
Integrated Amino Acid Profiling and 4D-DIA Proteomics Reveal Protein Quality Divergence and Metabolic Adaptation in Cordyceps Species

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Determination of Crude Protein and Amino Acid
2.3. Proteomics Profiling
2.4. Bioinformatics Analysis
2.5. Statistical Analysis
3. Results
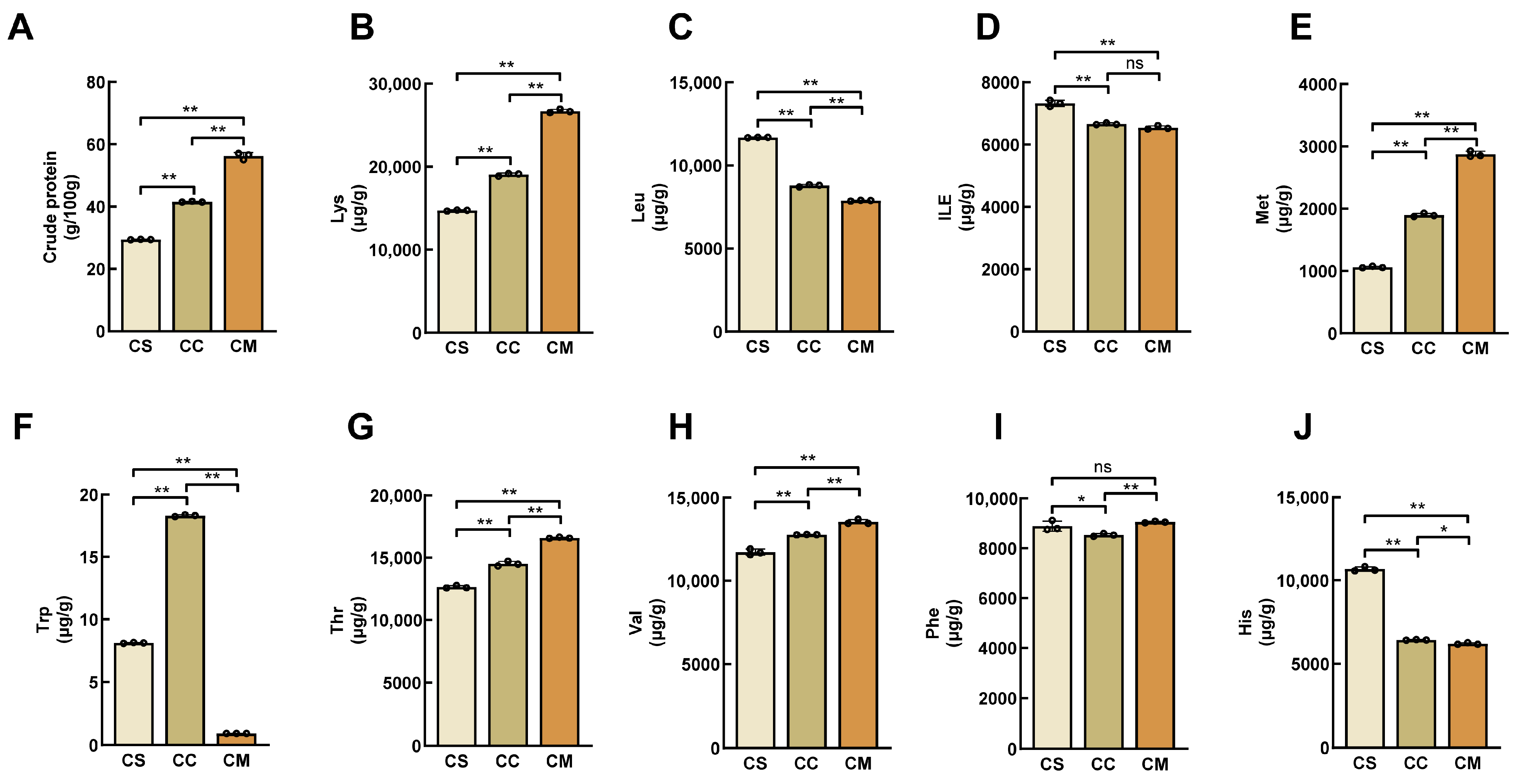
3.1. Determination of Crude Protein and Amino Acids
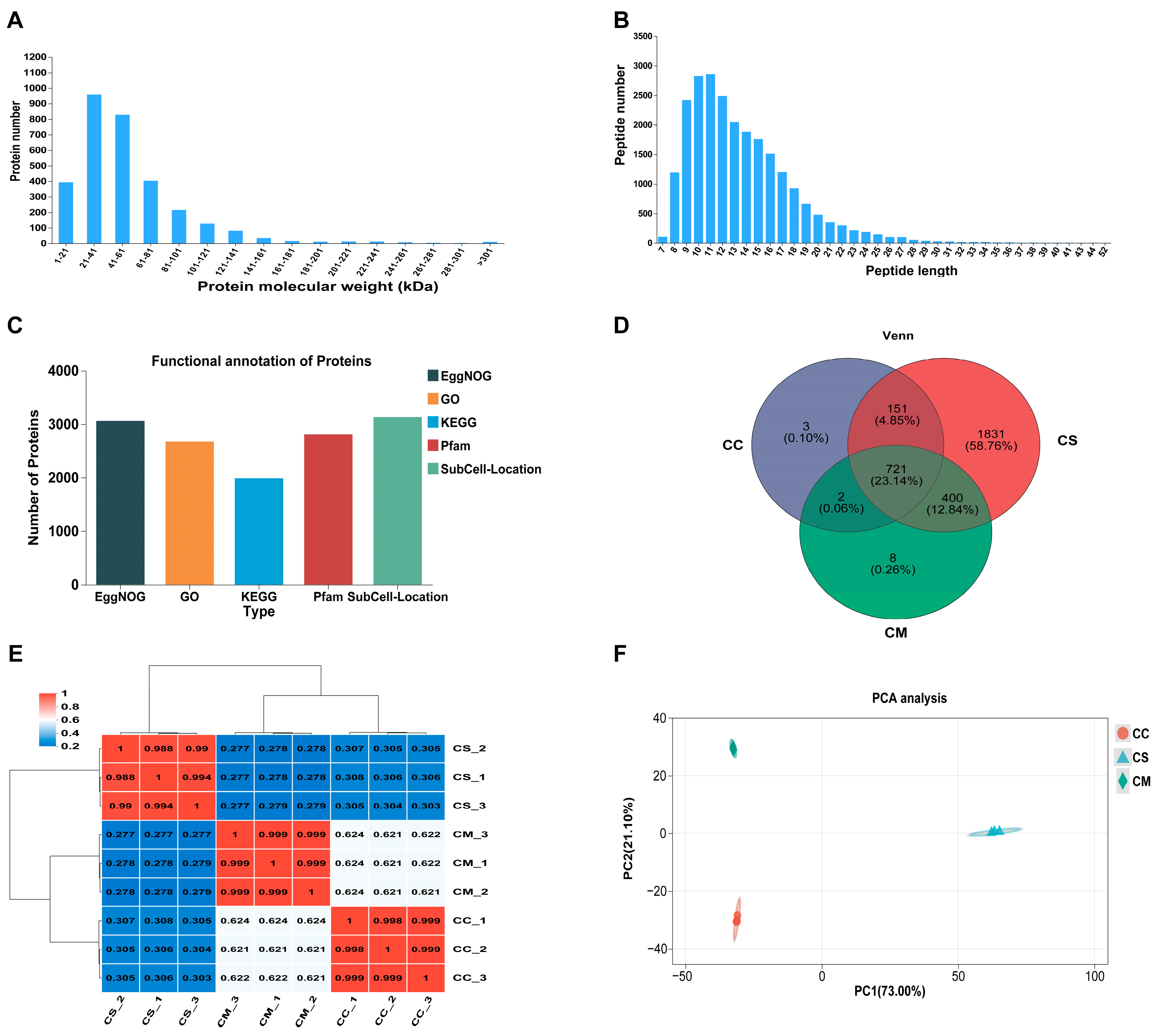
3.2. Comprehensive Analysis of Proteomic Profiles
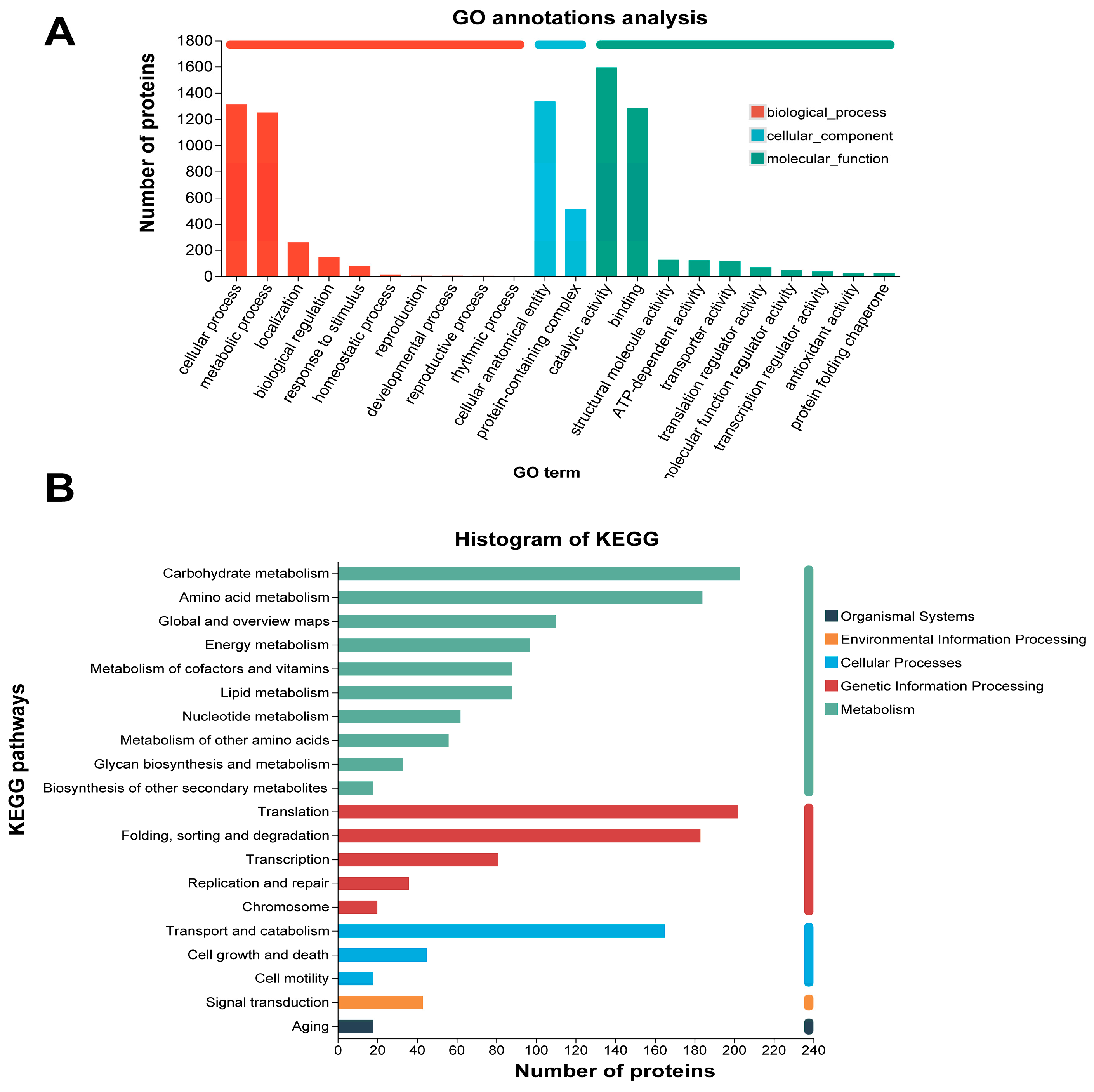
3.3. Annotation Analysis of Total Protein
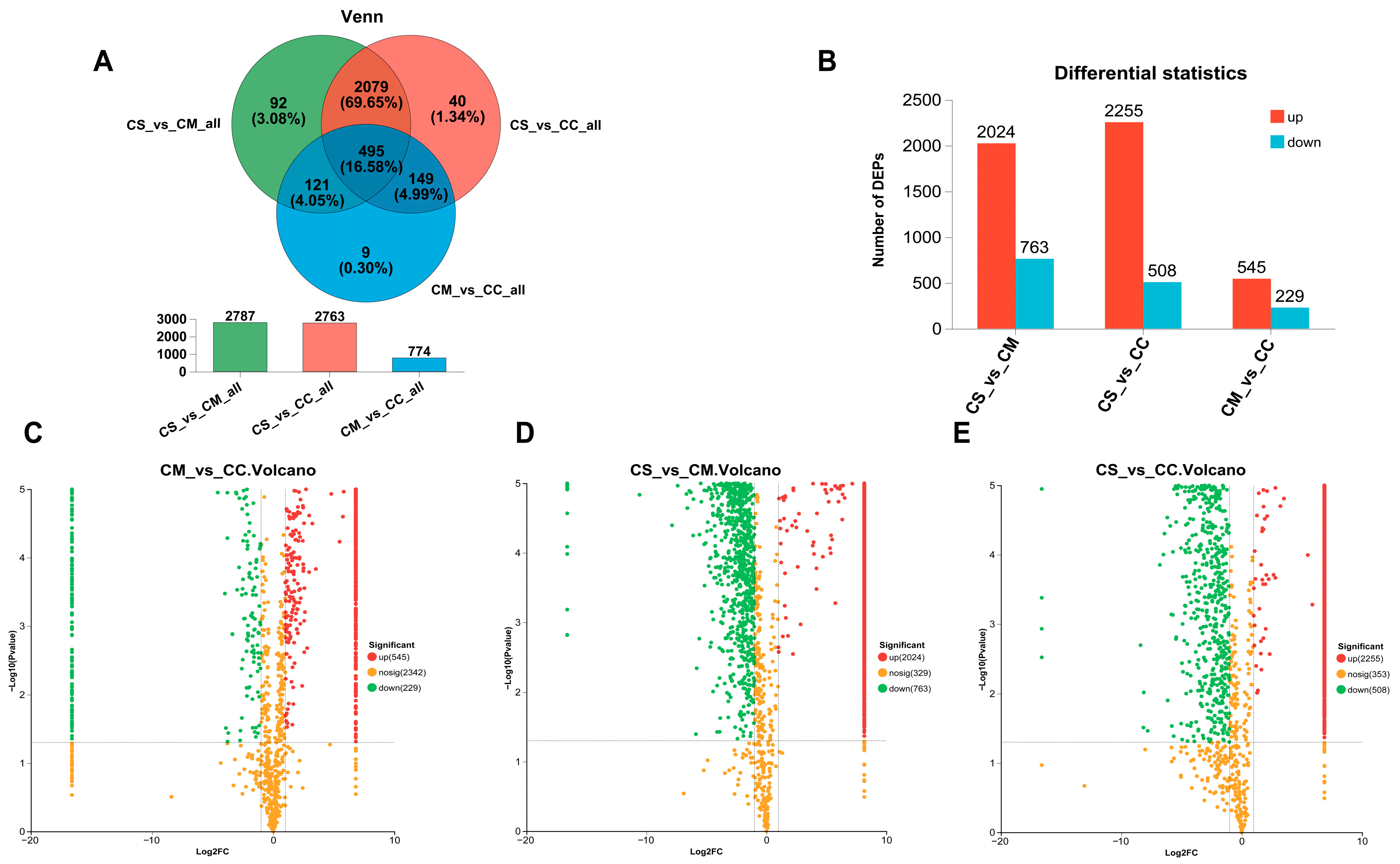
3.4. Screening and Analysis of Differential Proteins
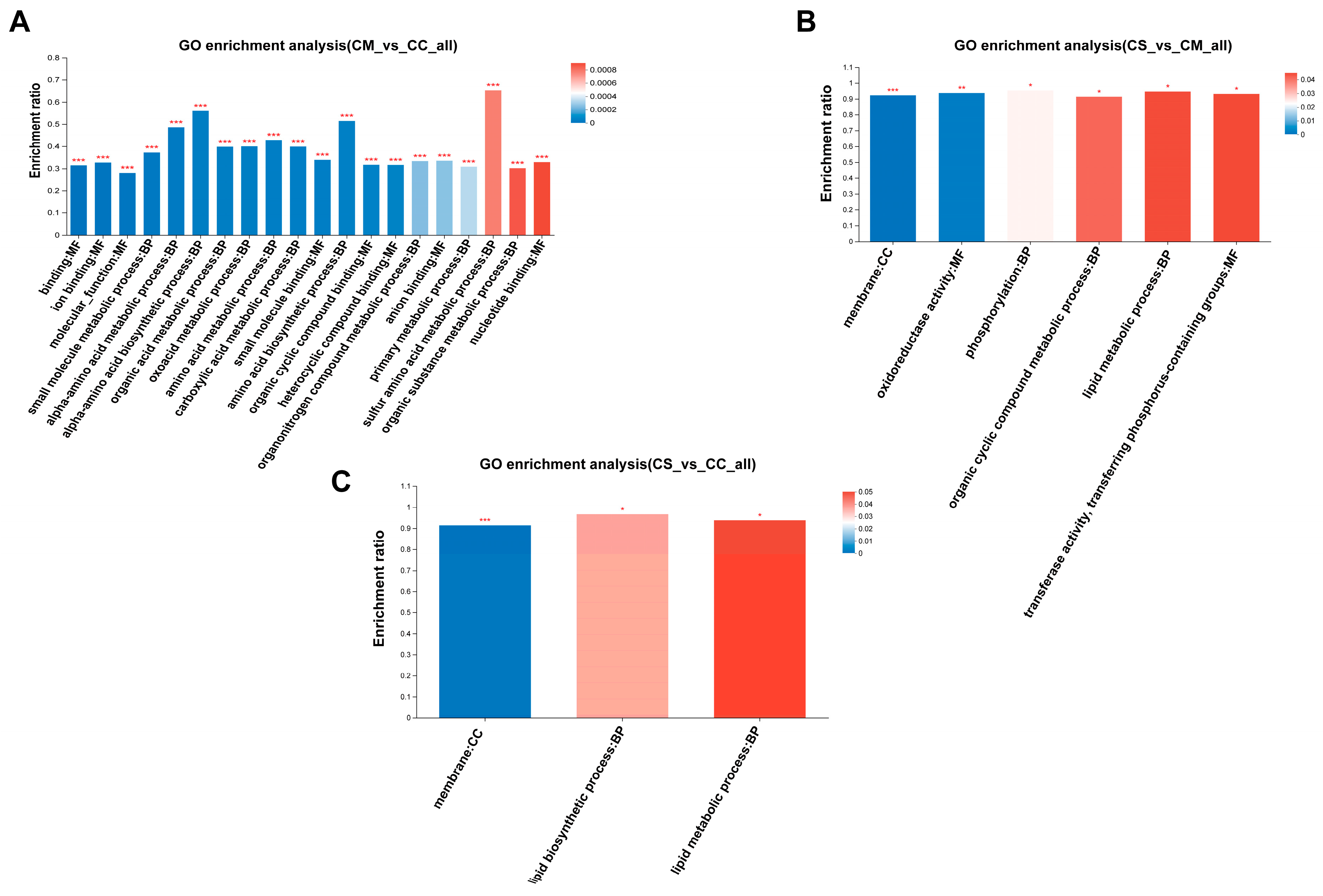
3.5. GO Enrichment Analysis of DEPs
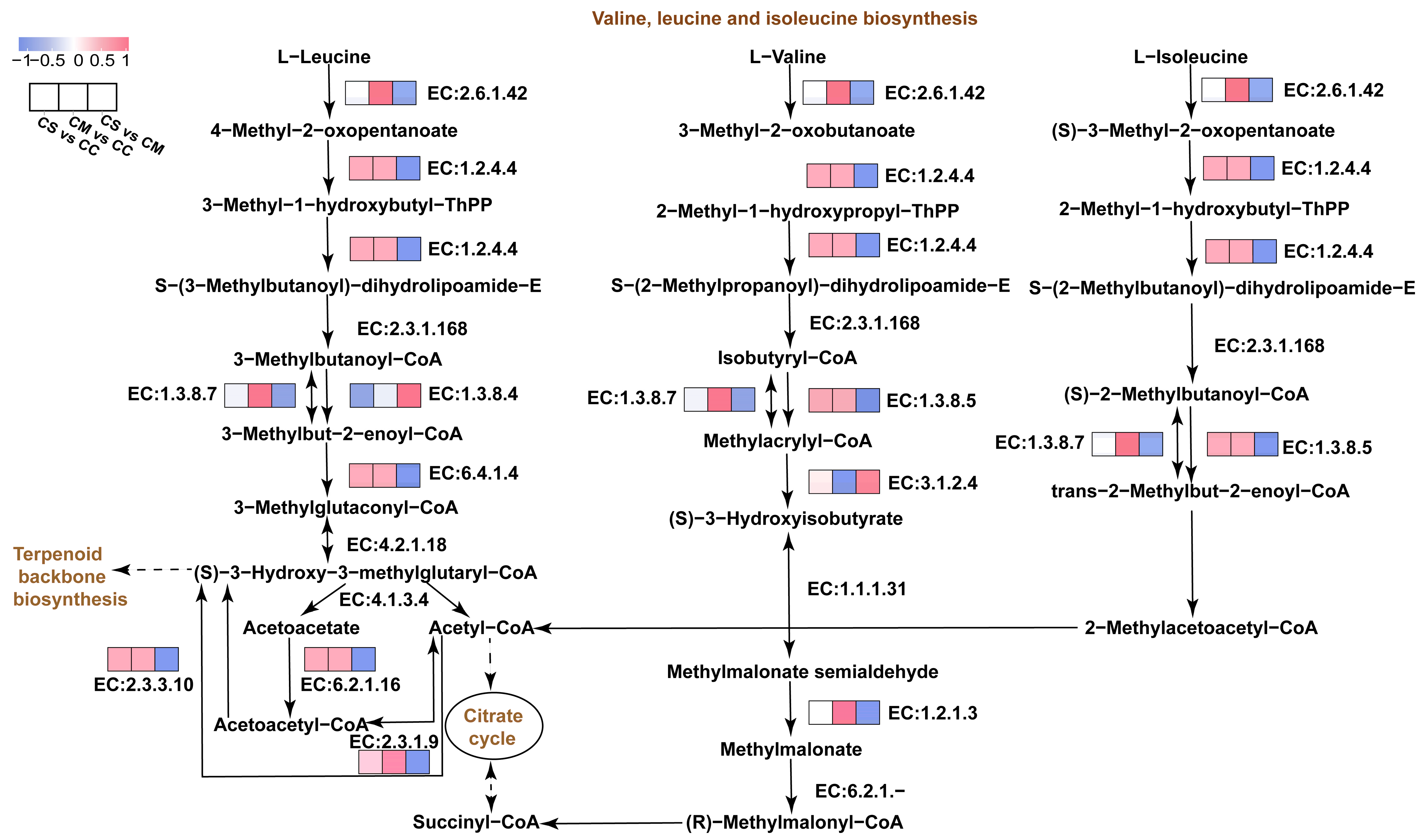
3.6. Analysis of Enrichment Pathways for Key DEPs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Cordyceps sinensis | CS |
| Cordyceps militaris | CM |
| Cordyceps cicadae | CC |
| Four-dimensional data-independent acquisition | 4D-DIA |
| Differentially expressed proteins | DEPs |
| Essential amino acids | EAAs |
| Non-essential amino acids | NEAAs |
| Carboxypeptidase A1 | CPA1 |
| Argininosuccinate synthase 1 | ASS1 |
| Branched-chain keto acid dehydrogenase E1 subunit alpha | BCKDHA |
| Acyl-CoA dehydrogenase short/branched chain | ACADSB |
| Heat shock proteins | HSPs |
| Liquid chromatography | LC |
| Data-independent acquisition | DIA |
| Mass spectrometry | MS |
| Maximum Entropy | MaxEnt |
| Gene Ontology | GO |
| Kyoto Encyclopedia of Genes and Genomes | KEGG |
| Pyruvate carboxylase | PC |
| Fumarate hydratase | FH |
| Dihydrolipoyl dehydrogenase | DLD |
| Deoxyuridine 5′-triphosphate nucleotidohydrolase | DUT |
| Uracil phosphoribosyltransferase | UPRT |
| Principal component analysis | PCA |
| Branched-chain amino acids | BCAAs |
| Branched-chain-amino-acid aminotransferase | BCAT |
| Branched-chain dihydrolipoamide acyltransferase | BCDAT |
| Medium-chain specific acyl-CoA dehydrogenase | ACADM |
| 3-hydroxyisobutyryl-CoA hydrolase | HIBCH |
| Acetyl-CoA acetyltransferase | ACAT |
| Glutamate oxalacetate transaminases 2 | GOT2 |
| Glutamate dehydrogenase | GDH |
| Aspartate transcarbamoylase | ATCase |
| Pancreatic ductal adenocarcinoma | PDAC |
| Acute myocardial ischemia | AMI |
References
- Holliday, J.; Cleaver, M.P. Medicinal Value of the Caterpillar Fungi Species of the Genus Cordyceps (Fr.) Link (Ascomycetes). A Review. Int. J. Med. Mushrooms 2008, 10, 219–234. [Google Scholar] [CrossRef]
- Liu, J.-X.; Cai, Q.-F.; Lin, X.-Z.; Gao, T.-T.; Luan, Y.-X. Research progress on active components, functional characteristics and applications of edible fungi. Food Med. Homol. 2025, 3, 9420087. [Google Scholar] [CrossRef]
- Zhang, G.-P.; Pan, Y.-M.; Ye, S.-M.; Lu, Y.-C.; Fan, X.-J.; Zhang, A.-Q. Bioactive components of Ganoderma lucidum and their efficacy and application in cosmetics. Food Med. Homol. 2025, 2, 9420044. [Google Scholar] [CrossRef]
- Cui, Y.-L.; Li, B. Hypoglycemic effects of edible fungus polysaccharides: A mini review. Food Med. Homol. 2025, 2, 9420046. [Google Scholar] [CrossRef]
- Xu, J.; Shen, R.; Jiao, Z.; Chen, W.; Peng, D.; Wang, L.; Yu, N.; Peng, C.; Cai, B.; Song, H.; et al. Current Advancements in Antitumor Properties and Mechanisms of Medicinal Components in Edible Mushrooms. Nutrients 2022, 14, 2622. [Google Scholar] [CrossRef]
- Niego, A.G.; Rapior, S.; Thongklang, N.; Raspé, O.; Jaidee, W.; Lumyong, S.; Hyde, K.D. Macrofungi as a Nutraceutical Source: Promising Bioactive Compounds and Market Value. J. Fungi 2021, 7, 397. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Sar, T.; Mahboubi, A.; Fristedt, R.; Taherzadeh, M.J.; Undeland, I. In vitro protein digestibility of edible filamentous fungi compared to common food protein sources. Food Biosci. 2023, 54, 102862. [Google Scholar] [CrossRef]
- Saeed, F.; Afzaal, M.; Khalid, A.; Shah, Y.A.; Ateeq, H.; Islam, F.; Akram, N.; Ejaz, A.; Nayik, G.A.; Shah, M.A. Role of mycoprotein as a non-meat protein in food security and sustainability: A review. Int. J. Food Prop. 2023, 26, 683–695. [Google Scholar] [CrossRef]
- Maruca, A.; Moraca, F.; Rocca, R.; Molisani, F.; Alcaro, F.; Gidaro, M.C.; Alcaro, S.; Costa, G.; Ortuso, F. Chemoinformatic Database Building and in Silico Hit-Identification of Potential Multi-Targeting Bioactive Compounds Extracted from Mushroom Species. Molecules 2017, 22, 1571. [Google Scholar] [CrossRef]
- Lin, T.-Y.; Hua, W.-J.; Yeh, H.; Tseng, A.-J. Functional proteomic analysis reveals that fungal immunomodulatory protein reduced expressions of heat shock proteins correlates to apoptosis in lung cancer cells. Phytomedicine 2021, 80, 153384. [Google Scholar] [CrossRef]
- Chen, X.; Ding, Z.-Y.; Wang, W.-Q.; Siu, K.-C.; Wu, J.-Y. An antioxidative galactomannan–protein complex isolated from fermentation broth of a medicinal fungus Cs-HK1. Carbohydr. Polym. 2014, 112, 469–474. [Google Scholar] [CrossRef]
- Kong, B.H.; Yap, C.A.; Razif, M.F.M.; Ng, S.T.; Tan, C.S.; Fung, S.Y. Antioxidant and Cytotoxic Effects and Identification of Ophiocordyceps sinensis Bioactive Proteins Using Shotgun Proteomic Analysis. Food Technol. Biotechnol. 2021, 59, 201–208. [Google Scholar] [CrossRef]
- Olatunji, O.J.; Tang, J.; Tola, A.; Auberon, F.; Oluwaniyi, O.; Ouyang, Z. The genus Cordyceps: An extensive review of its traditional uses, phytochemistry and pharmacology. Fitoterapia 2018, 129, 293–316. [Google Scholar] [CrossRef]
- Dong, C.; Guo, S.; Wang, W.; Liu, X. Cordyceps industry in China. Mycology 2015, 6, 121–129. [Google Scholar] [CrossRef]
- Yan, Y.; Li, Y.; Wang, W.-J.; He, J.-S.; Yang, R.-H.; Wu, H.-J.; Wang, X.-L.; Jiao, L.; Tang, Z.; Yao, Y.-J. Range shifts in response to climate change of Ophiocordyceps sinensis, a fungus endemic to the Tibetan Plateau. Biol. Conserv. 2017, 206, 143–150. [Google Scholar] [CrossRef]
- Tang, C.; Li, X.; Wang, T.; Wang, J.; Xiao, M.; He, M.; Chang, X.; Fan, Y.; Li, Y. Characterization of Metabolite Landscape Distinguishes Medicinal Fungus Cordyceps sinensis and other Cordyceps by UHPLC-Q Exactive HF-X Untargeted Metabolomics. Molecules 2023, 28, 7745. [Google Scholar] [CrossRef]
- Li, C.R.; Wang, Y.Q.; Cheng, W.M.; Chen, Z.A.; Hywel-Jones, N.; Li, Z.Z. Review on Research Progress and Prospects of Cicada Flower, Isaria cicadae (Ascomycetes). Int. J. Med. Mushrooms 2021, 23, 81–91. [Google Scholar] [CrossRef]
- Jędrejko, K.; Kała, K.; Sułkowska-Ziaja, K.; Krakowska, A.; Zięba, P.; Marzec, K.; Szewczyk, A.; Sękara, A.; Pytko-Polończyk, J.; Muszyńska, B. Cordyceps militaris-Fruiting Bodies, Mycelium, and Supplements: Valuable Component of Daily Diet. Antioxidants 2022, 11, 1861. [Google Scholar] [CrossRef]
- Zhang, M.; Sun, X.; Miao, Y.; Li, M.; Huang, L. Cordyceps cicadae and Cordyceps gunnii have closer species correlation with Cordyceps sinensis: From the perspective of metabonomic and MaxEnt models. Sci. Rep. 2022, 12, 20469. [Google Scholar] [CrossRef]
- Nxumalo, W.; Elateeq, A.A.; Sun, Y. Can Cordyceps cicadae be used as an alternative to Cordyceps militaris and Cordyceps sinensis?—A review. J. Ethnopharmacol. 2020, 257, 112879. [Google Scholar] [CrossRef]
- Xu, C.; Wu, F.; Zou, Z.; Mao, L.; Lin, S. Discovery of the chemical constituents, structural characteristics, and pharmacological functions of Chinese caterpillar fungus. Open Chem. 2023, 21, 20220337. [Google Scholar] [CrossRef]
- Yu, F.; Teo, G.C.; Kong, A.T.; Fröhlich, K.; Li, G.X.; Demichev, V.; Nesvizhskii, A.I. Analysis of DIA proteomics data using MSFragger-DIA and FragPipe computational platform. Nat. Commun. 2023, 14, 4154. [Google Scholar] [CrossRef]
- Huang, Y.; Ren, S.; Liu, Y.; Zhou, Y.; Wan, J.; Liu, L.; Zhu, Q. Studies of the binding mechanism between liquid smoke from tea tree branches and proteins in dry-cured tenderloin using 4D-DIA proteomics, synergistic multispectral analysis, and molecular docking techniques. Food Chem. 2025, 463, 141198. [Google Scholar] [CrossRef]
- Shen, H.P.; Dong, X.; Li, Z.B.; Wu, J.Z.; Zheng, C.M.; Hu, X.J.; Qian, C.; Wang, S.P.; Zhao, Y.L.; Li, J.C. Protein Profiles and Novel Molecular Biomarkers of Schizophrenia Based on 4D-DIA Proteomics. J. Proteome Res. 2024, 23, 2376–2385. [Google Scholar] [CrossRef]
- Teng, J.; Chen, L.; Yang, F.; Gao, P.; Yu, P.; Jiang, Q.; Xu, Y.; Xia, W.; Yu, D. Selection of texture-associated biomarkers in chilled and iced grass carp (Ctenopharyngodon idella) fillets via DIA-based proteomics. Food Res. Int. 2024, 188, 114505. [Google Scholar] [CrossRef]
- Liu, J.Y.; Chang, M.C.; Meng, J.L.; Feng, C.P.; Zhao, H.; Zhang, M.L. Comparative Proteome Reveals Metabolic Changes during the Fruiting Process in Flammulina velutipes. J. Agric. Food Chem. 2017, 65, 5091–5100. [Google Scholar] [CrossRef]
- GB_T5009.5-2016; National Food Safety Standard—Determination of Protein in Foods. National Health Commission of the People’s Republic of China: Beijing, China, 2016.
- Kamizake, N.K.K.; Gonçalves, M.M.; Zaia, C.T.B.V.; Zaia, D.A.M. Determination of total proteins in cow milk powder samples: A comparative study between the Kjeldahl method and spectrophotometric methods. J. Food Compos. Anal. 2003, 16, 507–516. [Google Scholar] [CrossRef]
- Jiang, Z.; Wang, Y.; Bai, S.; Bai, C.; Tu, Z.; Li, H.; Guo, P.; Liao, T.; Qiu, L. The viable but non-culturable (VBNC) status of Shewanella putrefaciens (S. putrefaciens) with thermosonication (TS) treatment. Ultrason. Sonochem. 2024, 109, 107008. [Google Scholar] [CrossRef]
- Abdul-Mumeen, I.; Zakpaa, H.D.; Mills-Robertson, F.C.; Samuel, T.L.; Duwiejuah, A.B. Amino acid profile and potential biomass conversion of Vitellaria paradoxa fruit pulp. Sci. Afr. 2024, 24, e02183. [Google Scholar] [CrossRef]
- Wang, X.; Xie, L.; Fang, J.; Pang, Y.; Xu, J.; Li, Z. Identification of Candidate Genes that Affect the Contents of 17 Amino Acids in the Rice Grain Using a Genome-Wide Haplotype Association Study. Rice 2023, 16, 40. [Google Scholar] [CrossRef]
- Riera-Ruiz, C.; Moriyama, H. Enzyme kinetics of deoxyuridine triphosphatase from Western corn rootworm. BMC Res. Notes 2023, 16, 336. [Google Scholar] [CrossRef]
- Jia, Q.; Zhang, J.; Wang, S.; Xu, F. Proteomic Analysis Reveals Differentially Expressed Proteins in Cordyceps militaris Cultured with Different Media. Curr. Microbiol. 2024, 82, 29. [Google Scholar] [CrossRef]
- Quan, Q.M.; Chen, L.L.; Wang, X.; Li, S.; Yang, X.L.; Zhu, Y.G.; Wang, M.; Cheng, Z. Genetic diversity and distribution patterns of host insects of Caterpillar Fungus Ophiocordyceps sinensis in the Qinghai-Tibet Plateau. PLoS ONE 2014, 9, e92293. [Google Scholar] [CrossRef]
- Wang, T.; Tang, C.; Chen, J.; Liang, J.; Li, Y.; Li, X. Accumulation Characteristics of Natural Ophiocordyceps sinensis Metabolites Driven by Environmental Factors. Metabolites 2024, 14, 414. [Google Scholar] [CrossRef]
- Choudhary, S.; Thakur, S.; Majeed, A.; Bhardwaj, P. Adaptability of Rhododendrons in high altitude habitats. J. For. Res. 2021, 32, 449–460. [Google Scholar] [CrossRef]
- Sun, Q.; Wu, F.; Wu, W.; Yu, W.; Zhang, G.; Huang, X.; Hao, Y.; Luo, L. Identification and quality evaluation of Lushan Yunwu tea from different geographical origins based on metabolomics. Food Res. Int. 2024, 186, 114379. [Google Scholar] [CrossRef]
- Gaur, P.; Prasad, S.; Kumar, B.; Sharma, S.K.; Vats, P. High-altitude hypoxia induced reactive oxygen species generation, signaling, and mitigation approaches. Int. J. Biometeorol. 2021, 65, 601–615. [Google Scholar] [CrossRef]
- Holeček, M. Histidine in Health and Disease: Metabolism, Physiological Importance, and Use as a Supplement. Nutrients 2020, 12, 848. [Google Scholar] [CrossRef]
- Soon, J.W.; Manca, M.A.; Laskowska, A.; Starkova, J.; Rohlenova, K.; Rohlena, J. Aspartate in tumor microenvironment and beyond: Metabolic interactions and therapeutic perspectives. Biochim. Biophys. Acta Mol. Basis Dis. 2024, 1870, 167451. [Google Scholar] [CrossRef]
- Tan, Q.; Zhao, X.; He, H.; Zhang, J.; Yi, T. Carbamoyl phosphate synthetase subunit Cpa1 interacting with Dut1, controls development, arginine biosynthesis, and pathogenicity of Colletotrichum gloeosporioides. Fungal Biol. 2021, 125, 184–190. [Google Scholar] [CrossRef]
- Sagawa, C.H.D.; Assis, R.A.B.; Zaini, P.A.; Saxe, H.; Wilmarth, P.A.; Salemi, M.; Phinney, B.S.; Dandekar, A.M. De Novo Arginine Synthesis Is Required for Full Virulence of Xanthomonas arboricola pv. juglandis During Walnut Bacterial Blight Disease. Phytopathology 2022, 112, 1500–1512. [Google Scholar] [CrossRef]
- Ge, F.; Sun, J.; Ren, Y.; He, B.; Li, J.; Yang, S.; Li, W. Transcriptomic and enzymatic analysis reveals the roles of glutamate dehydrogenase in Corynebacterium glutamicum. AMB Express 2022, 12, 161. [Google Scholar] [CrossRef]
- Nwosu, Z.C.; Pasca di Magliano, M. GOT2: An Unexpected Mediator of Immunosuppression in Pancreatic Cancer. Cancer Discov. 2022, 12, 2237–2239. [Google Scholar] [CrossRef]
- Witt, H.; Beer, S.; Rosendahl, J.; Chen, J.M.; Chandak, G.R.; Masamune, A.; Bence, M.; Szmola, R.; Oracz, G.; Macek, M., Jr.; et al. Variants in CPA1 are strongly associated with early onset chronic pancreatitis. Nat. Genet. 2013, 45, 1216–1220. [Google Scholar] [CrossRef]
- Song, Y.; Wang, Q.; Wang, D.; Junqiang, L.; Yang, J.; Li, H.; Wang, X.; Jin, X.; Jing, R.; Yang, J.H.; et al. Label-Free Quantitative Proteomics Unravels Carboxypeptidases as the Novel Biomarker in Pancreatic Ductal Adenocarcinoma. Transl. Oncol. 2018, 11, 691–699. [Google Scholar] [CrossRef]
- Chen, Q.; Wang, C.; Sun, Y.; Chen, S.; Zhou, J.; Han, T.; Wang, J.; Harpaz, S. An integrated analysis of transcriptome and metabolome reveals three BCAAs relieve lipid accumulation by inhibiting lipid synthesis and promoting lipid oxidation in the liver of largemouth bass (Micropterus salmoides). Aquaculture 2024, 581, 740384. [Google Scholar] [CrossRef]
- Choi, I.; Son, H.; Baek, J.H. Tricarboxylic Acid (TCA) Cycle Intermediates: Regulators of Immune Responses. Life 2021, 11, 69. [Google Scholar] [CrossRef]
- Lee, J.H.; Cho, Y.R.; Kim, J.H.; Kim, J.; Nam, H.Y.; Kim, S.W.; Son, J. Branched-chain amino acids sustain pancreatic cancer growth by regulating lipid metabolism. Exp. Mol. Med. 2019, 51, 1–11. [Google Scholar] [CrossRef]
- Tao, H.; Yang, X.; Wang, W.; Yue, S.; Pu, Z.; Huang, Y.; Shi, X.; Chen, J.; Zhou, G.; Chen, Y.; et al. Regulation of serum lipidomics and amino acid profiles of rats with acute myocardial ischemia by Salvia miltiorrhiza and Panax notoginseng herb pair. Phytomedicine 2020, 67, 153162. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, C.; Fan, Y.; Wang, T.; Wang, J.; Xiao, M.; He, M.; Chang, X.; Li, Y.; Li, X. Integrated Amino Acid Profiling and 4D-DIA Proteomics Reveal Protein Quality Divergence and Metabolic Adaptation in Cordyceps Species. J. Fungi 2025, 11, 365. https://doi.org/10.3390/jof11050365
Tang C, Fan Y, Wang T, Wang J, Xiao M, He M, Chang X, Li Y, Li X. Integrated Amino Acid Profiling and 4D-DIA Proteomics Reveal Protein Quality Divergence and Metabolic Adaptation in Cordyceps Species. Journal of Fungi. 2025; 11(5):365. https://doi.org/10.3390/jof11050365
Chicago/Turabian StyleTang, Chuyu, Yuejun Fan, Tao Wang, Jie Wang, Mengjun Xiao, Min He, Xiyun Chang, Yuling Li, and Xiuzhang Li. 2025. "Integrated Amino Acid Profiling and 4D-DIA Proteomics Reveal Protein Quality Divergence and Metabolic Adaptation in Cordyceps Species" Journal of Fungi 11, no. 5: 365. https://doi.org/10.3390/jof11050365
APA StyleTang, C., Fan, Y., Wang, T., Wang, J., Xiao, M., He, M., Chang, X., Li, Y., & Li, X. (2025). Integrated Amino Acid Profiling and 4D-DIA Proteomics Reveal Protein Quality Divergence and Metabolic Adaptation in Cordyceps Species. Journal of Fungi, 11(5), 365. https://doi.org/10.3390/jof11050365