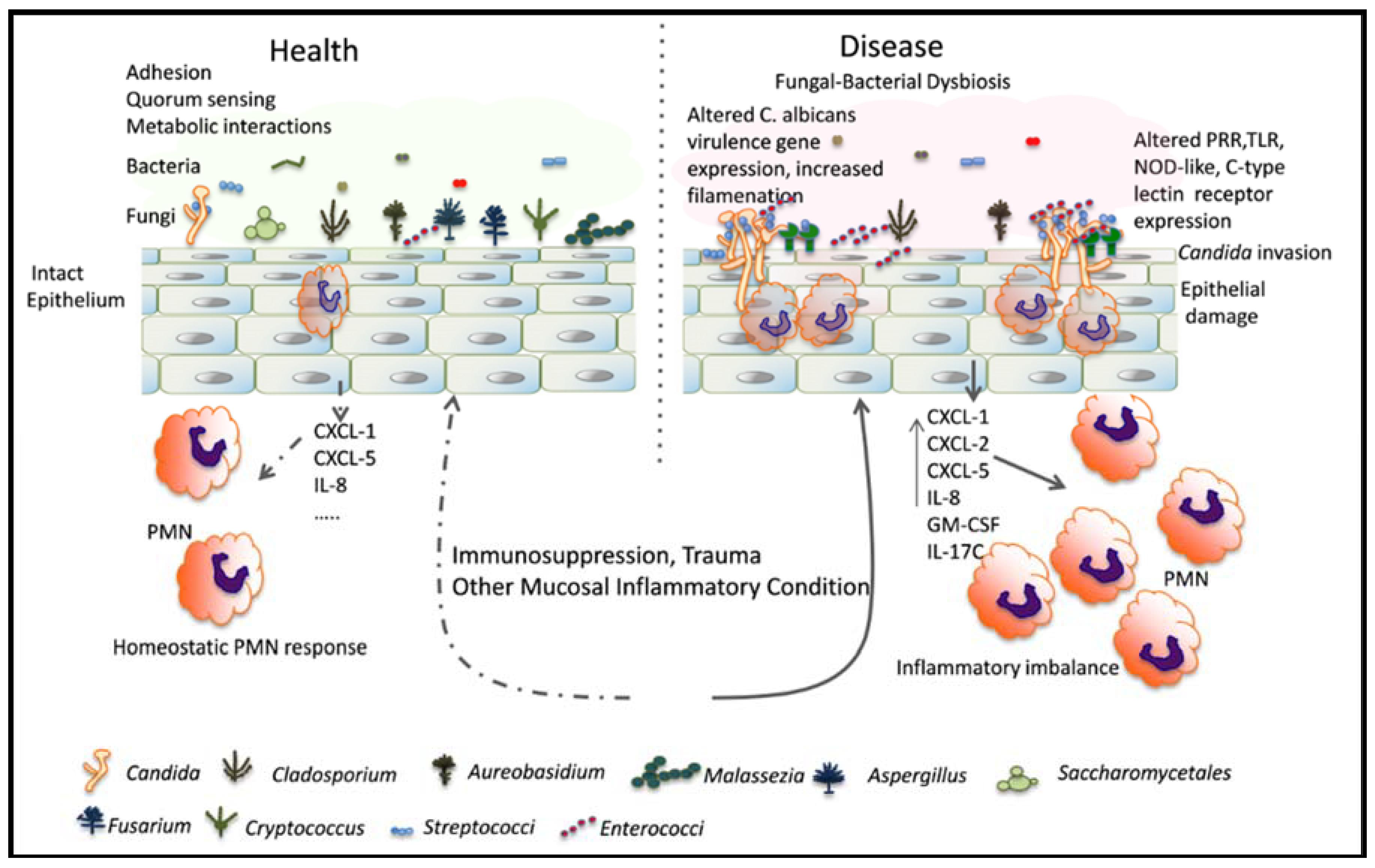
2. Mucosal Microbiota Act as an Innate Host Factor That Controls the Switch from Fungal Commensalism to Infection
2.1. Effect of Indigenous Bacteria in Mammalian Host Colonization Models
2.2. Effect of Indigenous Bacteria in Mucosal Candidiasis
3. Bacterial Signals Trigger Distinct Phenotypic Responses in C. albicans Associated with Commensalism or Virulence
3.1. Effect of Bacterial Interactions on Filamentation
3.2. Effect of Bacterial Interactions on C. albicans Metabolism, Growth, and Biofilm Formation
4. Bacteria Influence C. albicans Gene Expression and Site-Specific Adaptation in Mammalian Host Niches
5. C. albicans Has Reciprocal Effects on the Mucosal Bacterial Microbiota
6. Unanswered Questions on Fungal-Bacterial Interactions in Human Health and Disease
Author Contributions
Funding
Conflicts of Interest
References
- Richardson, J.P.; Moyes, D.L.; Ho, J.; Naglik, J.R. Candida innate immunity at the mucosa. Semin. Cell Dev. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Neville, B.A.; d’Enfert, C.; Bougnoux, M.E. Candida albicans commensalism in the gastrointestinal tract. FEMS Yeast Res. 2015, 15. [Google Scholar] [CrossRef] [PubMed]
- Cruz, M.R.; Graham, C.E.; Gagliano, B.C.; Lorenz, M.C.; Garsin, D.A. Enterococcus faecalis inhibits hyphal morphogenesis and virulence of Candida albicans. Infect. Immun. 2013, 81, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Graham, C.E.; Cruz, M.R.; Garsin, D.A.; Lorenz, M.C. Enterococcus faecalis bacteriocin entv inhibits hyphal morphogenesis, biofilm formation, and virulence of Candida albicans. Proc. Natl. Acad. Sci. USA 2017, 114, 4507–4512. [Google Scholar] [CrossRef] [PubMed]
- Tampakakis, E.; Peleg, A.Y.; Mylonakis, E. Interaction of Candida albicans with an intestinal pathogen, salmonella enterica serovar typhimurium. Eukaryot. Cell 2009, 8, 732–737. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Mylonakis, E. Killing of Candida albicans filaments by salmonella enterica serovar typhimurium is mediated by sopb effectors, parts of a type iii secretion system. Eukaryot. Cell 2011, 10, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Dutzan, N.; Abusleme, L.; Bridgeman, H.; Greenwell-Wild, T.; Zangerle-Murray, T.; Fife, M.E.; Bouladoux, N.; Linley, H.; Brenchley, L.; Wemyss, K.; et al. On-going mechanical damage from mastication drives homeostatic th17 cell responses at the oral barrier. Immunity 2017, 46, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.; Coughlin, L.A.; Neubauer, M.M.; Kim, J.; Kim, M.S.; Zhan, X.; Simms-Waldrip, T.R.; Xie, Y.; Hooper, L.V.; Koh, A.Y. Activation of hif-1alpha and ll-37 by commensal bacteria inhibits Candida albicans colonization. Nat. Med. 2015, 21, 808–814. [Google Scholar] [CrossRef] [PubMed]
- Shankar, J.; Solis, N.V.; Mounaud, S.; Szpakowski, S.; Liu, H.; Losada, L.; Nierman, W.C.; Filler, S.G. Using bayesian modelling to investigate factors governing antibiotic-induced Candida albicans colonization of the gi tract. Sci. Rep. 2015, 5, 8131. [Google Scholar] [CrossRef] [PubMed]
- Mason, K.L.; Erb Downward, J.R.; Falkowski, N.R.; Young, V.B.; Kao, J.Y.; Huffnagle, G.B. Interplay between the gastric bacterial microbiota and Candida albicans during postantibiotic recolonization and gastritis. Infect. Immun. 2012, 80, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Mason, K.L.; Erb Downward, J.R.; Mason, K.D.; Falkowski, N.R.; Eaton, K.A.; Kao, J.Y.; Young, V.B.; Huffnagle, G.B. Candida albicans and bacterial microbiota interactions in the cecum during recolonization following broad-spectrum antibiotic therapy. Infect. Immun. 2012, 80, 3371–3380. [Google Scholar] [CrossRef] [PubMed]
- Henry-Stanley, M.J.; Garni, R.M.; Alice Johnson, M.; Bendel, C.M.; Wells, C.L. Comparative abilities of Candida glabrata and Candida albicans to colonize and translocate from the intestinal tract of antibiotic-treated mice. Microb. Ecol. Health Dis. 2005, 17, 129–137. [Google Scholar] [CrossRef]
- Koh, A.Y. Murine models of Candida gastrointestinal colonization and dissemination. Eukaryot. Cell 2013, 12, 1416–1422. [Google Scholar] [CrossRef] [PubMed]
- Koh, A.Y.; Kohler, J.R.; Coggshall, K.T.; Van Rooijen, N.; Pier, G.B. Mucosal damage and neutropenia are required for Candida albicans dissemination. PLoS Pathog. 2008, 4, e35. [Google Scholar] [CrossRef] [PubMed]
- Dongari-Bagtzoglou, A.; Fidel, P.L., Jr. The host cytokine responses and protective immunity in oropharyngeal candidiasis. J. Dent. Res. 2005, 84, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Sobue, T.; Thompson, A.; Xie, Z.; Poon, K.; Ricker, A.; Cervantes, J.; Diaz, P.I.; Dongari-Bagtzoglou, A. Streptococcal co-infection augments Candida pathogenicity by amplifying the mucosal inflammatory response. Cell. Microbiol. 2014, 16, 214–231. [Google Scholar] [CrossRef] [PubMed]
- Kong, E.F.; Kucharikova, S.; Van Dijck, P.; Peters, B.M.; Shirtliff, M.E.; Jabra-Rizk, M.A. Clinical implications of oral candidiasis: Host tissue damage and disseminated bacterial disease. Infect. Immun. 2015, 83, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Peters, B.M.; Noverr, M.C. Candida albicans-Staphylococcus aureus polymicrobial peritonitis modulates host innate immunity. Infect. Immun. 2013, 81, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, A.C.; Seman, B.G.; Hammond, J.H.; Archambault, L.S.; Hogan, D.A.; Wheeler, R.T. Candida and pseudomonas interact to enhance virulence of mucosal infection in transparent zebrafish. Infect. Immun. 2017. [Google Scholar] [CrossRef] [PubMed]
- Sjovall, J.; Huitfeldt, B.; Magni, L.; Nord, C.E. Effect of beta-lactam prodrugs on human intestinal microflora. Scand. J. Infect. Dis. Suppl. 1986, 49, 73–84. [Google Scholar] [PubMed]
- Ponnuvel, K.M.; Rajkumar, R.; Menon, T.; Sankaranarayanan, V.S. Role of Candida in indirect pathogenesis of antibiotic associated diarrhoea in infants. Mycopathologia 1996, 135, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Krause, R.; Schwab, E.; Bachhiesl, D.; Daxbock, F.; Wenisch, C.; Krejs, G.J.; Reisinger, E.C. Role of Candida in antibiotic-associated diarrhea. J. Infect. Dis. 2001, 184, 1065–1069. [Google Scholar] [CrossRef] [PubMed]
- Gligorov, J.; Bastit, L.; Gervais, H.; Henni, M.; Kahila, W.; Lepille, D.; Luporsi, E.; Sasso, G.; Varette, C.; Azria, D.; et al. Prevalence and treatment management of oropharyngeal candidiasis in cancer patients: Results of the french candidoscope study. Int. J. Radiat. Oncol. Biol. Phys. 2011, 80, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.; Lilly, E.; Barousse, M.; Fidel, P.L., Jr. Epithelial cell-derived s100 calcium-binding proteins as key mediators in the hallmark acute neutrophil response during Candida vaginitis. Infect. Immun. 2010, 78, 5126–5137. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, L.E.; Millhouse, E.; Sherry, L.; Kean, R.; Malcolm, J.; Nile, C.J.; Ramage, G. Polymicrobial Candida biofilms: Friends and foe in the oral cavity. FEMS Yeast Res. 2015, 15. [Google Scholar] [CrossRef]
- Pammi, M.; Zhong, D.; Johnson, Y.; Revell, P.; Versalovic, J. Polymicrobial bloodstream infections in the neonatal intensive care unit are associated with increased mortality: A case-control study. BMC Infect. Dis. 2014, 14, 390. [Google Scholar] [CrossRef] [PubMed]
- Maeda, Y.; Elborn, J.S.; Parkins, M.D.; Reihill, J.; Goldsmith, C.E.; Coulter, W.A.; Mason, C.; Millar, B.C.; Dooley, J.S.; Lowery, C.J.; et al. Population structure and characterization of viridans group streptococci (vgs) including streptococcus pneumoniae isolated from adult patients with cystic fibrosis (cf). J. Cyst. Fibros. 2011, 10, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Garsin, D.A.; Lorenz, M.C. Candida albicans and Enterococcus faecalis in the gut: Synergy in commensalism? Gut Microbes 2013, 4, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Hermann, C.; Hermann, J.; Munzel, U.; Ruchel, R. Bacterial flora accompanying Candida yeasts in clinical specimens. Mycoses 1999, 42, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Kovac, J.; Kovac, D.; Slobodnikova, L.; Kotulova, D. Enterococcus faecalis and Candida albicans in the dental root canal and periapical infections. Bratisl. Lek. Listy 2013, 114, 716–720. [Google Scholar] [PubMed]
- Dahlen, G.; Blomqvist, S.; Almstahl, A.; Carlen, A. Virulence factors and antibiotic susceptibility in enterococci isolated from oral mucosal and deep infections. J. Oral Microbiol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Bohm, L.; Torsin, S.; Tint, S.H.; Eckstein, M.T.; Ludwig, T.; Perez, J.C. The yeast form of the fungus Candida albicans promotes persistence in the gut of gnotobiotic mice. PLoS Pathog. 2017, 13, e1006699. [Google Scholar] [CrossRef] [PubMed]
- Riggle, P.J.; Andrutis, K.A.; Chen, X.; Tzipori, S.R.; Kumamoto, C.A. Invasive lesions containing filamentous forms produced by a Candida albicans mutant that is defective in filamentous growth in culture. Infect. Immun. 1999, 67, 3649–3652. [Google Scholar] [PubMed]
- Xu, H.; Sobue, T.; Bertolini, M.; Thompson, A.; Vickerman, M.; Nobile, C.J.; Dongari-Bagtzoglou, A.S. Oralis activates the efg1 filamentation pathway in C. albicans to promote cross-kingdom interactions and mucosal biofilms. Virulence 2017, 8, 1602–1617. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, M.M.; Xu, H.; Sobue, T.; Nobile, C.J.; Del Bel Cury, A.A.; Dongari-Bagtzoglou, A. Candida-streptococcal mucosal biofilms display distinct structural and virulence characteristics depending on growth conditions and hyphal morphotypes. Mol. Oral Microbiol. 2015, 30, 307–322. [Google Scholar] [CrossRef] [PubMed]
- McAlester, G.; O’Gara, F.; Morrissey, J.P. Signal-mediated interactions between Pseudomonas aeruginosa and Candida albicans. J. Med. Microbiol. 2008, 57, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Trejo-Hernandez, A.; Andrade-Dominguez, A.; Hernandez, M.; Encarnacion, S. Interspecies competition triggers virulence and mutability in Candida albicans-Pseudomonas aeruginosa mixed biofilms. ISME J. 2014, 8, 1974–1988. [Google Scholar] [CrossRef] [PubMed]
- Bamford, C.V.; d’Mello, A.; Nobbs, A.H.; Dutton, L.C.; Vickerman, M.M.; Jenkinson, H.F. Streptococcus gordonii modulates Candida albicans biofilm formation through intergeneric communication. Infect. Immun. 2009, 77, 3696–3704. [Google Scholar] [CrossRef] [PubMed]
- Bachtiar, E.W.; Bachtiar, B.M.; Jarosz, L.M.; Amir, L.R.; Sunarto, H.; Ganin, H.; Meijler, M.M.; Krom, B.P. Ai-2 of aggregatibacter actinomycetemcomitans inhibits Candida albicans biofilm formation. Front. Cell. Infect. Microbiol. 2014, 4, 94. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.R.; Taylor, G.W.; Rutman, A.; Hoiby, N.; Cole, P.J.; Wilson, R. Pseudomonas aeruginosa pyocyanin and 1-hydroxyphenazine inhibit fungal growth. J. Clin. Pathol. 1999, 52, 385–387. [Google Scholar] [CrossRef] [PubMed]
- Jack, A.A.; Daniels, D.E.; Jepson, M.A.; Vickerman, M.M.; Lamont, R.J.; Jenkinson, H.F.; Nobbs, A.H. Streptococcus gordonii comcde (competence) operon modulates biofilm formation with Candida albicans. Microbiology 2015, 161, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Jarosz, L.M.; Deng, D.M.; van der Mei, H.C.; Crielaard, W.; Krom, B.P. Streptococcus mutans competence-stimulating peptide inhibits Candida albicans hypha formation. Eukaryot. Cell 2009, 8, 1658–1664. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.L.; Lee, R.T.; Fang, H.M.; Wang, Y.M.; Li, R.; Zou, H.; Zhu, Y.; Wang, Y. Bacterial peptidoglycan triggers Candida albicans hyphal growth by directly activating the adenylyl cyclase cyr1p. Cell Host Microbe 2008, 4, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Bandara, H.M.; BP, K.C.; Watt, R.M.; Jin, L.J.; Samaranayake, L.P. Pseudomonas aeruginosa lipopolysaccharide inhibits Candida albicans hyphae formation and alters gene expression during biofilm development. Mol. Oral Microbiol. 2013, 28, 54–69. [Google Scholar] [CrossRef] [PubMed]
- Barnard, J.P.; Stinson, M.W. The alpha-hemolysin of Streptococcus gordonii is hydrogen peroxide. Infect. Immun. 1996, 64, 3853–3857. [Google Scholar] [PubMed]
- Nasution, O.; Srinivasa, K.; Kim, M.; Kim, Y.J.; Kim, W.; Jeong, W.; Choi, W. Hydrogen peroxide induces hyphal differentiation in Candida albicans. Eukaryot. Cell 2008, 7, 2008–2011. [Google Scholar] [CrossRef] [PubMed]
- Noverr, M.C.; Huffnagle, G.B. Regulation of Candida albicans morphogenesis by fatty acid metabolites. Infect. Immun. 2004, 72, 6206–6210. [Google Scholar] [CrossRef] [PubMed]
- Vylkova, S.; Carman, A.J.; Danhof, H.A.; Collette, J.R.; Zhou, H.; Lorenz, M.C. The fungal pathogen Candida albicans autoinduces hyphal morphogenesis by raising extracellular pH. MBio 2011, 2, e00055-00011. [Google Scholar] [CrossRef] [PubMed]
- Peleg, A.Y.; Tampakakis, E.; Fuchs, B.B.; Eliopoulos, G.M.; Moellering, R.C., Jr.; Mylonakis, E. Prokaryote-eukaryote interactions identified by using caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2008, 105, 14585–14590. [Google Scholar] [CrossRef] [PubMed]
- Pande, K.; Chen, C.; Noble, S.M. Passage through the mammalian gut triggers a phenotypic switch that promotes Candida albicans commensalism. Nat. Genet. 2013, 45, 1088–1091. [Google Scholar] [CrossRef] [PubMed]
- Pierce, J.V.; Dignard, D.; Whiteway, M.; Kumamoto, C.A. Normal adaptation of Candida albicans to the murine gastrointestinal tract requires efg1p-dependent regulation of metabolic and host defense genes. Eukaryot. Cell 2013, 12, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Mailander-Sanchez, D.; Braunsdorf, C.; Grumaz, C.; Muller, C.; Lorenz, S.; Stevens, P.; Wagener, J.; Hebecker, B.; Hube, B.; Bracher, F.; et al. Antifungal defense of probiotic lactobacillus rhamnosus gg is mediated by blocking adhesion and nutrient depletion. PLoS ONE 2017, 12, e0184438. [Google Scholar] [CrossRef] [PubMed]
- Dutton, L.C.; Paszkiewicz, K.H.; Silverman, R.J.; Splatt, P.R.; Shaw, S.; Nobbs, A.H.; Lamont, R.J.; Jenkinson, H.F.; Ramsdale, M. Transcriptional landscape of trans-kingdom communication between Candida albicans and Streptococcus gordonii. Mol. Oral Microbiol. 2016, 31, 136–161. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Lopez, C.; Collette, J.R.; Brothers, K.M.; Shepardson, K.M.; Cramer, R.A.; Wheeler, R.T.; Lorenz, M.C. Candida albicans induces arginine biosynthetic genes in response to host-derived reactive oxygen species. Eukaryot. Cell 2013, 12, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Jenkinson, H.F.; Lala, H.C.; Shepherd, M.G. Coaggregation of streptococcus sanguis and other streptococci with Candida albicans. Infect. Immun. 1990, 58, 1429–1436. [Google Scholar] [PubMed]
- Purschke, F.G.; Hiller, E.; Trick, I.; Rupp, S. Flexible survival strategies of Pseudomonas aeruginosa in biofilms result in increased fitness compared with Candida albicans. Mol. Cell. Proteomics 2012, 11, 1652–1669. [Google Scholar] [CrossRef] [PubMed]
- Ovchinnikova, E.S.; Krom, B.P.; van der Mei, H.C.; Busscher, H.J. Force microscopic and thermodynamic analysis of the adhesion between Pseudomonas aeruginosa and Candida albicans. Soft Matter 2012, 8, 6454–6461. [Google Scholar] [CrossRef]
- Silverman, R.J.; Nobbs, A.H.; Vickerman, M.M.; Barbour, M.E.; Jenkinson, H.F. Interaction of Candida albicans cell wall als3 protein with Streptococcus gordonii sspb adhesin promotes development of mixed-species communities. Infect. Immun. 2010, 78, 4644–4652. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, L.L.; Oh, S.H.; Jones, R.; Cota, E. A proposed mechanism for the interaction between the Candida albicans als3 adhesin and streptococcal cell wall proteins. Front. Microbiol. 2014, 5, 564. [Google Scholar] [CrossRef] [PubMed]
- Gregoire, S.; Xiao, J.; Silva, B.B.; Gonzalez, I.; Agidi, P.S.; Klein, M.I.; Ambatipudi, K.S.; Rosalen, P.L.; Bauserman, R.; Waugh, R.E.; et al. Role of glucosyltransferase b in interactions of Candida albicans with Streptococcus mutans and with an experimental pellicle on hydroxyapatite surfaces. Appl. Environ. Microbiol. 2011, 77, 6357–6367. [Google Scholar] [CrossRef] [PubMed]
- Bruno, V.M.; Shetty, A.C.; Yano, J.; Fidel, P.L., Jr.; Noverr, M.C.; Peters, B.M. Transcriptomic analysis of vulvovaginal candidiasis identifies a role for the nlrp3 inflammasome. MBio 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Fox, E.P.; Cowley, E.S.; Nobile, C.J.; Hartooni, N.; Newman, D.K.; Johnson, A.D. Anaerobic bacteria grow within Candida albicans biofilms and induce biofilm formation in suspension cultures. Curr. Biol. 2014, 24, 2411–2416. [Google Scholar] [CrossRef] [PubMed]
- Villar, C.C.; Kashleva, H.; Nobile, C.J.; Mitchell, A.P.; Dongari-Bagtzoglou, A. Mucosal tissue invasion by Candida albicans is associated with e-cadherin degradation, mediated by transcription factor rim101p and protease sap5p. Infect. Immun. 2007, 75, 2126–2135. [Google Scholar] [CrossRef] [PubMed]
- Cavalcanti, Y.W.; Morse, D.J.; da Silva, W.J.; Del-Bel-Cury, A.A.; Wei, X.; Wilson, M.; Milward, P.; Lewis, M.; Bradshaw, D.; Williams, D.W. Virulence and pathogenicity of Candida albicans is enhanced in biofilms containing oral bacteria. Biofouling 2015, 31, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, P.; Thompson, A.; Xie, Z.; Kashleva, H.; Ganguly, S.; Mitchell, A.P.; Dongari-Bagtzoglou, A. Role of bcr1-activated genes hwp1 and hyr1 in Candida albicans oral mucosal biofilms and neutrophil evasion. PLoS ONE 2011, 6, e16218. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.I.; Dolben, E.F.; Okegbe, C.; Harty, C.E.; Golub, Y.; Thao, S.; Ha, D.G.; Willger, S.D.; O’Toole, G.A.; Harwood, C.S.; et al. Candida albicans ethanol stimulates Pseudomonas aeruginosa wspr-controlled biofilm formation as part of a cyclic relationship involving phenazines. PLoS Pathog. 2014, 10, e1004480. [Google Scholar] [CrossRef] [PubMed]
- Ader, F.; Jawhara, S.; Nseir, S.; Kipnis, E.; Faure, K.; Vuotto, F.; Chemani, C.; Sendid, B.; Poulain, D.; Guery, B. Short term Candida albicans colonization reduces Pseudomonas aeruginosa-related lung injury and bacterial burden in a murine model. Crit. Care 2011, 15, R150. [Google Scholar] [CrossRef] [PubMed]
- Mear, J.B.; Gosset, P.; Kipnis, E.; Faure, E.; Dessein, R.; Jawhara, S.; Fradin, C.; Faure, K.; Poulain, D.; Sendid, B.; et al. Candida albicans airway exposure primes the lung innate immune response against Pseudomonas aeruginosa infection through innate lymphoid cell recruitment and interleukin-22-associated mucosal response. Infect. Immun. 2014, 82, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Medina, E.; Fan, D.; Coughlin, L.A.; Ho, E.X.; Lamont, I.L.; Reimmann, C.; Hooper, L.V.; Koh, A.Y. Candida albicans inhibits Pseudomonas aeruginosa virulence through suppression of pyochelin and pyoverdine biosynthesis. PLoS Pathog. 2015, 11, e1005129. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.; Bowen, W.H. Candida albicans and Streptococcus mutans: A potential synergistic alliance to cause virulent tooth decay in children. Future Microbiol. 2014, 9, 1295–1297. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Jenkinson, H.F.; Dongari-Bagtzoglou, A. Innocent until proven guilty: Mechanisms and roles of streptococcus-candida interactions in oral health and disease. Mol. Oral Microbiol. 2014, 29, 99–116. [Google Scholar] [CrossRef] [PubMed]
- Falsetta, M.L.; Klein, M.I.; Colonne, P.M.; Scott-Anne, K.; Gregoire, S.; Pai, C.H.; Gonzalez-Begne, M.; Watson, G.; Krysan, D.J.; Bowen, W.H.; et al. Symbiotic relationship between Streptococcus mutans and Candida albicans synergizes virulence of plaque biofilms in vivo. Infect. Immun. 2014, 82, 1968–1981. [Google Scholar] [CrossRef] [PubMed]
- Lambooij, J.M.; Hoogenkamp, M.A.; Brandt, B.W.; Janus, M.M.; Krom, B.P. Fungal mitochondrial oxygen consumption induces the growth of strict anaerobic bacteria. Fungal Genet. Biol. 2017, 109, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Diaz, P.I.; Xie, Z.; Sobue, T.; Thompson, A.; Biyikoglu, B.; Ricker, A.; Ikonomou, L.; Dongari-Bagtzoglou, A. Synergistic interaction between Candida albicans and commensal oral streptococci in a novel in vitro mucosal model. Infect. Immun. 2012, 80, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Staib, P.; Kretschmar, M.; Nichterlein, T.; Hof, H.; Morschhauser, J. Differential activation of a Candida albicans virulence gene family during infection. Proc. Natl. Acad. Sci. USA 2000, 97, 6102–6107. [Google Scholar] [CrossRef] [PubMed]
- Rosenbach, A.; Dignard, D.; Pierce, J.V.; Whiteway, M.; Kumamoto, C.A. Adaptations of Candida albicans for growth in the mammalian intestinal tract. Eukaryot. Cell 2010, 9, 1075–1086. [Google Scholar] [CrossRef] [PubMed]
- Takakura, N.; Sato, Y.; Ishibashi, H.; Oshima, H.; Uchida, K.; Yamaguchi, H.; Abe, S. A novel murine model of oral candidiasis with local symptoms characteristic of oral thrush. Microbiol. Immunol. 2003, 47, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Zakikhany, K.; Naglik, J.R.; Schmidt-Westhausen, A.; Holland, G.; Schaller, M.; Hube, B. In vivo transcript profiling of Candida albicans identifies a gene essential for interepithelial dissemination. Cell. Microbiol. 2007, 9, 2938–2954. [Google Scholar] [CrossRef] [PubMed]
- Hebecker, B.; Vlaic, S.; Conrad, T.; Bauer, M.; Brunke, S.; Kapitan, M.; Linde, J.; Hube, B.; Jacobsen, I.D. Dual-species transcriptional profiling during systemic candidiasis reveals organ-specific host-pathogen interactions. Sci. Rep. 2016, 6, 36055. [Google Scholar] [CrossRef] [PubMed]
- Andes, D.; Lepak, A.; Pitula, A.; Marchillo, K.; Clark, J. A simple approach for estimating gene expression in Candida albicans directly from a systemic infection site. J. Infect. Dis. 2005, 192, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Allert, S.; Brunke, S.; Hube, B. In vivo transcriptional profiling of human pathogenic fungi during infection: Reflecting the real life? PLoS Pathog. 2016, 12, e1005471. [Google Scholar] [CrossRef] [PubMed]
- Boktour, M.R.; Kontoyiannis, D.P.; Hanna, H.A.; Hachem, R.Y.; Girgawy, E.; Bodey, G.P.; Raad, I.I. Multiple-species candidemia in patients with cancer. Cancer 2004, 101, 1860–1865. [Google Scholar] [CrossRef] [PubMed]
- Puig-Asensio, M.; Ruiz-Camps, I.; Fernandez-Ruiz, M.; Aguado, J.M.; Munoz, P.; Valerio, M.; Delgado-Iribarren, A.; Merino, P.; Bereciartua, E.; Fortun, J.; et al. Epidemiology and outcome of candidaemia in patients with oncological and haematological malignancies: Results from a population-based surveillance in Spain. Clin. Microbiol. Infect. 2015, 21, 491. [Google Scholar] [CrossRef] [PubMed]
- Holler, E.; Butzhammer, P.; Schmid, K.; Hundsrucker, C.; Koestler, J.; Peter, K.; Zhu, W.; Sporrer, D.; Hehlgans, T.; Kreutz, M.; et al. Metagenomic analysis of the stool microbiome in patients receiving allogeneic stem cell transplantation: Loss of diversity is associated with use of systemic antibiotics and more pronounced in gastrointestinal graft-versus-host disease. Biol. Blood Marrow Transplant. 2014, 20, 640–645. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ranjan, A.; Dongari-Bagtzoglou, A. Tipping the Balance: C. albicans Adaptation in Polymicrobial Environments. J. Fungi 2018, 4, 112. https://doi.org/10.3390/jof4030112
Ranjan A, Dongari-Bagtzoglou A. Tipping the Balance: C. albicans Adaptation in Polymicrobial Environments. Journal of Fungi. 2018; 4(3):112. https://doi.org/10.3390/jof4030112
Chicago/Turabian StyleRanjan, Amit, and Anna Dongari-Bagtzoglou. 2018. "Tipping the Balance: C. albicans Adaptation in Polymicrobial Environments" Journal of Fungi 4, no. 3: 112. https://doi.org/10.3390/jof4030112
APA StyleRanjan, A., & Dongari-Bagtzoglou, A. (2018). Tipping the Balance: C. albicans Adaptation in Polymicrobial Environments. Journal of Fungi, 4(3), 112. https://doi.org/10.3390/jof4030112