
Integrated Transcriptomics and Metabolomics Provide Insight into Degeneration-Related Molecular Mechanisms of Morchella importuna During Repeated Subculturing

 , , , , , , ,
, , , , , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. Metabolite Extraction
2.3. UHPLC-MS/MS Analysis
2.4. Metabolite Identification and Quantification
2.5. RNA Extraction and Sequencing
2.6. Transcriptomic Analysis
2.7. Correlation Analysis Between the Metabolome and Transcriptome Data
2.8. Extraction of Biosynthetic Gene Clusters (BGCs) from Genome Sequence Data of M. importuna
2.9. Quantitative Real-Time PCR (qRT-PCR) Validation
2.10. Quantitative Analysis of Flavonoids
3. Results
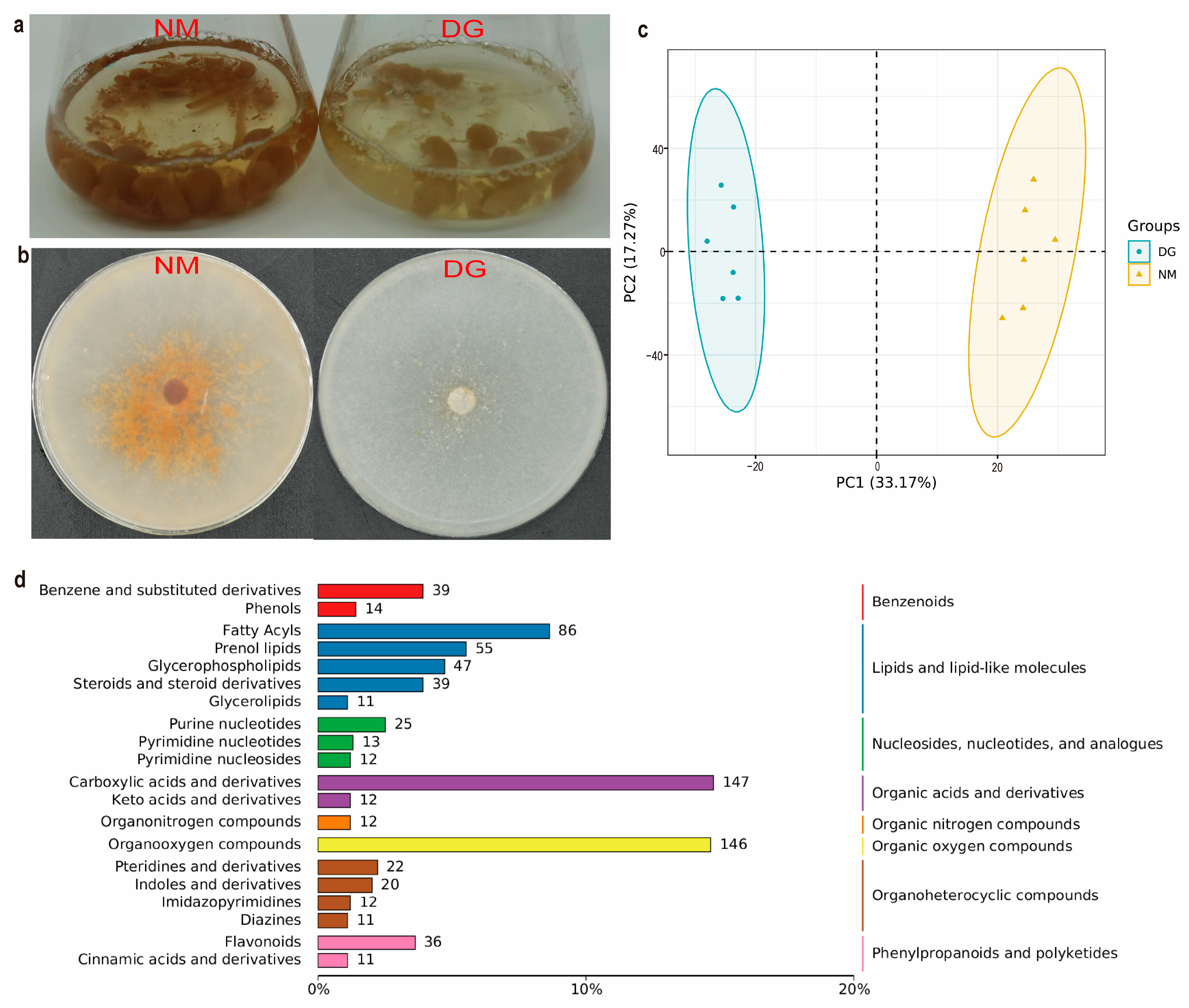
3.1. Obtaining of the Degenerate Strain and Metabolomics Analysis of Normal and Degenerate Mycelia of M. importuna
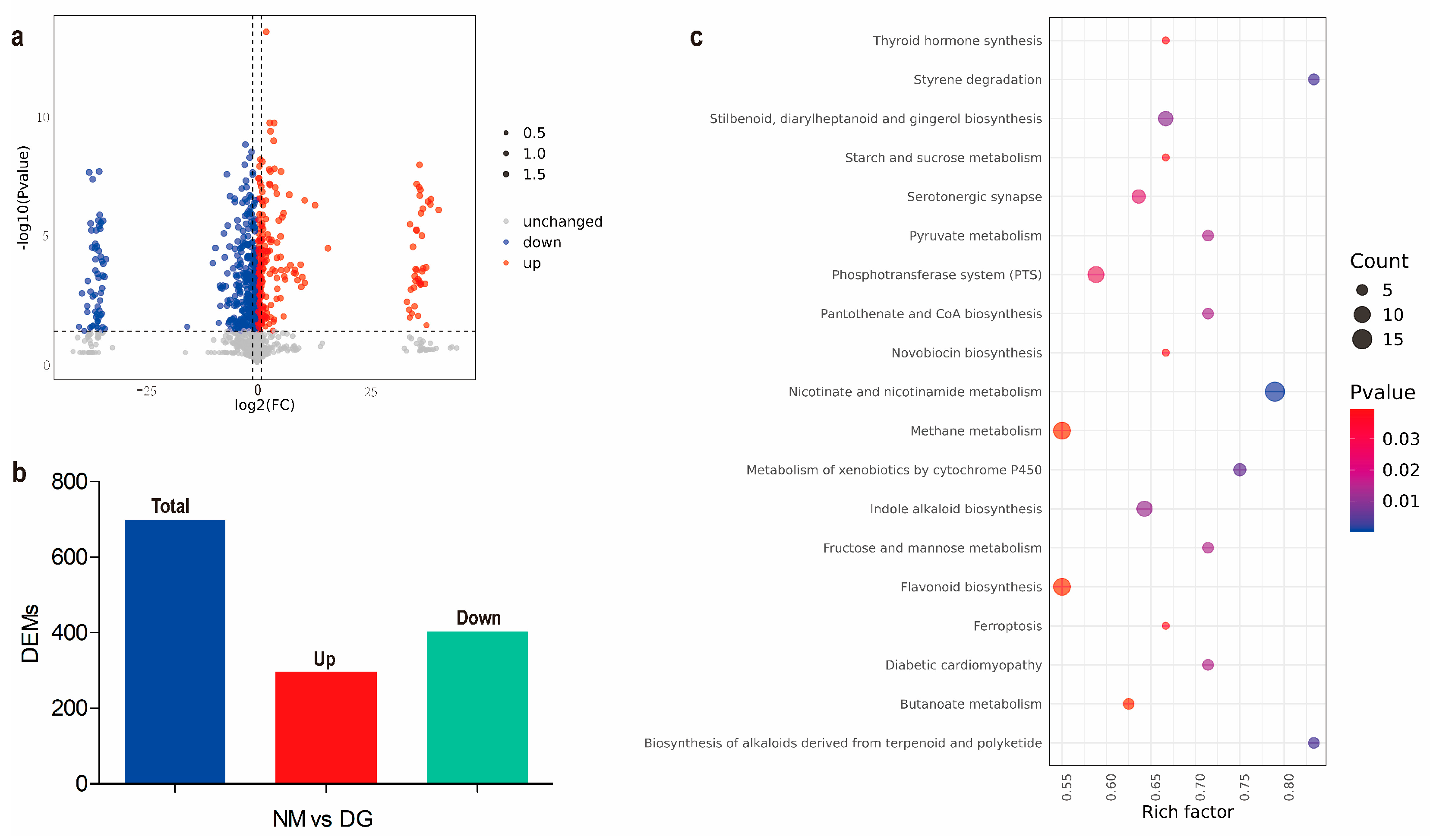
3.2. Transcriptomics Analysis Between Normal and Degenerate Mycelia of M. importuna
3.3. Integrated Metabolomics and Transcriptomics Analysis
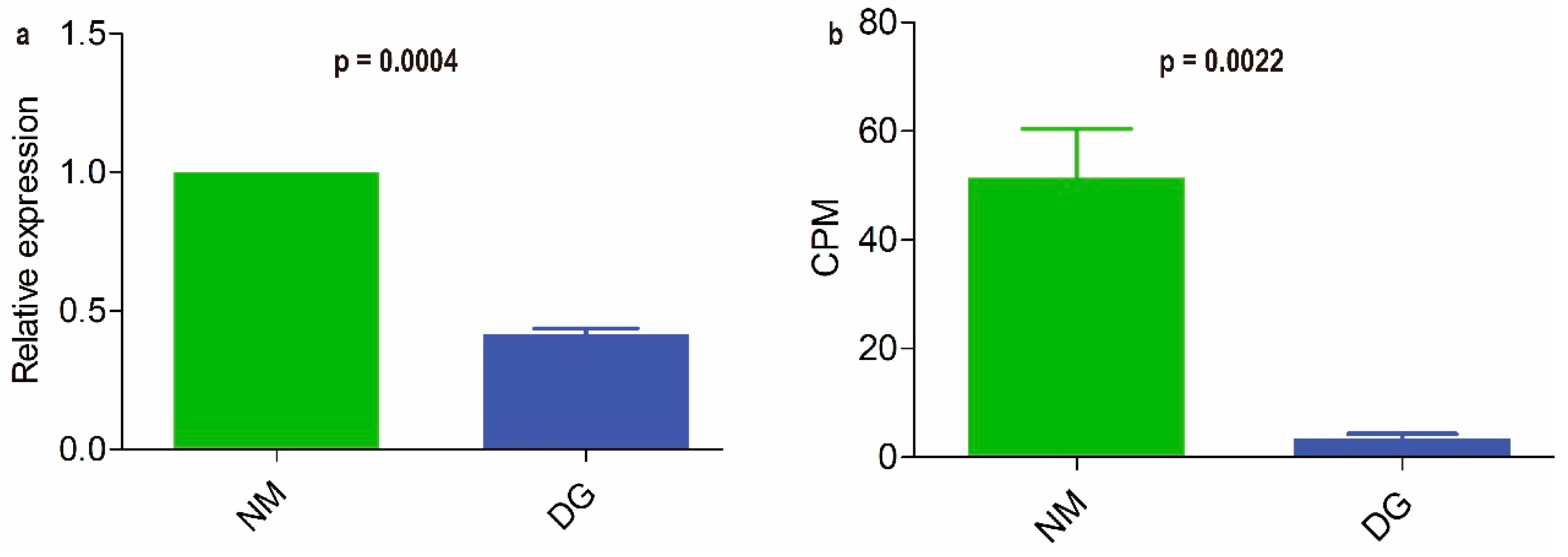
3.4. Extraction of BGCs and qRT-PCR Analysis of NR-PKS Gene
3.5. Determination of Flavonoid Content
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, X.; Liu, X.; Cui, B.; Miao, W.; Cheng, W.; Zhao, F. Characteristics analysis reveals the progress of Volvariella volvacea mycelium subculture degeneration. Front. Microbiol. 2019, 10, 2045. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Xin, X.; Weng, Y.; Gui, Z. Transcriptome-wide analysis reveals the progress of Cordyceps militaris subculture degeneration. PLoS ONE 2017, 12, e0186279. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Li, Y.; Liu, Y.; Gao, Y.; Qi, Y.; Shen, J. Particle and naked RNA mycoviruses in industrially cultivated mushroom Pleurotus ostreatus in China. Fungal Biol. 2010, 114, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Ma, D.; Ge, W.; Buswell, J.A. Induction of laccase activity in the edible straw mushroom, Volvariella volvacea. Fems Microbiol. Lett. 2003, 218, 143–148. [Google Scholar] [CrossRef]
- Chen, S.; Ge, W.; Buswell, J.A. Molecular cloning of a new laccase from the edible straw mushroom Volvariella volvacea. FEMS Microbiol. Lett. 2004, 230, 171–176. [Google Scholar] [CrossRef]
- Zhang, Z. The Degeneration of Volvariella volvacea Spawn Preservation by Mycelium Subculture; Kunming University of Science and Technology: Kunming, China, 2016. [Google Scholar]
- Xiong, C.; Xia, Y.; Zheng, P.; Wang, C. Increasing oxidative stress tolerance and subculturing stability of Cordyceps militaris by overexpression of a glutathione peroxidase gene. Appl. Microbiol. Biotechnol. 2013, 97, 2009–2015. [Google Scholar] [CrossRef]
- Lou, H.; Lin, J.; Guo, L.; Wang, X.; Tian, S.; Liu, C.; Zhao, Y.; Zhao, R. Advances in research on Cordyceps militaris degeneration. Appl. Microbiol. Biotechnol. 2019, 103, 7835–7841. [Google Scholar] [CrossRef]
- Pérez, G.; Lopez-Moya, F.; Chuina, E.; Ibañez-Vea, M.; Garde, E.; López-Llorca, L.V.; Pisabarro, A.G.; Ramírez, L. Strain degeneration in Pleurotus ostreatus: A genotype dependent oxidative stress process which triggers oxidative stress, cellular detoxifying and cell wall reshaping genes. J. Fungi 2021, 7, 862. [Google Scholar] [CrossRef]
- Wang, Y.L.; Wang, Z.X.; Liu, C.; Wang, S.B.; Huang, B. Genome-wide analysis of DNA methylation in the sexual stage of the insect pathogenic fungus Cordyceps militaris. Fungal Biol. 2015, 119, 1246–1254. [Google Scholar] [CrossRef]
- Wang, X.; Hong, S.; Tang, G.; Wang, C. Accumulation of the spontaneous and random mutations is causative of fungal culture degeneration. Fundam. Res. 2024, 18, 7. [Google Scholar] [CrossRef]
- Chen, Y.; Cao, X.; Xie, L.; Tang, J.; Liu, L.; Wang, D.; Wu, X.; Liu, T.; Yu, Y.; Wang, Y.; et al. Comparative transcriptomics and metabolomics provide insight into degeneration-related physiological mechanisms of Morchella importuna after long-term preservation. Microb. Biotechnol. 2025, 18, e70045. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Li, L.; Wu, W.; Pu, K.; Qi, W.; Qi, J.; Li, M. Enhancing Morchella mushroom yield and quality through the amendment of soil physicochemical properties and microbial community with wood ash. Microorganisms 2024, 12, 2406. [Google Scholar] [CrossRef] [PubMed]
- Dunn, W.B.; Broadhurst, D.; Begley, P.; Zelena, E.; Francis-McIntyre, S.; Anderson, N.; Brown, M.; Knowles, J.D.; Halsall, A.; Haselden, J.N.; et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2011, 6, 1060–1083. [Google Scholar] [CrossRef] [PubMed]
- Want, E.J.; Wilson, I.D.; Gika, H.; Theodoridis, G.; Plumb, R.S.; Shockcor, J.; Holmes, E.; Nicholson, J.K. Global metabolic profiling procedures for urine using, UPLC–MS. Nat. Protoc. 2010, 5, 1005–1018. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, T.; Shen, X.; Liu, J.; Zhao, D.; Sun, Y.; Wang, L.; Liu, Y.; Gong, X.; Liu, Y.; et al. Serum metabolomics for early diagnosis of esophageal squamous cell carcinoma by UHPLC-QTOF/MS. Metabolomics 2016, 12, 116. [Google Scholar] [CrossRef]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef]
- Wishart, D.S.; Djoumbou Feunang, Y.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef]
- Fahy, E.; Sud, M.; Cotter, D.; Subramaniam, S. LIPID MAPS online tools for lipid research. Nucleic Acids Res. 2007, 35, W606–W612. [Google Scholar] [CrossRef]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: An R package for multivariate analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Oxford Nanopore Technologies. Pinfish: A Toolset for Long-Read Transcriptomics Data Analysis; GitHub: San Francisco, CA, USA, 2024. [Google Scholar]
- Deng, Y.; Li, J.; Wu, S.; Zhu, Y.; Chen, Y.; He, F. Integrated nr database in protein annotation system and its localization. Comput. Eng. 2006, 32, 71–72. [Google Scholar] [CrossRef]
- UniProt Consortium. UniProt: The Universal Protein Knowledgebase in 2025. Nucleic Acids Res. 2025, 53, D609–D617. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Krylov, D.M.; Makarova, K.S.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; Rao, B.S.; et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004, 5, R7. [Google Scholar] [CrossRef]
- Paysan-Lafosse, T.; Andreeva, A.; Blum, M.; Chuguransky, S.R.; Grego, T.; Lazaro Pinto, B.; Salazar, G.A.; Bileschi, M.L.; Llinares-López, F.; Meng-Papaxanthos, L.; et al. The Pfam protein families database: Embracing, AI/ML. Nucleic Acids Res. 2025, 53, D523–D534. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef]
- Hernández-Plaza, A.; Szklarczyk, D.; Botas, J.; Cantalapiedra, C.P.; Giner-Lamia, J.; Mende, D.R.; Kirsch, R.; Rattei, T.; Letunic, I.; Jensen, L.J.; et al. eggNOG 6.0: Enabling comparative genomics across 12535 organisms. Nucleic Acids Res. 2023, 51, D389–D394. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar] [CrossRef]
- Tang, J.; Matsuda, Y. Discovery of fungal onoceroid triterpenoids through domainless enzyme-targeted global genome mining. Nat. Commun. 2024, 15, 4312. [Google Scholar] [CrossRef]
- Liu, Q.; He, G.; Wei, J.; Dong, C. Comparative transcriptome analysis of cells from different areas reveals ROS responsive mechanism at sclerotial initiation stage in Morchella importuna. Sci. Rep. 2021, 11, 9418. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Liu, W.; Cai, Y.; Lan, A.; Bian, Y. Validation of internal control genes for quantitative real-time PCR gene expression analysis in Morchella. Molecules 2018, 23, 2331. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.; Sumner, B.W.; Bhatia, A.; Sarma, S.J.; Sumner, L.W. UHPLC-MS analyses of plant flavonoids. Curr. Protoc. Mol. Biol. 2019, 4, e20085. [Google Scholar] [CrossRef]
- Wang, T.; Xiao, J.; Hou, H.; Li, P.; Yuan, Z.; Xu, H.; Liu, R.; Li, Q.; Bi, K. Development of an ultra-fast liquid chromatography-tandem mass spectrometry method for simultaneous determination of seven flavonoids in rat plasma: Application to a comparative pharmacokinetic investigation of Ginkgo biloba extract and single pure ginkgo flavonoids after oral administration. J. Chromatogr. B-Anal. Technol. Biomed. Life Sci. 2017, 1060, 173–181. [Google Scholar] [CrossRef]
- Villiers, A.; Venter, P.; Pasch, H. Recent advances and trends in the liquid-chromatography-mass spectrometry analysis of flavonoids. J. Chromatogr. A 2016, 1430, 16–78. [Google Scholar] [CrossRef]
- Feng, P.; Shang, Y.; Cen, K.; Wang, C. Fungal biosynthesis of the bibenzoquinone oosporein to evade insect immunity. Proc. Natl. Acad. Sci. USA 2015, 112, 11365–11370. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, X.; Feng, D.; Liang, Y.; Wu, Z.; Du, S.; Zhou, Y.; Geng, C.; Men, P.; Fu, C.; et al. Discovery of a Unique Flavonoid Biosynthesis Mechanism in Fungi by Genome Mining. Angew. Chem. Int. Ed. 2023, 62, e202215529. [Google Scholar] [CrossRef]
- Dias, M.C.; Pinto, D.; Silva, A.M.S. Plant flavonoids: Chemical characteristics and biological activity. Molecules 2021, 26, 5377. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Content (ng/g) | Fold Change | p Value | |
|---|---|---|---|---|
| NM | DG | |||
| Chrysin | 402.6 ± 5.604 | 29.99 ± 1.903 | 13.424 | *** |
| Liquiritigenin | 381.7 ± 10.13 | 51.95 ± 0.998 | 7.347 | *** |
| Apigenin | 0.592 ± 0.04 | 0.71 ± 0.036 | 0.834 | * |
| Naringenin | 54.7 ± 0.685 | 12.15 ± 0.635 | 4.502 | *** |
| Luteolin | 0.673 ± 0.091 | 0.255 ± 0.016 | 2.639 | * |
| L-Epicatechin | 102.4 ± 3.569 | ND | ND | |
| Kaempferide | 1.38 ± 0.052 | 0.495 ± 0.029 | 2.788 | *** |
| Quercetin | 138.4 ± 6.425 | 36.08 ± 2.34 | 3.836 | *** |
| Dihydromyricetin | 150.9 ± 3.777 | 47.5 ± 1.575 | 3.177 | *** |
| Vitexin | 1.326 ± 0.055 | 0.387 ± 0.018 | 3.426 | *** |
| Astragalin | 15.91 ± 0.777 | 18.78 ± 0.565 | 0.847 | ** |
| Quercitrin | 8.374 ± 0.606 | 0.245 ± 0.011 | 34.180 | *** |
| Cynaroside | 1.791 ± 0.077 | ND | ND | |
| Quercetin 3-glucoside | 7.514 ± 0.496 | 1.611 ± 0.107 | 4.664 | *** |
| Naringin | 212.2 ± 4.018 | 41.38 ± 2.199 | 5.128 | *** |
| Diosmin | 3.226 ± 0.079 | ND | ND | |
| Rutin | 342.5 ± 2.849 | 76.86 ± 0.248 | 4.456 | *** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huo, W.; He, X.; Liu, Y.; Zhang, L.; Dai, L.; Qi, P.; Qiao, T.; Hu, S.; Lu, P.; Li, J. Integrated Transcriptomics and Metabolomics Provide Insight into Degeneration-Related Molecular Mechanisms of Morchella importuna During Repeated Subculturing. J. Fungi 2025, 11, 420. https://doi.org/10.3390/jof11060420
Huo W, He X, Liu Y, Zhang L, Dai L, Qi P, Qiao T, Hu S, Lu P, Li J. Integrated Transcriptomics and Metabolomics Provide Insight into Degeneration-Related Molecular Mechanisms of Morchella importuna During Repeated Subculturing. Journal of Fungi. 2025; 11(6):420. https://doi.org/10.3390/jof11060420
Chicago/Turabian StyleHuo, Wenyan, Xuelian He, Yu Liu, Liguang Zhang, Lu Dai, Peng Qi, Ting Qiao, Suying Hu, Pengpeng Lu, and Junzhi Li. 2025. "Integrated Transcriptomics and Metabolomics Provide Insight into Degeneration-Related Molecular Mechanisms of Morchella importuna During Repeated Subculturing" Journal of Fungi 11, no. 6: 420. https://doi.org/10.3390/jof11060420
APA StyleHuo, W., He, X., Liu, Y., Zhang, L., Dai, L., Qi, P., Qiao, T., Hu, S., Lu, P., & Li, J. (2025). Integrated Transcriptomics and Metabolomics Provide Insight into Degeneration-Related Molecular Mechanisms of Morchella importuna During Repeated Subculturing. Journal of Fungi, 11(6), 420. https://doi.org/10.3390/jof11060420