
Evolutionary Dynamics and Functional Bifurcation of the C2H2 Gene Family in Basidiomycota


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Retrieval and Quality Assessment
2.2. Identification of C2H2 Gene Family Members
2.3. Gene Structure Analysis of C2H2 Family
2.4. Phylogenetic and Evolutionary Analysis of C2H2 Genes
2.5. Promoter Cis-Regulatory Element Analysis
2.6. Expression of C2H2 Zinc Finger Genes in Transcriptomes of Sarcomyxa edulis
3. Results
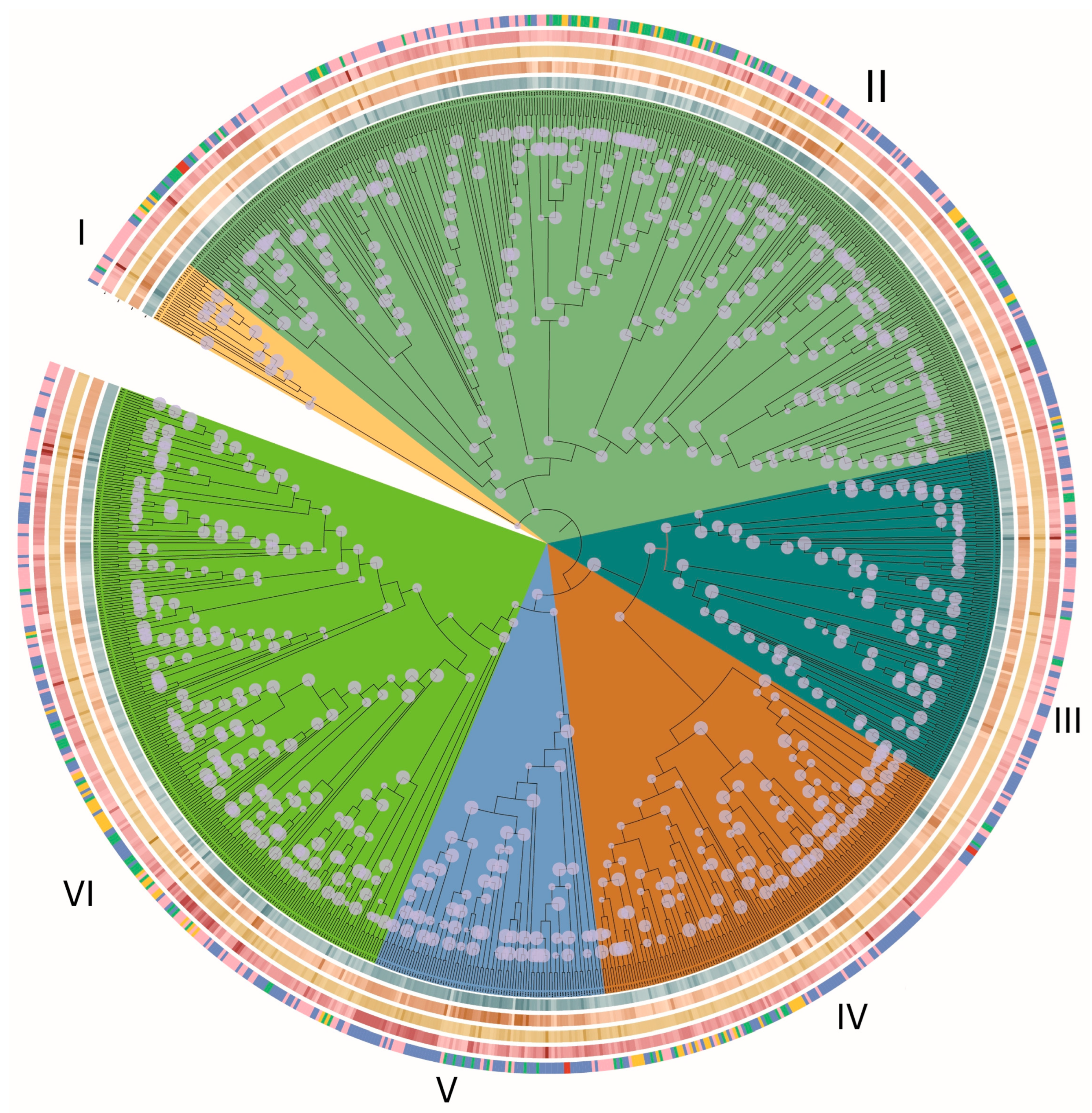
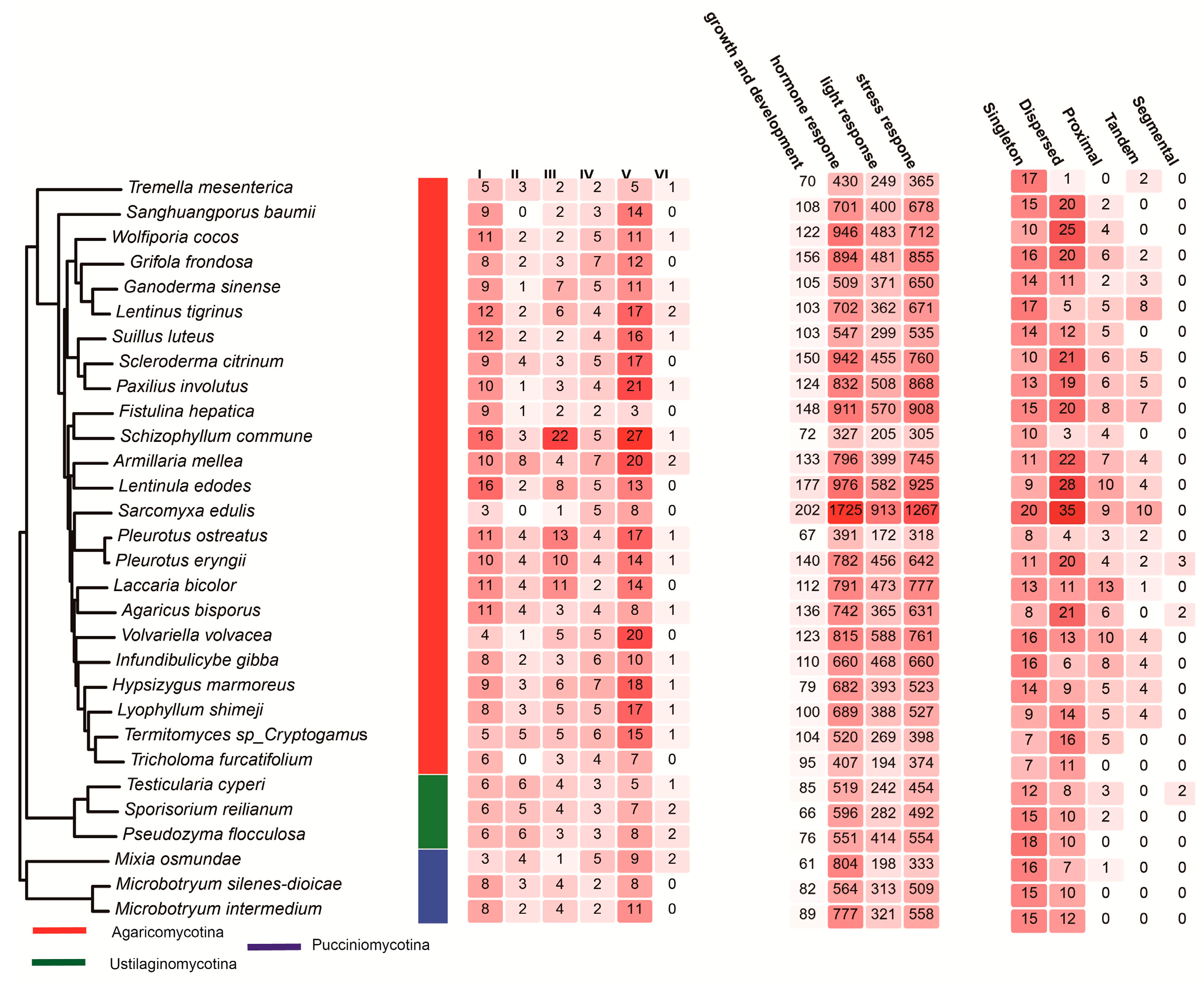
3.1. Systematic Identification and Classification of C2H2 Genes in Basidiomycota
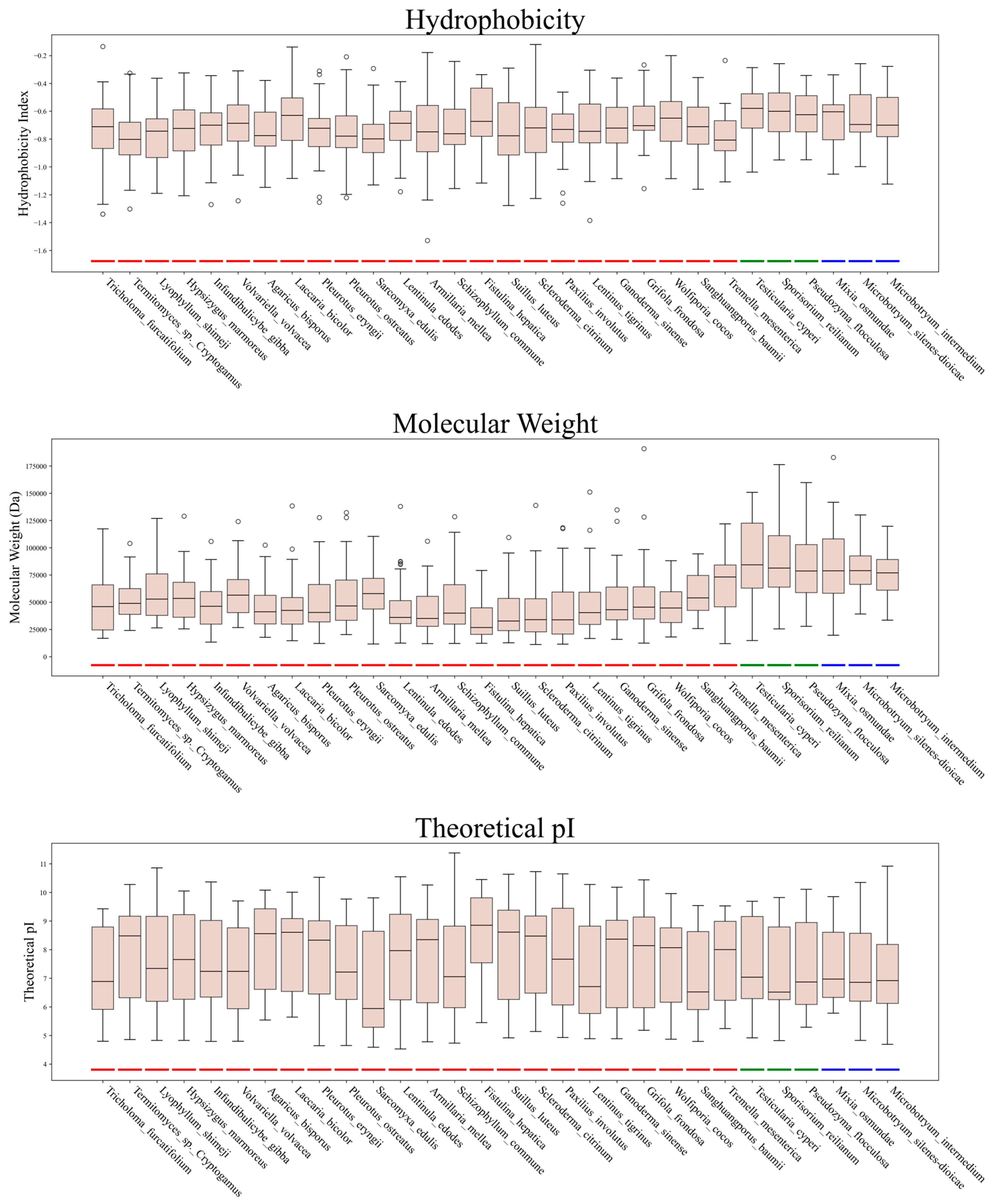
3.2. Physicochemical Properties of C2H2 Zinc Finger Proteins
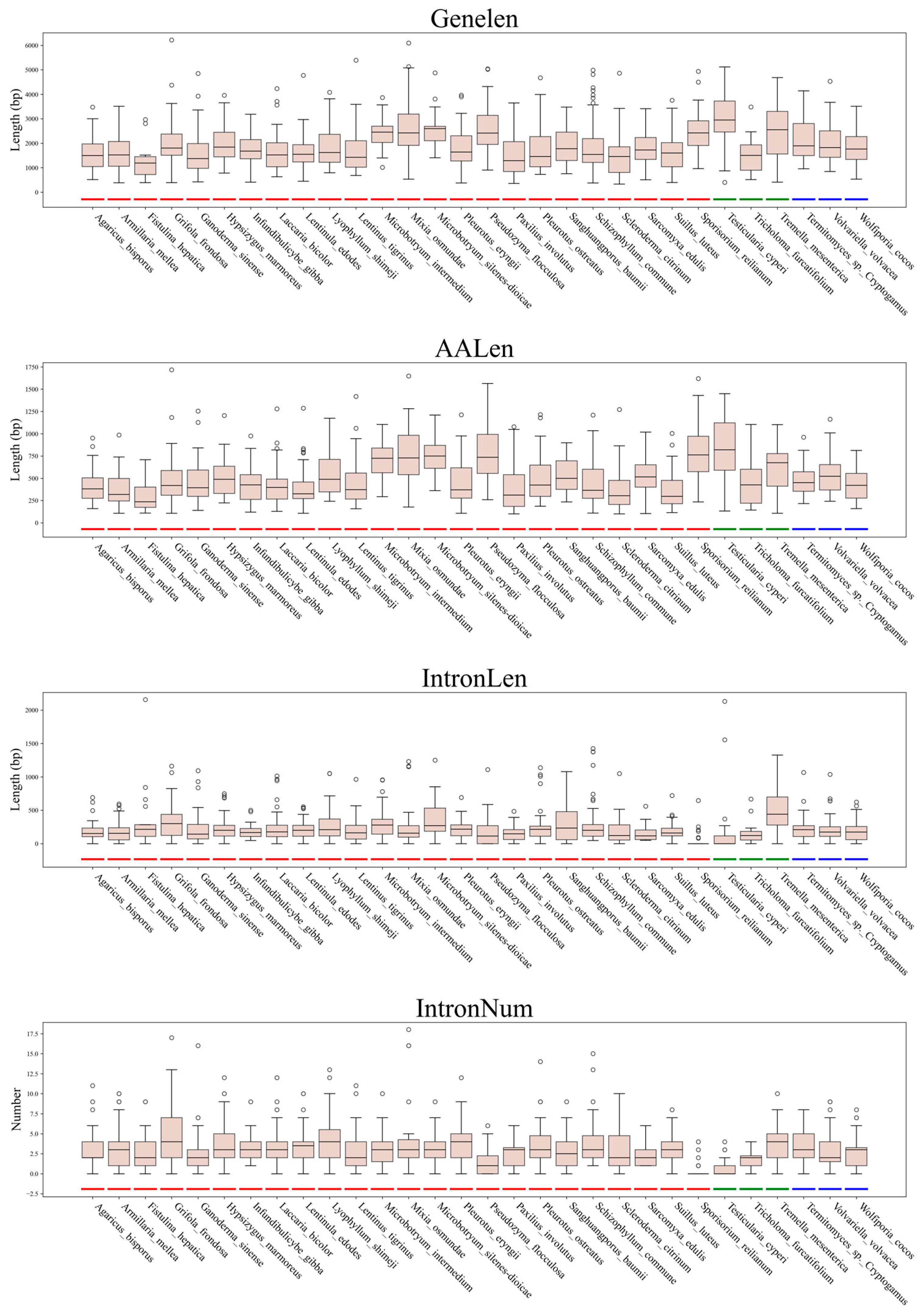
3.3. Gene Architecture Variations Among Subphyla
3.4. Cis-Regulatory Element Distribution in Promoters
3.5. Gene Duplication Modes
3.6. Synteny and Evolutionary Conservation
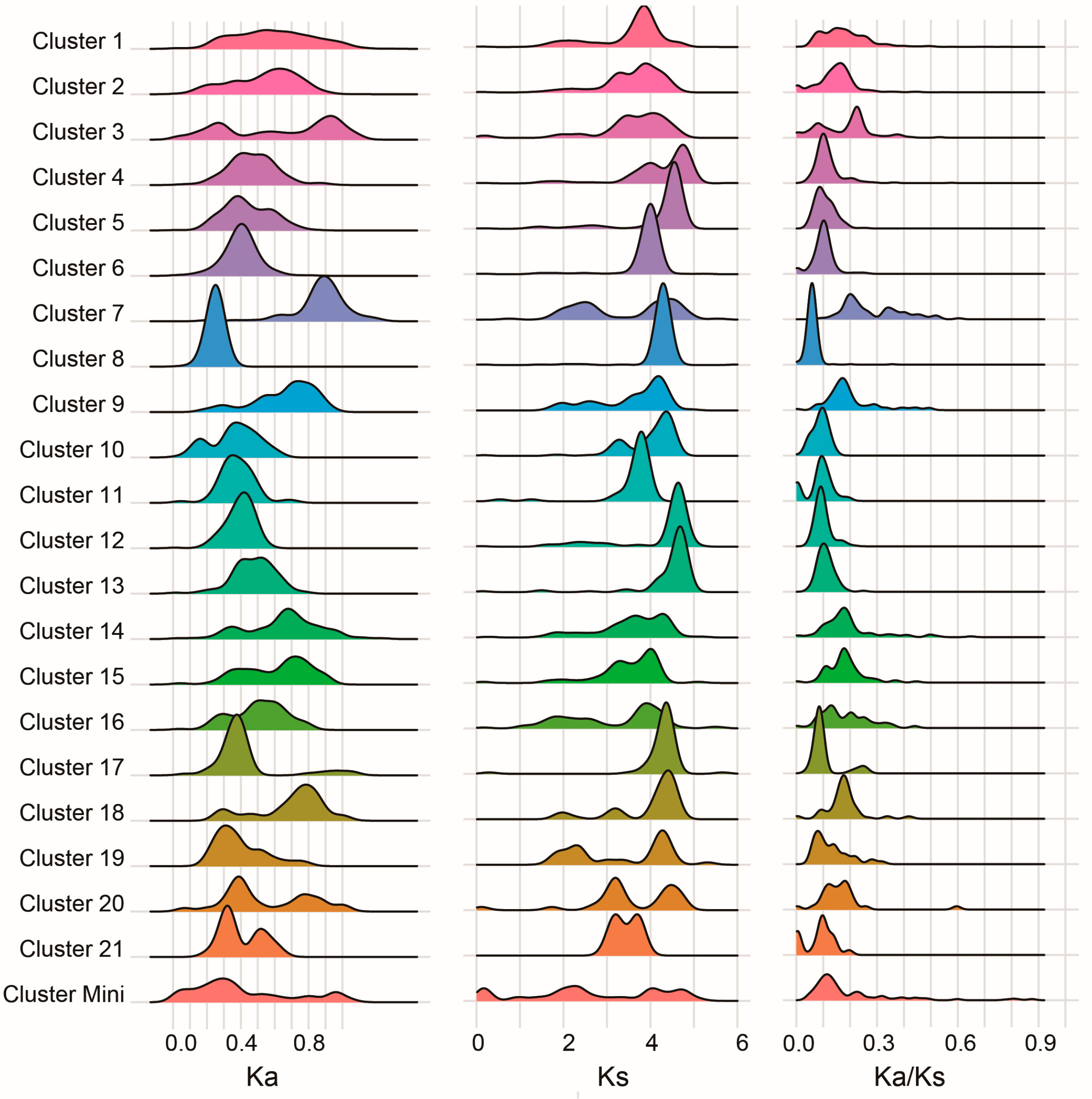
3.7. Evolutionary Constraints on C2H2 Clusters
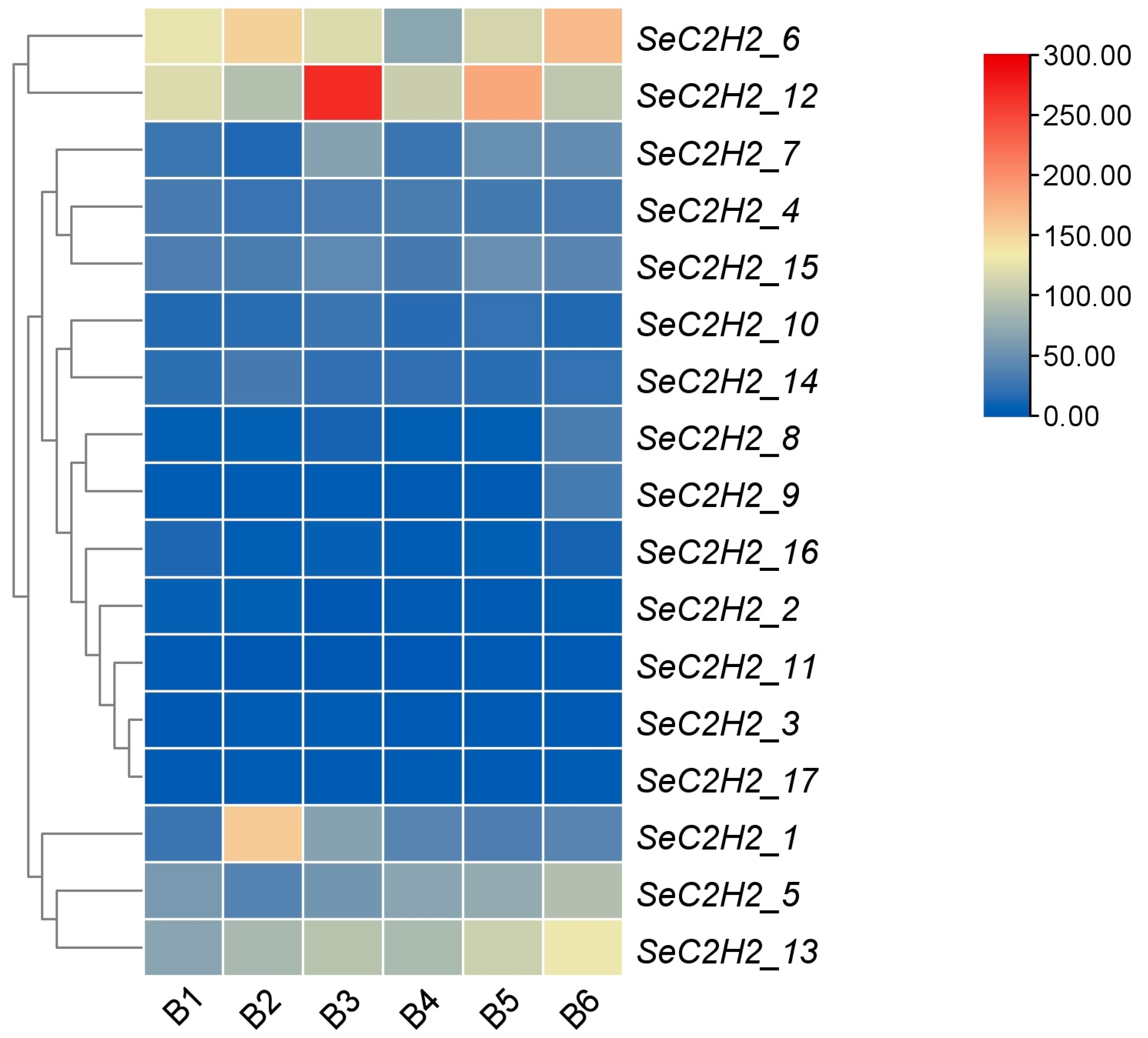
3.8. Expression Patterns of Individual C2H2 Zinc Finger Genes in Transcriptomes of S. edulis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Seetharam, A.; Stuart, G.W. A study on the distribution of 37 well conserved families of C2H2 zinc finger genes in eukaryotes. BMC Genom. 2013, 14, 420. [Google Scholar] [CrossRef] [PubMed]
- Ohm, R.A.; Jong, J.F.D.; Bekker, C.D.; Wösten, H.A.; Lugones, L.G. Transcription factor genes of Schizophyllum commune involved in regulation of mushroom formation. Mol. Microbiol. 2011, 81, 1433–1445. [Google Scholar] [CrossRef]
- Xiong, D.; Wang, Y.; Deng, C.; Hu, R.; Tian, C. Phylogenic analysis revealed an expanded C2H2-homeobox subfamily and expression profiles of C2H2 zinc finger gene family in Verticillium dahliae. Gene 2015, 562, 169–179. [Google Scholar] [CrossRef]
- Floudas, D.; Binder, M.; Riley, R.; Barry, K.; Blanchette, R.A.; Henrissat, B.; Hibbett, D.S. The Paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science 2012, 336, 1715–1719. [Google Scholar] [CrossRef]
- Kubicek, C.P.; Herrera-Estrella, A.; Seidl-Seiboth, V.; Martinez, D.A.; Druzhinina, I.S.; Thon, M.; Grigoriev, I.V. Comparative genome sequence analysis underscores mycoparasitism as the ancestral life style of Trichoderma. Genome Biol. 2011, 12, R40. [Google Scholar] [CrossRef]
- Kijpornyongpan, T.; Schwartz, A.; Yaguchi, A.; Salvachúa, D. Genome-scale analysis of evolutionary patterns and gene content in plant-pathogenic Ustilaginomycotina fungi. Genome Biol. Evol. 2018, 10, 3196–3210. [Google Scholar]
- Spanu, P.D.; Abbott, J.C.; Amselem, J.; Burgis, T.A.; Soanes, D.M.; Stüber, K.; Themaat, E.V.L.; Brown, J.K.M.; Butcher, S.A.; Gurr, S.J.; et al. Genome expansion and gene loss in powdery mildew fungi reveal tradeoffs in extreme parasitism. Science 2010, 330, 1543–1546. [Google Scholar] [CrossRef] [PubMed]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO Update: Novel and Streamlined Workflows. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef]
- Eddy, S.R. Accelerated profile HMM searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef] [PubMed]
- Shimeld, S.M. C2H2 zinc finger genes of the Gli, Zic, KLF, SP, Wilms’ tumour, Huckebein, Snail, Ovo, Spalt, Odd, Blimp-1, Fez and related gene families from Branchiostoma floridae. Dev. Genes Evol. 2008, 218, 639–649. [Google Scholar] [CrossRef]
- Stein, L.D.; Mungall, C.; Shu, S.; Caudy, M.; Mangone, M.; Day, A.; Nickerson, E.; Stajich, J.E.; Harris, T.W.; Arva, A.; et al. The Generic Genome Browser: A Building Block for a Model Organism System Database. Genome Res. 2002, 12, 1599–1610. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Tang, H.; Bowers, J.E.; Wang, X.; Ming, R.; Alam, M.; Paterson, A.H. Synteny and Collinearity in Plant Genomes. Science 2008, 320, 486–488. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, J.; Zhao, X.Q.; Wang, J.; Wong, G.K.S.; Yu, J. KaKs_Calculator: Calculating Ka and Ks Through Model Selection and Model Averaging. Genom. Proteom. Bioinform. 2006, 4, 259–263. [Google Scholar] [CrossRef]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Duan, C.; Tian, F.-H.; Yao, L.; Lv, J.-H.; Jia, C.-W.; Li, C.-T. Comparative transcriptome and WGCNA reveal key genes involved in lignocellulose degradation in Sarcomyxa edulis. Sci. Rep. 2022, 12, 18379. [Google Scholar] [CrossRef]
- Qhanya, L.B.; Matowane, G.; Chen, W.; Sun, Y.; Letsimo, E.M.; Parvez, M.; Yu, J.H.; Mashele, S.S.; Syed, K. Genome-Wide Annotation and Comparative Analysis of Cytochrome P450 Monooxygenases in Basidiomycete Biotrophic Plant Pathogens. PLoS ONE 2015, 10, e0142100. [Google Scholar] [CrossRef]
- Riley, R.; Salamov, A.A.; Brown, D.W.; Nagy, L.G.; Floudas, D.; Held, B.W.; Levasseur, A.; Lombard, D.; Morin, E.; Otillar, R.; et al. Extensive sampling of basidiomycete genomes demonstrates inadequacy of the white-rot/brown-rot paradigm for wood decay fungi. Proc. Natl. Acad. Sci. USA 2014, 111, 9923–9928. [Google Scholar] [CrossRef]
- Mohanta, T.K.; Mishra, A.K.; Khan, A.; Hashem, A.; Abd-Allah, E.F.; Al-Harrasi, A. Virtual 2-D map of the fungal proteome. Sci. Rep. 2021, 11, 6676. [Google Scholar] [CrossRef] [PubMed]
- Kiraga, J.; Mackiewicz, P.; Mackiewicz, D.; Kowalczuk, M.; Biecek, P.; Polak, N.; Smolarczyk, K.; Dudek, M.R.; Cebrat, S. The relationships between the isoelectric point and: Length of proteins, taxonomy and ecology of organisms. BMC Genom. 2007, 8, 163. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Chen, B.; Xu, S.; Zuo, J.; Xu, Y.; Xiong, S.; Chen, F. Investigation of crop straw for edible and medicinal fungi cultivation: Assessment of lignocellulose preprocessing and spent substrate biofuel properties. Ind. Crops Prod. 2025, 223, 120004. [Google Scholar] [CrossRef]
- Freeling, M. Bias in Plant Gene Content Following Different Sorts of Duplication: Tandem, Whole-Genome, Segmental, or by Transposition. Annu. Rev. Plant Biol. 2009, 60, 433–453. [Google Scholar] [CrossRef]
- Leister, D. Tandem and segmental gene duplication and recombination in the evolution of plant disease resistance gene. Trends Genet. 2004, 20, 116–122. [Google Scholar] [CrossRef]
- Maere, S.; Bodt, S.D.; Raes, J.; Casneuf, T.; Montagu, M.V.; Kuiper, M.; Van de Peer, Y. Modeling gene and genome duplications in eukaryotes. Proc. Natl. Acad. Sci. USA 2005, 102, 5454–5459. [Google Scholar] [CrossRef]
- Mondragon-Palomino, M.; Gaut, B.S. Gene conversion and the evolution of three leucine-rich repeat gene families in Arabidopsis thaliana. Mol. Biol. Evol. 2005, 22, 2444–2456. [Google Scholar] [CrossRef]
- Kim, J.; Lee, J.; Choi, J.P.; Park, I.; Yang, K.; Kim, M.K.; Lee, Y.H.; Nou, I.S.; Kim, D.S.; Min, S.R.; et al. Functional innovations of three chronological mesohexaploid Brassica rapa genomes. BMC Genom. 2014, 15, 606. [Google Scholar] [CrossRef]
- Li, L.L.; Xiao, Y.; Wang, X.; He, Z.H.; Lv, Y.W.; Hu, X.S. The Ka /Ks and πa /πs Ratios under Different Models of Gametophytic and Sporophytic Selection. Genome Biol. Evol. 2023, 15, evad151. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, J.; Yang, Z.E.; Chen, E.Y.; Zhang, C.J.; Zhang, X.Y.; Li, F.G. Genome-wide analysis of GRAS transcription factor gene family in Gossypium hirsutum L. BMC Genom. 2018, 19, 348. [Google Scholar] [CrossRef]
- Marand, A.P.; Eveland, A.L.; Kaufmann, K.; Springer, N.M. cis-Regulatory Elements in Plant Development, Adaptation, and Evolution. Annu. Rev. Plant Biol. 2023, 74, 111–137. [Google Scholar] [CrossRef] [PubMed]
- Tong, Z.; Han, X.; Duan, X.; Lin, J.; Chen, J.; Xiao, J.; Gan, Y.; Gan, B.; Yan, J. Genome-Wide Identification and Expression Analysis of the Cys2His2 Zinc Finger Protein Gene Family in Flammulina filiformis. J. Fungi 2024, 10, 644. [Google Scholar] [CrossRef]
- Martínez-Pastor, M.T.; Marchler, G.; Schüller, C.; Marchler-Bauer, A.; Ruis, H.; Estruch, F. The Saccharomyces cerevisiae zinc finger proteins Msn2p and Msn4p are required for transcriptional induction through the stress response element (STRE). EMBO J. 1996, 15, 2227–2235. [Google Scholar] [CrossRef] [PubMed]
- Bayram, Ö.; Braus, G.H. Coordination of secondarymetabolism and development in fungi: The velvet familyof regulatory proteins. FEMS Microbiol. Rev. 2012, 36, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Schoberle, T.J.; Nguyen-Coleman, C.K.; Herold, J.; Yang, A.; Weirauch, M.; Hughes, T.R.; McMurray, J.S.; May, G.S.; Butler, G. A Novel C2H2 Transcription Factor that Regulates gliA Expression Interdependently with GliZ in Aspergillus fumigatus. PLoS Genet. 2014, 10, e1004336. [Google Scholar] [CrossRef]
- Pelkmans, J.F.; Vos, A.M.; Scholtmeijer, K.; Hendrix, E.; Baars, J.J.P.; Gehrmann, T.; Reinders, M.J.T.; Lugones, L.G.; Wösten, H.A.B. The transcriptional regulator c2h2 accelerates mushroom formation in Agaricus bisporus. Appl. Microbiol. Biotechnol. 2016, 100, 7151–7159. [Google Scholar] [CrossRef]
- Ding, Q.; Zhao, H.; Zhu, P.; Jiang, X.; Nie, F.; Li, G. Genome-wide identification and expression analyses of C2H2 zinc finger transcription factors in Pleurotus ostreatus. PeerJ 2022, 10, e12654. [Google Scholar] [CrossRef]
- Li, S.; Li, Y.; Cai, Q.; Li, X.; Sun, Y.; Yu, T.; Yang, J.; Zhang, J. Analysis of the C2H2 Gene Family in Maize (Zea mays L.) under Cold Stress: Identification and Expression. Life 2023, 13, 122. [Google Scholar] [CrossRef]
- Murad, A.M.A.; Leng, P.; Straffon, M.; Wishart, J.; Macaskill, S.; MacCallum, D.; Schnell, N.; Talibi, D.; Marechal, D.; Tekaia, F.; et al. NRG1 represses yeast–hypha morphogenesis and hypha-specific gene expression in Candida albicans. EMBO J. 2014, 20, 4742–4752. [Google Scholar] [CrossRef] [PubMed]
- Braun, B.R.; Kadosh, D.; Johnson, A.D. NRG1, a repressor of filamentous growth in C.albicans, is down-regulated during filament induction. EMBO J. 2001, 20, 4753. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duan, C.; Yang, J. Evolutionary Dynamics and Functional Bifurcation of the C2H2 Gene Family in Basidiomycota. J. Fungi 2025, 11, 487. https://doi.org/10.3390/jof11070487
Duan C, Yang J. Evolutionary Dynamics and Functional Bifurcation of the C2H2 Gene Family in Basidiomycota. Journal of Fungi. 2025; 11(7):487. https://doi.org/10.3390/jof11070487
Chicago/Turabian StyleDuan, Chao, and Jie Yang. 2025. "Evolutionary Dynamics and Functional Bifurcation of the C2H2 Gene Family in Basidiomycota" Journal of Fungi 11, no. 7: 487. https://doi.org/10.3390/jof11070487
APA StyleDuan, C., & Yang, J. (2025). Evolutionary Dynamics and Functional Bifurcation of the C2H2 Gene Family in Basidiomycota. Journal of Fungi, 11(7), 487. https://doi.org/10.3390/jof11070487