
Integrative Transcriptome and Metabolome Analysis Reveals Candidate Genes Related to Terpenoid Synthesis in Amylostereum areolatum (Russulales: Amylostereaceae)

 , , ,
, , ,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
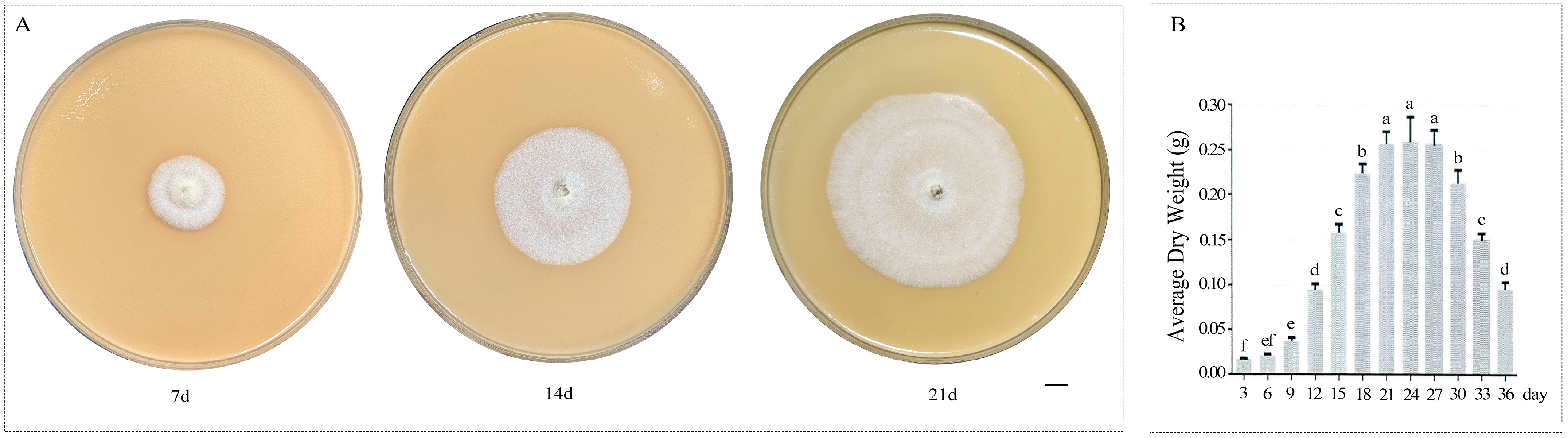
2.1. Strains and Growth Conditions
2.2. Detection and Analysis of Metabolites
2.3. RNA Extraction and Transcriptome Data Analysis
2.4. Comprehensive Analysis of the Metabolome and Transcriptome
2.5. Real-Time Quantitative PCR
3. Results
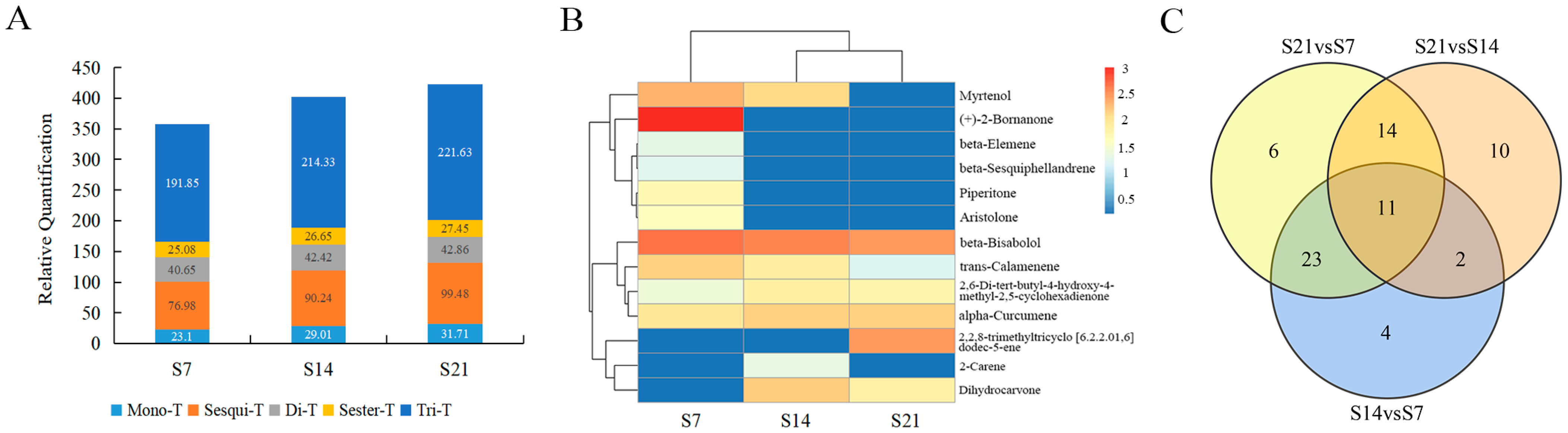
3.1. Analysis of Terpenoid Components in A. areolatum
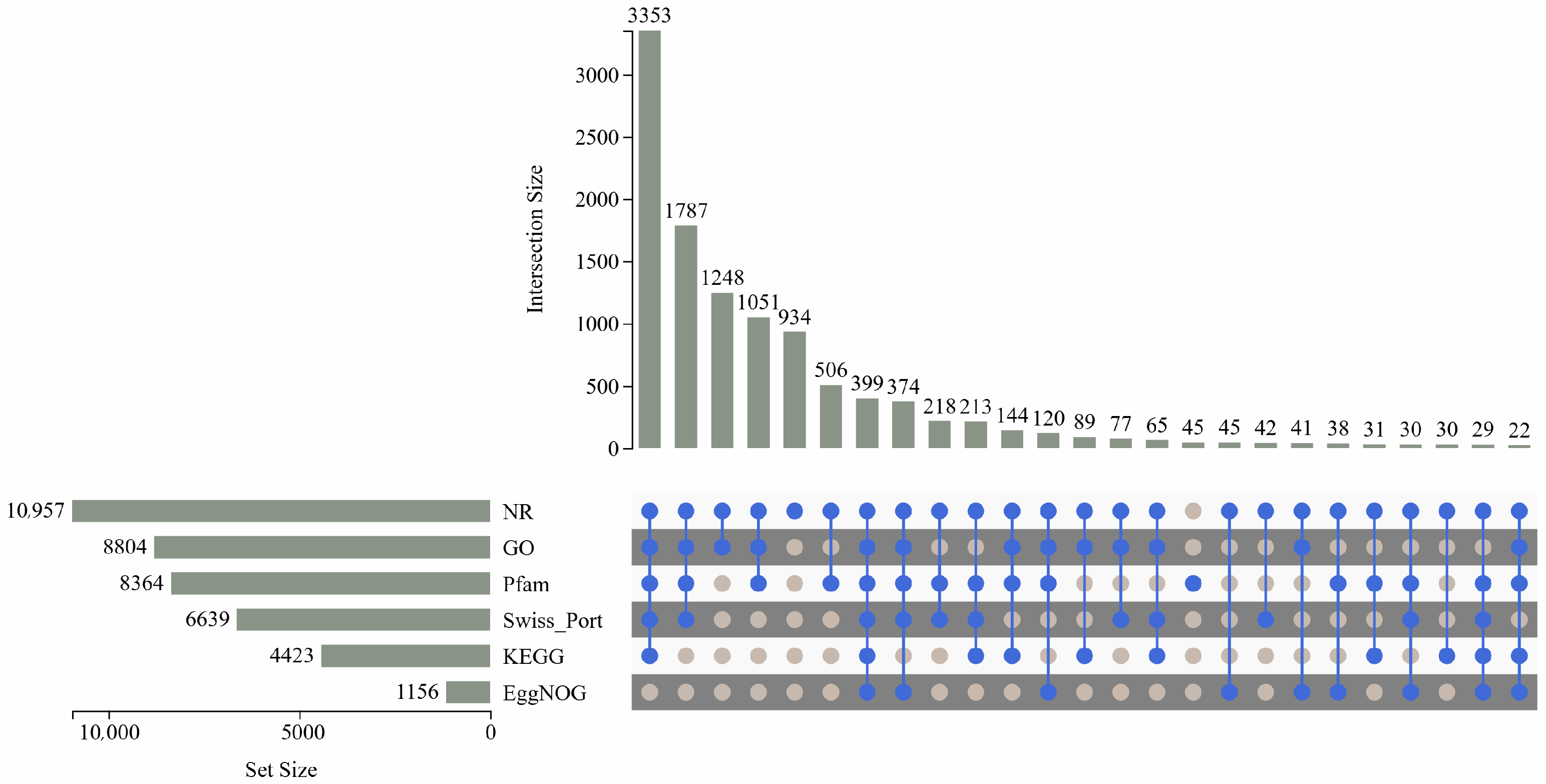
3.2. RNA-Seq and Transcriptomic Assembly
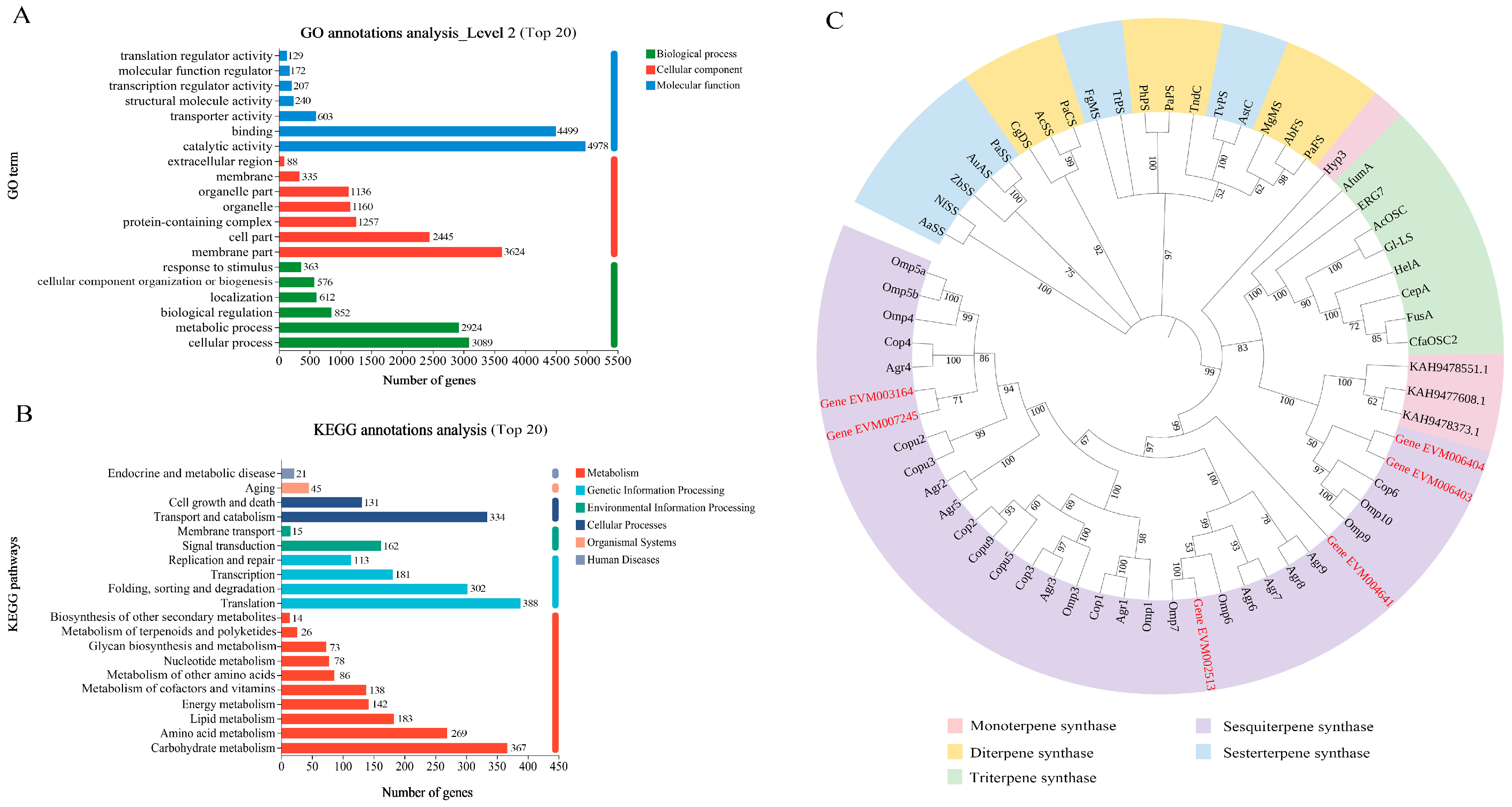
3.3. Gene Annotation and Functional Classification
3.4. Identification of Differentially Expressed Genes (DEGs)
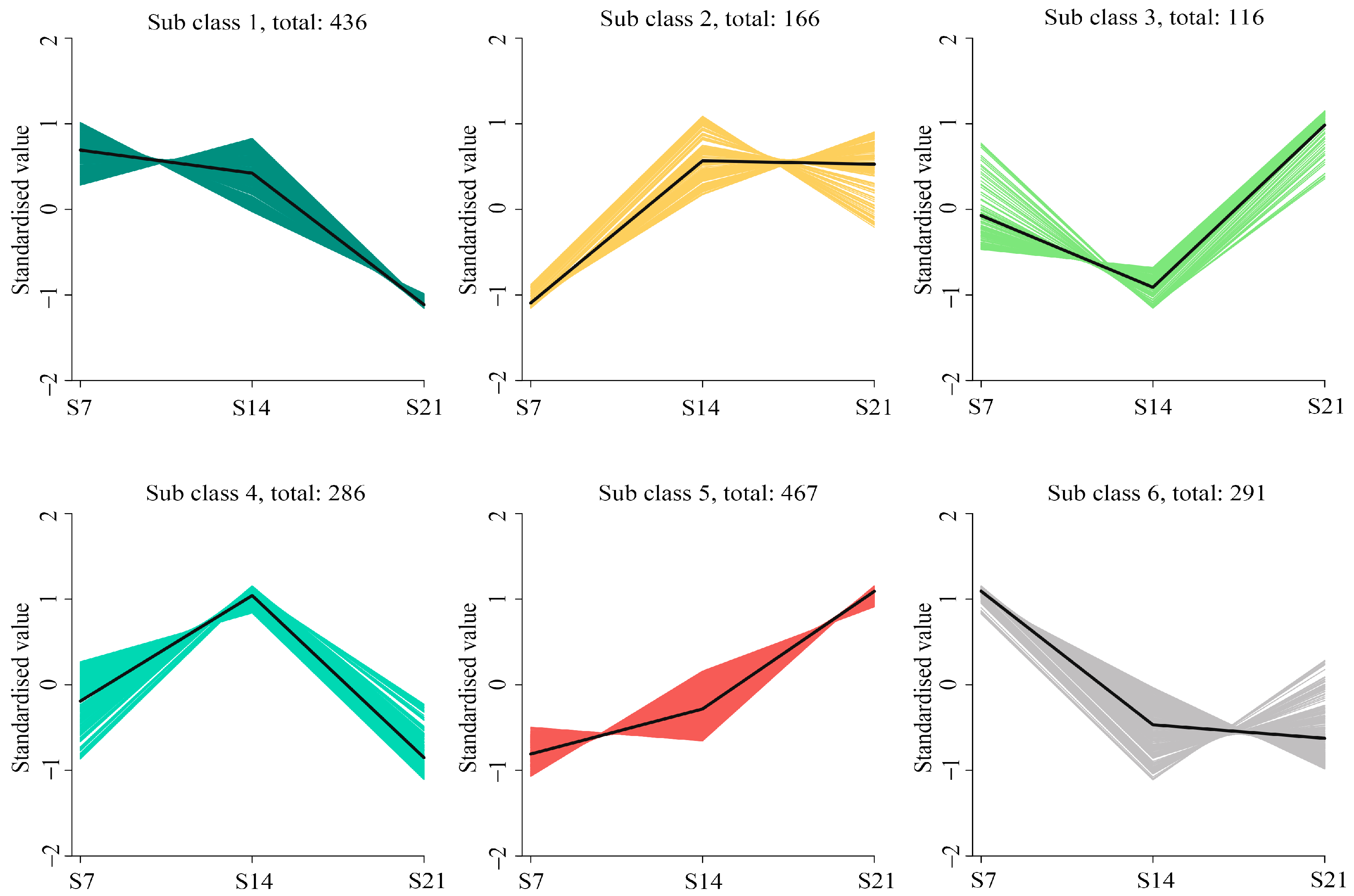
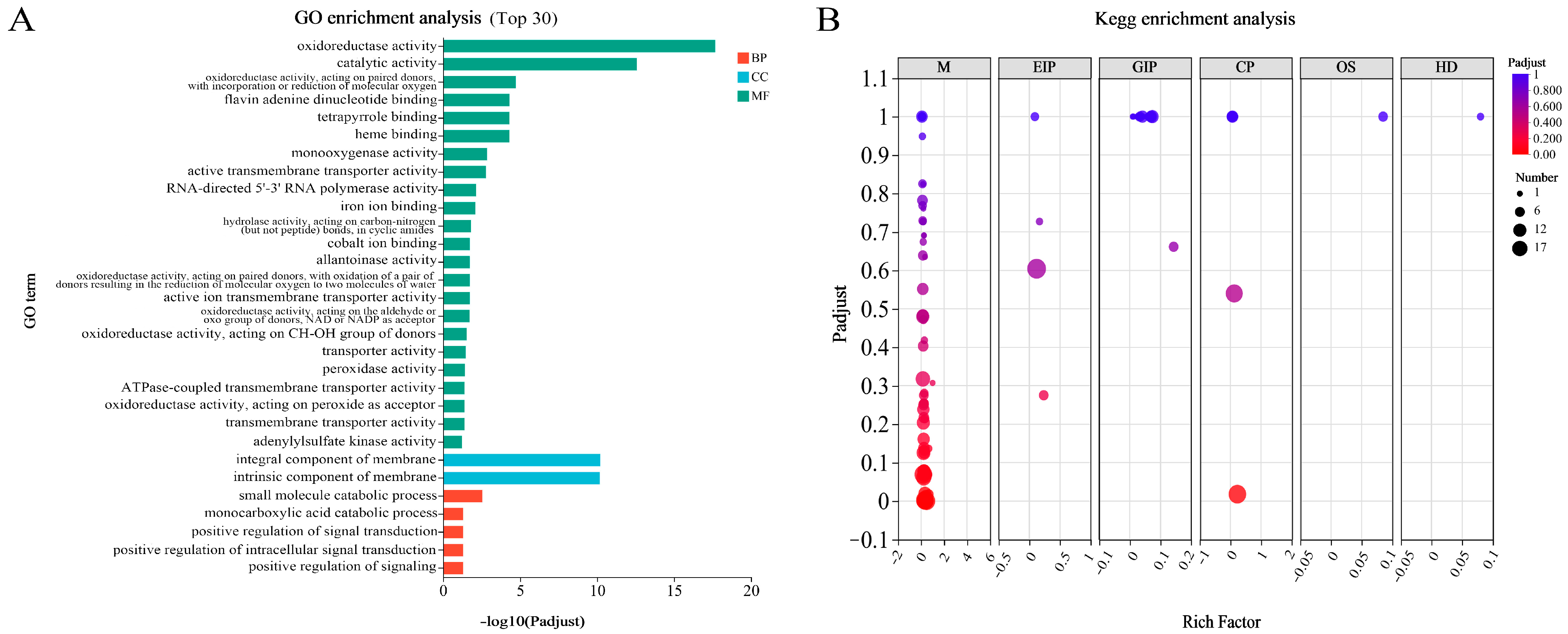
3.5. Functional Classification of DEGs
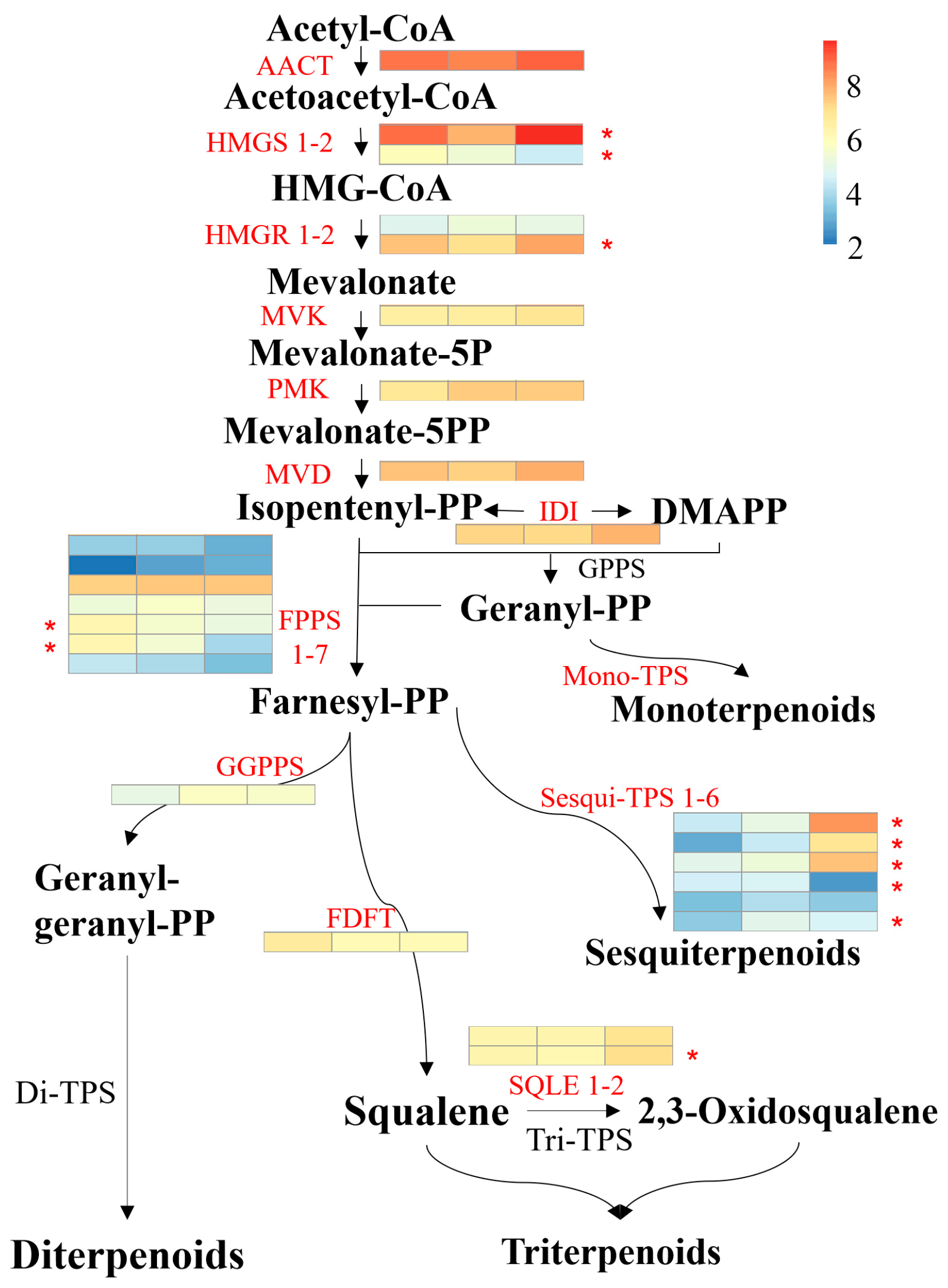
3.6. Genes Involved in Terpenoid Biosythesis
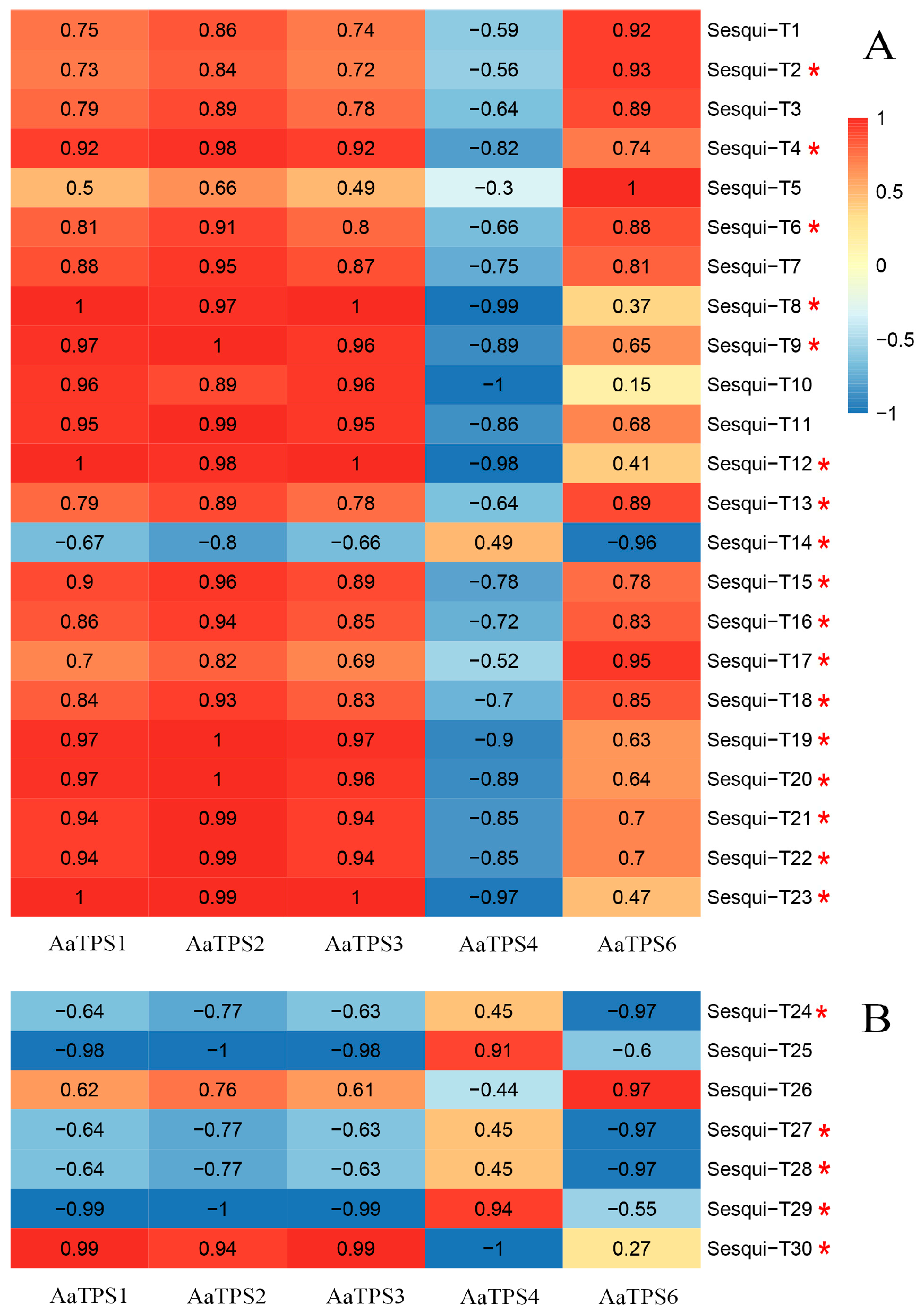
3.7. Correlation Analysis of Terpenoids and Differentially Expressed Terpene Synthase Genes
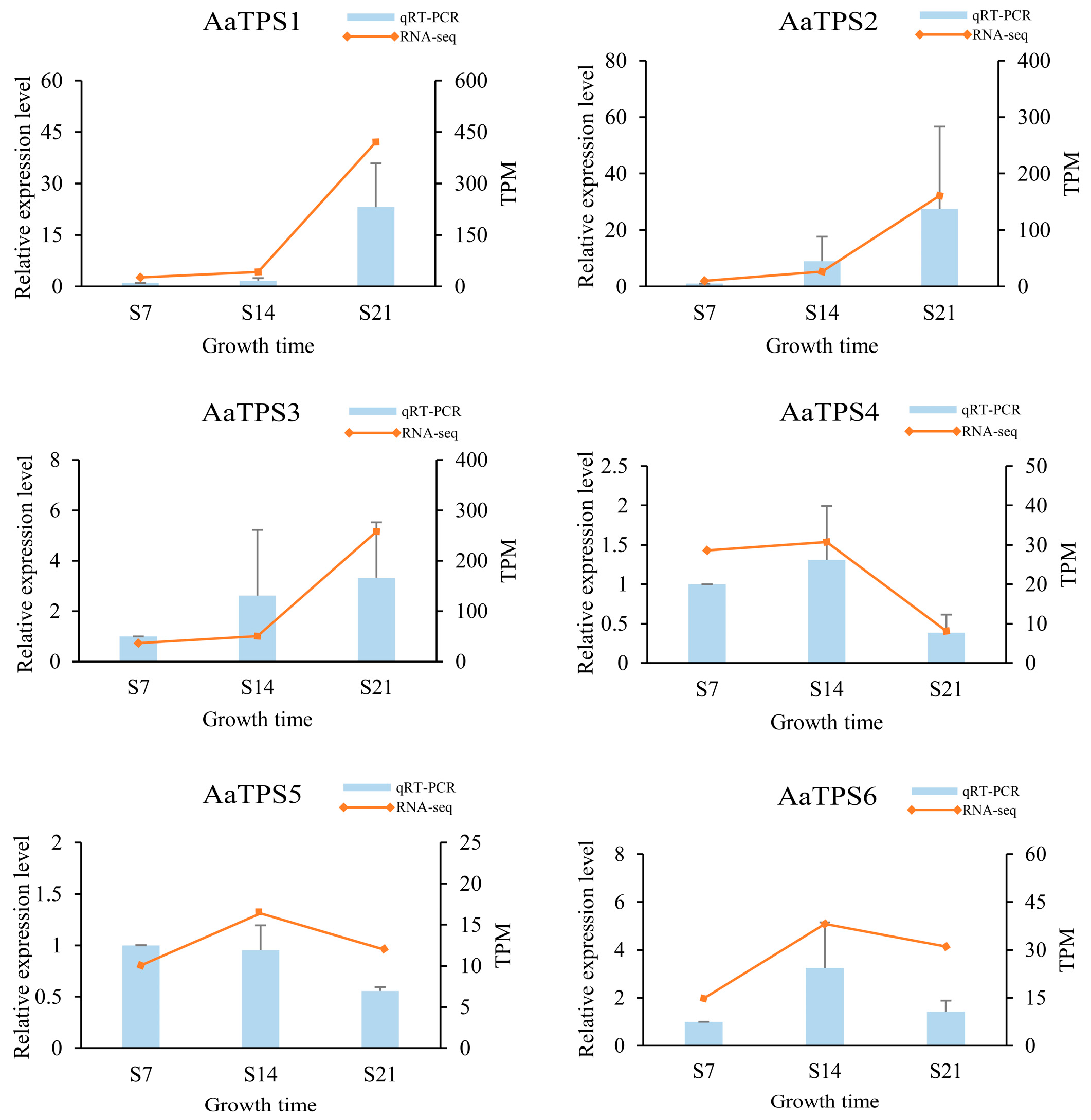
3.8. Verification of Gene Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Neumann, F.G.; Minko, G. The sirex wood wasp in Australian radiata pine plantations. Aust. For. 1981, 44, 46–63. [Google Scholar] [CrossRef]
- Haugen, D.A.; Underdown, M.G. Sirex noctilio control program in response to the 1987 Green Triangle outbreak. Aust. For. 1990, 53, 33–40. [Google Scholar] [CrossRef]
- Li, D.; Shi, J.; Lu, M.; Ren, L.; Zhen, C.; Luo, Y. Detection and Identification of the Invasive Sirex noctilio (Hymenoptera: Siricidae) Fungal Symbiont, Amylostereum areolatum (Russulales: Amylostereacea), in China and the Stimulating Effect of Insect Venom on Laccase Production by A. areolatum YQL03. J. Econ. Entomol. 2015, 108, 1136–1147. [Google Scholar] [CrossRef]
- Xu, Q.; Cao, L.; Ma, J.; Ren, L.; Sun, X.; Luo, Y.; Shi, J. Study on the morphology and damage characteristics of Sirex noctilio. J. Environ. Entomol. 2018, 40, 299–305. [Google Scholar]
- Sun, X.; Tao, J.; Ren, L.; Shi, J.; Luo, Y. Identification of Sirex noctilio (Hymenoptera: Siricidae) Using a Species-Specific Cytochrome C Oxidase Subunit I PCR Assay. J. Econ. Entomol. 2016, 109, 1424–1430. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, X.; Ren, L.; Shi, J.; Luo, Y. Variety of Endophytic Fungi Associated with Conifers in Mixed Conifer Forests Invaded by Sirex noctilio. Sci. Silvae Sin. 2017, 53, 81–89. [Google Scholar] [CrossRef]
- Francke-Grosmann, H. On the symbiosis of wood wasps (Siricinae) with fungi. Z. Angew. Entomol. 1939, 25, 647–679. Available online: https://www.fabinet.up.ac.za/src.fabinet.up.ac.za/sirexweb/sirexlit/Francke-Grosmann%201939,%20Rutland%20translation.pdf (accessed on 2 February 2025). [CrossRef]
- Parkin, E.A. Symbiosis in Larval Siricidæ (Hymenoptera). Nature 1941, 147, 329. [Google Scholar] [CrossRef]
- Thomsen, I.M. Characters of fruitbodies, basidiospores and cultures useful for recognizing Amylostereum areolatum and A. chailletii. Mycotaxon 1998, 69, 419–428. [Google Scholar]
- Couts, M. The mechanism of pathogenicity of Sirex noctilio on Pinus radiata I. Effects of thesymbiotic fungus Amylostereum sp. (Thelophoraceae). Aust. J. Biol. Sci. 1969, 22, 915–924. [Google Scholar] [CrossRef]
- Couts, M. The mechanism of pathogenicity of Sirex noctilio on Pinus radiata I. Effects of S. noctilio mucus. Aust. J. Biol. Sci. 1969, 22, 1153–1162. [Google Scholar] [CrossRef]
- Fernández Ajó, A.A.; Martínez, A.; Villacide, J.; Corley, J. Behavioural response of the woodwasp Sirex noctilio to volatile emissions of its fungal symbiont. J. Appl. Entomol. 2015, 139, 654–659. [Google Scholar] [CrossRef]
- Sarvary, M.A.; Hajek, A.E.; Böröczky, K.; Raguso, R.A.; Cooperband, M.F. Investigating the effects of symbiotic fungi on the flight behaviour of Sirex noctilio (Hymenoptera: Siricidae). Can. Entomol. 2016, 148, 543–551. [Google Scholar] [CrossRef]
- Faal, H.; Cha, D.H.; Hajek, A.E.; Teale, S.A. A double-edged sword: Amylostereum areolatum odors attract both Sirex noctilio (Hymenoptera: Siricidae) and its parasitoid, Ibalia leucospoides. Fungal Ecol. 2021, 54, 101108. [Google Scholar] [CrossRef]
- Masagué, S.; Fernández, P.C.; Devescovi, F.; Segura, D.F.; De La Vega, G.J.; Corley, J.C.; Villacide, J.M.; Martínez, A.S. Oviposition substrate location by the invasive woodwasp Sirex noctilio: The combined effect of chemical cues emitted by its obligate symbiont Amylostereum areolatum and different host-tree species. Pest Manag. Sci. 2023, 79, 3959–3969. [Google Scholar] [CrossRef]
- Wang, L.X.; Ren, L.L.; Liu, X.B.; Shi, J.; Wang, J.Z.; Luo, Y.Q. Effects of endophytic fungi in Mongolian pine on the selection behavior of woodwasp (Sirex noctilio) and the growth of its fungal symbiont. Pest Manag. Sci. 2018, 75, 492–505. [Google Scholar] [CrossRef]
- Wang, M.; Gao, C.; Xu, Q.; Fu, N.; Li, J.; Ren, L.; Luo, Y. Different genotypes and species of symbiotic fungi mediate the behavioral response of invasive Sirex noctilio fabricius (Hymenoptera: Siricidae). Front. Microbiol. 2024, 15, 1341646. [Google Scholar] [CrossRef] [PubMed]
- Bashford, R. The development of static trapping systems to monitor for wood-boring insects in forestry plantations. Aust. For. 2008, 71, 236–241. [Google Scholar] [CrossRef]
- Böröczky, K.; Zylstra, K.E.; Mastro, V.C.; Tumlinson, J.H. Volatile Profiles and Trap Catches of Two Pine-Host Species of Sirex noctilio. In Proceedings. 20th U.S. Department of Agriculture Interagency Research Forum on Invasive Species 2009; U.S. Department of Agriculture: New York, NY, USA, 2009; p. 10. [Google Scholar]
- Quin, M.B.; Flynn, C.M.; Schmidt-Dannert, C. Traversing the fungal terpenome. Nat. Prod. Rep. 2014, 31, 1449–1473. [Google Scholar] [CrossRef]
- Wang, Q.; Cao, R.; Zhang, Y.; Qi, P.; Wang, L.; Fang, S. Biosynthesis and regulation of terpenoids from basidiomycetes: Exploration of new research. AMB Express 2021, 11, 150. [Google Scholar] [CrossRef]
- Pan, L.; Huang, J.; Lv, J.; Wang, G.; Hu, D.; Gao, H. Biosynthesis of fungal terpenoids. Nat. Prod. Rep. 2024, 41, 748–783. [Google Scholar] [CrossRef]
- Wu, J.; Yang, X.; Duan, Y.; Wang, P.; Qi, J.; Gao, J.-M.; Liu, C. Biosynthesis of Sesquiterpenes in Basidiomycetes: A Review. J. Fungi 2022, 8, 913. [Google Scholar] [CrossRef]
- Zhang, P.; Qi, J.; Duan, Y.; Gao, J.-m.; Liu, C. Research Progress on Fungal Sesterterpenoids Biosynthesis. J. Fungi 2022, 8, 1080. [Google Scholar] [CrossRef]
- Liu, Z.; Tong, X.; Liu, R.; Zou, L. Metabolome and Transcriptome Profiling Reveal That Four Terpenoid Hormones Dominate the Growth and Development of Sanghuangporus baumii. J. Fungi 2022, 8, 648. [Google Scholar] [CrossRef]
- Xu, X.; Li, C.; Wu, F.; Zhao, S.; Chen, T.; You, H.; Lin, Y.; Zou, X. Integrated Transcriptomic and Targeted Metabolomic Analysis Reveals the Key Genes Involved in Triterpenoid Biosynthesis of Ganoderma lucidum. J. Fungi 2025, 11, 57. [Google Scholar] [CrossRef]
- Lu, M.J.; Fan, W.; Wang, W.; Chen, T.; Tang, Y.; Chu, F.; Chang, T.; Wang, S.; Li, M.; Chen, Y.; et al. Genomic and transcriptomic analyses of the medicinal fungus Antrodia cinnamomea for its metabolite biosynthesis and sexual development. Proc. Natl. Acad. Sci. USA 2014, 111, E4743–E4752. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Y.; Yuan, X.; Zhang, H.; Zheng, Y. Genomic Features of Taiwanofungus gaoligongensis and the Transcriptional Regulation of Secondary Metabolite Biosynthesis. J. Fungi 2024, 10, 826. [Google Scholar] [CrossRef]
- Meng, K.; Lv, J.; Zhang, T.; Liu, Y.; Zhang, P.; Zhang, Y.; Hu, B.; Huang, Q.; Xie, B.; Fu, J. Chromosome-Scale Genome and Transcriptomic Analyses Reveal Differential Regulation of Terpenoid Secondary Metabolites in Hericium coralloides. J. Fungi 2024, 10, 704. [Google Scholar] [CrossRef] [PubMed]
- Fu, N.; Wang, M.; Wang, L.; Luo, Y.; Ren, L. Genome Sequencing and Analysis of the Fungal Symbiont of Sirex noctilio, Amylostereum areolatum: Revealing the Biology of Fungus-Insect Mutualism. mSphere 2020, 5, e00301-20. [Google Scholar] [CrossRef]
- Wang, L.X.; Ren, L.L.; Luo, Y.Q.; Wang, M.; Zhan, Z.Y. The Invention Discloses a Culture Medium and a Culture Method of Amylostereum areolatum. CN Patent CN113475312A, 8 October 2021. Available online: https://kns.cnki.net/kcms2/article/abstract?v=IVqNFfq6ZnIPeVOhmPs3Ymv6tYChpJ01sT-JG_NhdBSlQWMuCQvMZ1v-9uOMsHjYkyJhqAXmcAK1wuiwHhKbgBcOAbhTlQ32LAgCDCmqJcfpQDWBUTQgI5R-ih0cFPc48rVxroA_AFgxImNy_BruZvWkNuYvMhbKm6_3y5uAKn9yP3745w6r6w==&uniplatform=NZKPT&language=CHS (accessed on 2 February 2025).
- Jofré, N.; Pildain, M.; Cirigliano, A.; Cabrera, G.; Corley, J.; Martínez, A. Host selection by Ibalia leucospoides based on temporal variations of volatiles from the hosts’ fungal symbiont. J. Appl. Entomol. 2016, 140, 736–743. [Google Scholar] [CrossRef]
- Li, D.P. Collaborative Damage Effect of the Invasive Sirex noctilio and its Fungal Symbiont Amylostereum areolatum on Host Tree; Beijing Forestry University: Beijing, China, 2015. [Google Scholar]
- Yu, S.; Yang, F.; Pu, J.; Guo, Q.; Zou, Q.; Zhang, H.; Liu, M.; Zheng, Y.; Wang, T. Integrated transcriptomic and metabolomic analyses reveal the characteristics of volatile oils in Chrysanthemum morifolium. Sci. Hortic. 2023, 315, 111983. [Google Scholar] [CrossRef]
- Li, C.; Xu, Y.; Li, Z.; Cheng, P.; Yu, G. Transcriptomic and metabolomic analysis reveals the potential mechanisms underlying the improvement of β-carotene and torulene production in Rhodosporidiobolus colostri under low temperature treatment. Food Res. Int. 2022, 156, 111158. [Google Scholar] [CrossRef] [PubMed]
- Fu, N.; Li, J.; Wang, M.; Ren, L.; Zong, S.; Luo, Y. Identification and validation of reference genes for gene expression analysis in different development stages of Amylostereum areolatum. Front. Microbiol. 2022, 12, 827241. [Google Scholar] [CrossRef] [PubMed]
- Liao, P.; Hemmerlin, A.; Bach, T.J.; Chye, M.L. The potential of the mevalonate pathway for enhanced isoprenoid production. Biotechnol. Adv. 2016, 34, 697–713. [Google Scholar] [CrossRef]
- Perez-Gil, J.; Rodriguez-Concepcion, M. Metabolic plasticity for isoprenoid biosynthesis in bacteria. Biochem. J. 2013, 452, 19–25. [Google Scholar] [CrossRef]
- Agger, S.; Lopez-Gallego, F.; Schmidt-Dannert, C. Diversity of sesquiterpene synthases in the basidiomycete Coprinus cinereus. Mol. Microbiol. 2009, 72, 1181–1195. [Google Scholar] [CrossRef]
- Wawrzyn, G.; Quin, M.; Choudhary, S.; López-Gallego, F.; Schmidt-Dannert, C. Draft Genome of Omphalotus olearius Provides a Predictive Framework for Sesquiterpenoid Natural Product Biosynthesis in Basidiomycota. Chem. Biol. 2012, 19, 772–783. [Google Scholar] [CrossRef]
- Zhang, C.; Chen, X.; Orban, A.; Shukal, S.; Birk, F.; Too, H.-P.; Rühl, M. Agrocybe aegerita Serves As a Gateway for Identifying Sesquiterpene Biosynthetic Enzymes in Higher Fungi. ACS Chem. Biol. 2020, 15, 1268–1277. [Google Scholar] [CrossRef]
- González-Hernández, R.A.; Valdez-Cruz, N.A.; Macías-Rubalcava, M.L.; Trujillo-Roldán, M.A. Overview of fungal terpene synthases and their regulation. World J. Microbiol. Biotechnol. 2023, 39, 194. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Clean Reads | Clean Bases (bp) | Error Rate (%) | GC Content (%) | Q20 (%) | Q30 (%) | Mapping Ratio (%) |
|---|---|---|---|---|---|---|---|
| S7_1 | 51,889,968 | 7,670,121,267 | 0.0253 | 57.46 | 97.82 | 93.98 | 89.37 |
| S7_2 | 43,616,060 | 6,412,761,653 | 0.0251 | 57.54 | 97.89 | 94.19 | 87.81 |
| S7_3 | 48,759,422 | 7,206,339,669 | 0.0252 | 57.23 | 97.85 | 94.02 | 88.63 |
| S14_1 | 46,067,288 | 6,825,290,603 | 0.0251 | 57.19 | 97.88 | 94.11 | 89.01 |
| S14_2 | 40,644,990 | 6,012,925,517 | 0.0251 | 57.4 | 97.88 | 94.16 | 89.61 |
| S14_3 | 46,154,948 | 6,849,932,991 | 0.0253 | 57.27 | 97.81 | 93.98 | 89.21 |
| S21_1 | 47,690,676 | 7,005,804,935 | 0.0249 | 57.45 | 97.98 | 94.38 | 91.95 |
| S21_2 | 47,365,588 | 6,963,548,962 | 0.0248 | 57.28 | 97.99 | 94.41 | 91.6 |
| S21_3 | 45,241,166 | 6,714,329,801 | 0.0251 | 57.31 | 97.89 | 94.19 | 90.66 |
| No. | Comparison | Gene ID | Name | Fold Change | Log2FC | p Value | p-Adjust | Type |
|---|---|---|---|---|---|---|---|---|
| 1 | S14 vs. S7 | Gene_EVM002515 | HMGS1 | 0.4172 | −1.2610 | 0.0000 | 0.0001 | down |
| 2 | Gene_EVM007970 | FPPS6 | 0.4899 | −1.0294 | 0.0034 | 0.0405 | down | |
| 3 | Gene_EVM006404 | AaTPS2 | 2.2842 | 1.1917 | 0.0016 | 0.0244 | up | |
| 4 | Gene_EVM007245 | AaTPS6 | 2.2338 | 1.1595 | 0.0000 | 0.0001 | up | |
| 5 | S21 vs. S14 | Gene_EVM002515 | HMGS1 | 4.4473 | 2.1529 | 0.0000 | 0.0000 | up |
| 6 | Gene_EVM010955 | HMGR2 | 2.3009 | 1.2022 | 0.0033 | 0.0312 | up | |
| 7 | Gene_EVM007970 | FPPS6 | 0.3928 | −1.3481 | 0.0012 | 0.0152 | down | |
| 8 | Gene_EVM008329 | SQLE2 | 2.3181 | 1.2130 | 0.0001 | 0.0025 | up | |
| 9 | Gene_EVM006403 | AaTPS1 | 11.2815 | 3.4959 | 0.0000 | 0.0000 | up | |
| 10 | Gene_EVM002513 | AaTPS3 | 5.8442 | 2.5470 | 0.0052 | 0.0435 | up | |
| 11 | Gene_EVM003164 | AaTPS4 | 0.2998 | −1.7378 | 0.0000 | 0.0000 | down | |
| 12 | S21 vs. S7 | Gene_EVM002681 | HMGS2 | 0.3376 | −1.5668 | 0.0000 | 0.0003 | down |
| 13 | Gene_EVM007969 | FPPS5 | 0.4732 | −1.0793 | 0.0030 | 0.0182 | down | |
| 14 | Gene_EVM007970 | FPPS6 | 0.1928 | −2.3752 | 0.0000 | 0.0000 | down | |
| 15 | Gene_EVM006403 | AaTPS1 | 15.7895 | 3.9809 | 0.0000 | 0.0000 | up | |
| 16 | Gene_EVM006404 | AaTPS2 | 16.7023 | 4.0620 | 0.0001 | 0.0009 | up | |
| 17 | Gene_EVM002513 | AaTPS3 | 6.9804 | 2.8033 | 0.0013 | 0.0090 | up | |
| 18 | Gene_EVM003164 | AaTPS4 | 0.2760 | −1.8573 | 0.0000 | 0.0000 | down | |
| 19 | Gene_EVM007245 | AaTPS6 | 2.0777 | 1.0550 | 0.0001 | 0.0011 | up |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Fu, N.; Wang, M.; Zhan, Z.; Luo, Y.; Wu, J.; Ren, L. Integrative Transcriptome and Metabolome Analysis Reveals Candidate Genes Related to Terpenoid Synthesis in Amylostereum areolatum (Russulales: Amylostereaceae). J. Fungi 2025, 11, 383. https://doi.org/10.3390/jof11050383
Wang L, Fu N, Wang M, Zhan Z, Luo Y, Wu J, Ren L. Integrative Transcriptome and Metabolome Analysis Reveals Candidate Genes Related to Terpenoid Synthesis in Amylostereum areolatum (Russulales: Amylostereaceae). Journal of Fungi. 2025; 11(5):383. https://doi.org/10.3390/jof11050383
Chicago/Turabian StyleWang, Lixia, Ningning Fu, Ming Wang, Zhongyi Zhan, Youqing Luo, Jianrong Wu, and Lili Ren. 2025. "Integrative Transcriptome and Metabolome Analysis Reveals Candidate Genes Related to Terpenoid Synthesis in Amylostereum areolatum (Russulales: Amylostereaceae)" Journal of Fungi 11, no. 5: 383. https://doi.org/10.3390/jof11050383
APA StyleWang, L., Fu, N., Wang, M., Zhan, Z., Luo, Y., Wu, J., & Ren, L. (2025). Integrative Transcriptome and Metabolome Analysis Reveals Candidate Genes Related to Terpenoid Synthesis in Amylostereum areolatum (Russulales: Amylostereaceae). Journal of Fungi, 11(5), 383. https://doi.org/10.3390/jof11050383