
Characterization of the High-Quality Genome Sequence and Virulence Factors of Fusarium oxysporum f. sp. vasinfectum Race 7


Abstract
1. Introduction
2. Materials and Methods
2.1. DNA Extraction and Genome Sequencing
2.2. Genome Assembly and Gene Prediction
2.3. Genome Assembly Using Hi-C
2.4. Evolutionary Analysis
2.5. Identification of LS Genomic Regions and Comparative Genomic Analysis
2.6. Identification of Secreted in Xylem (SIX) Protein of FOV7
2.7. Fungal Transformation
2.8. Fov Inoculation and Infection Assays
2.9. Transient Expression in N. benthamiana
2.10. Yeast Signal-Sequence Trap System
3. Results
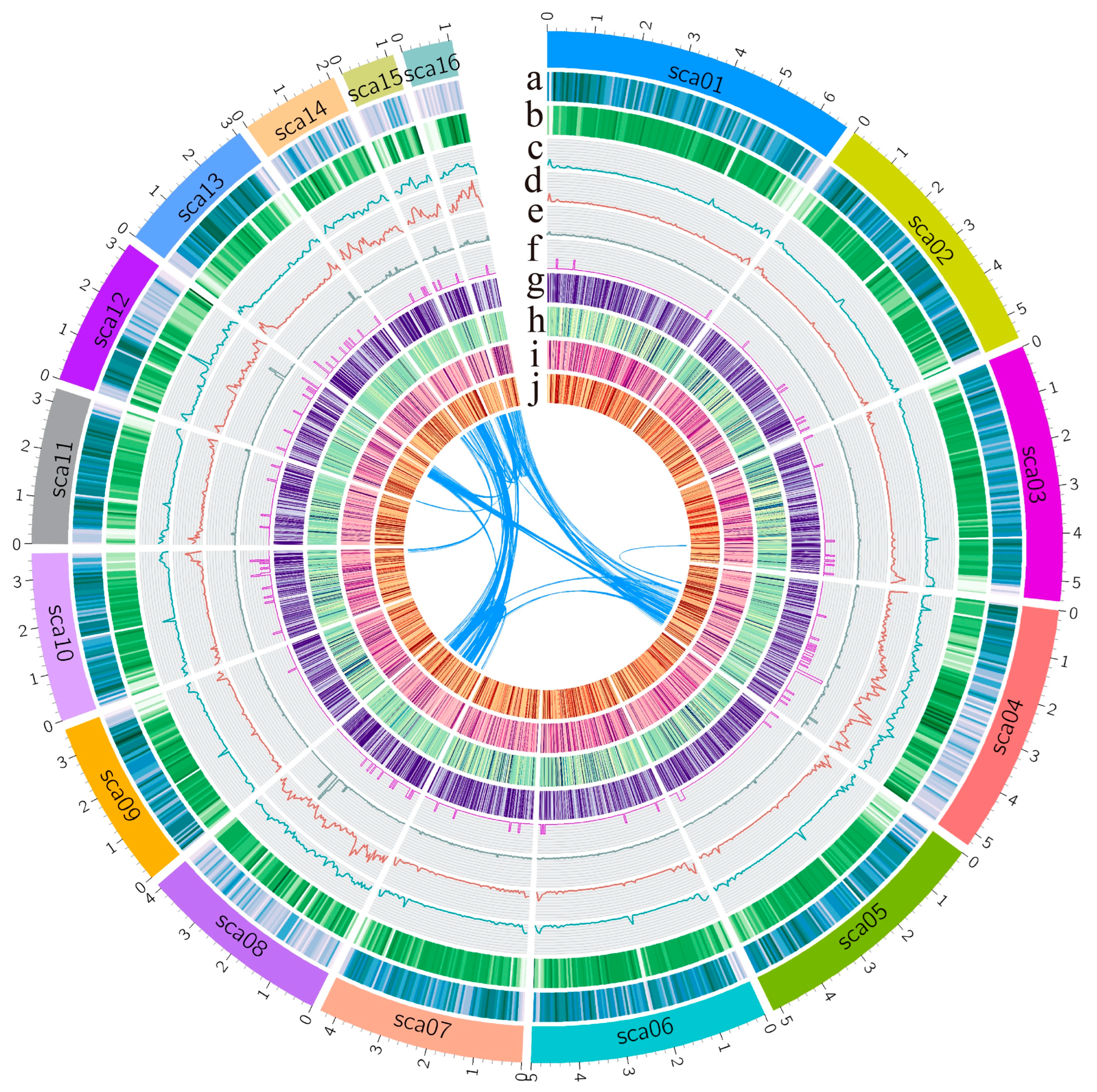
3.1. Genome Assembly and Annotation of Fusarium oxysporum f. sp. vasinfectum Race 7
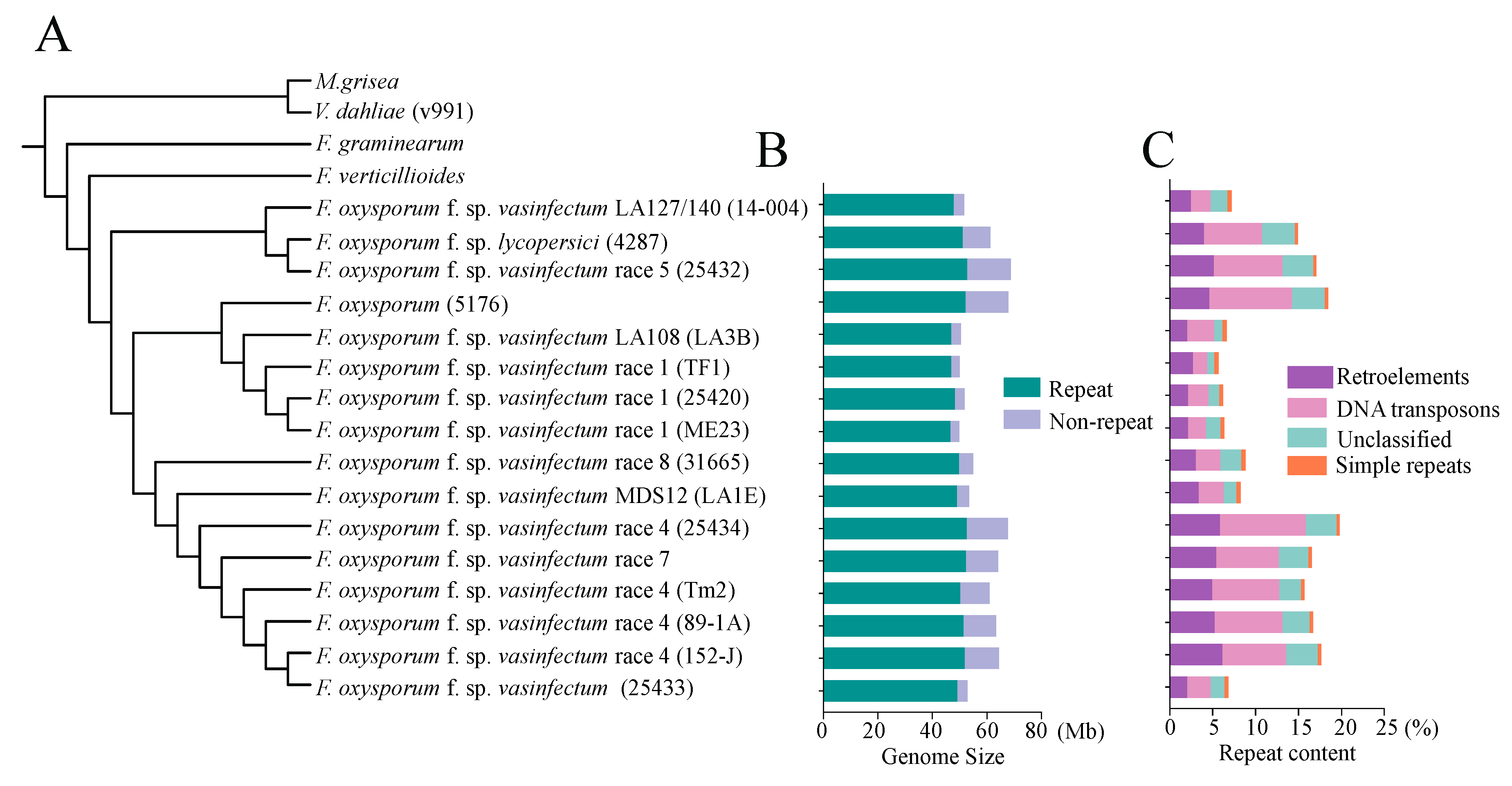
3.2. Phylogenomic Analysis Revealed the Divergence or Closeness in the Evolutionary Relationships among Different Races of Fov
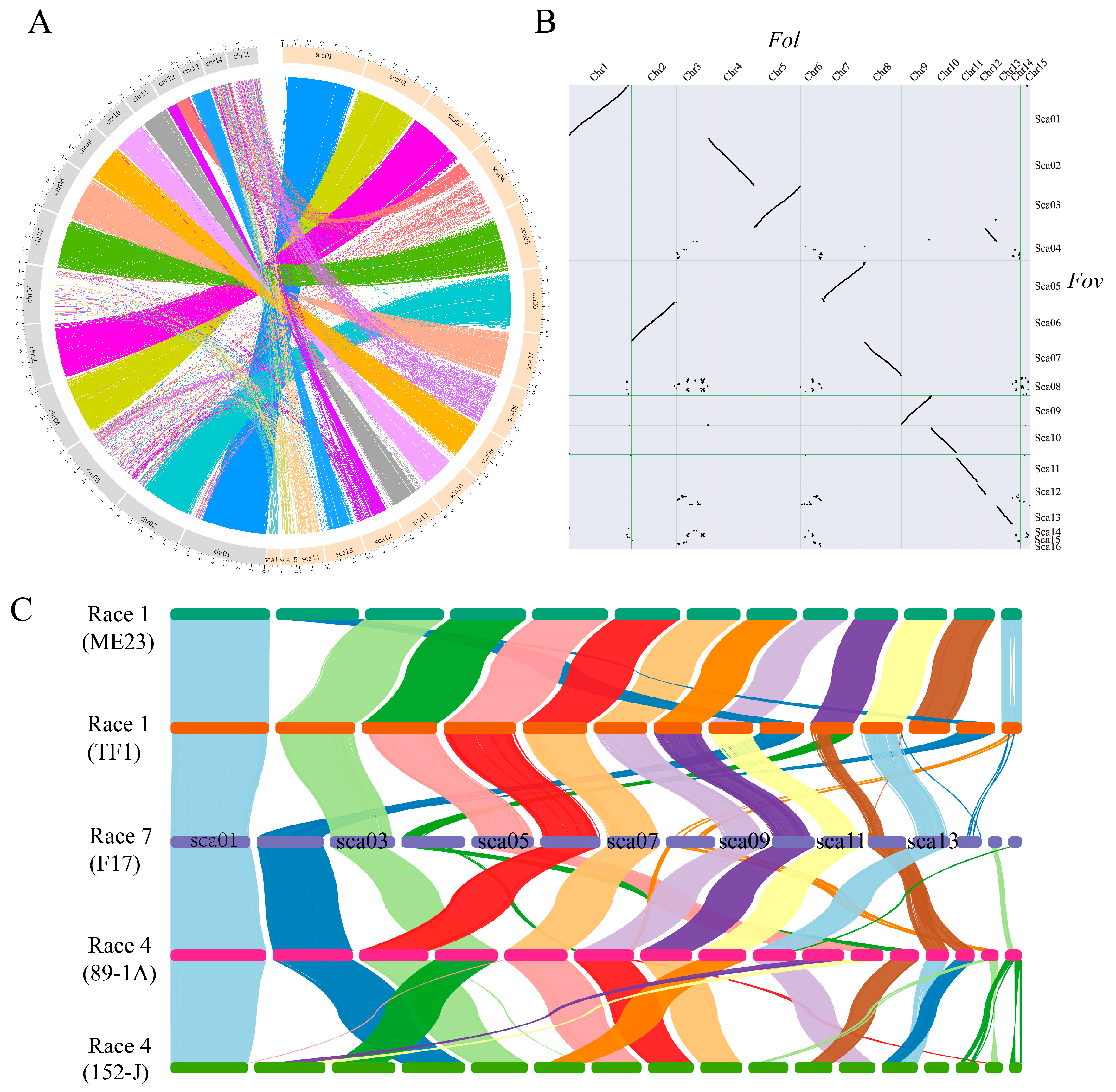
3.3. Comparative Genome Analysis Identified Lineage-Specific Regions of FOV7
3.4. Identification of Potential Effector Genes
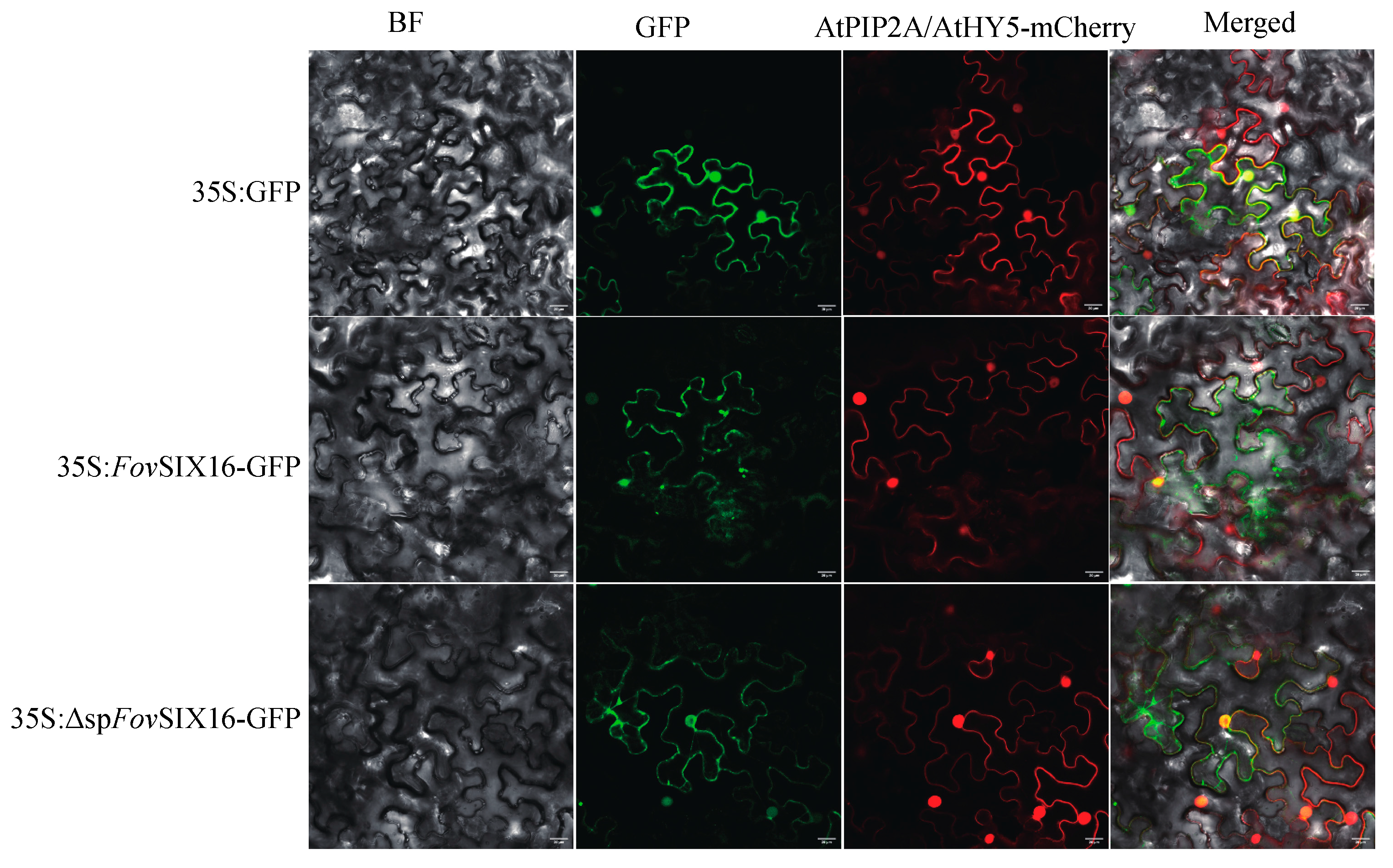
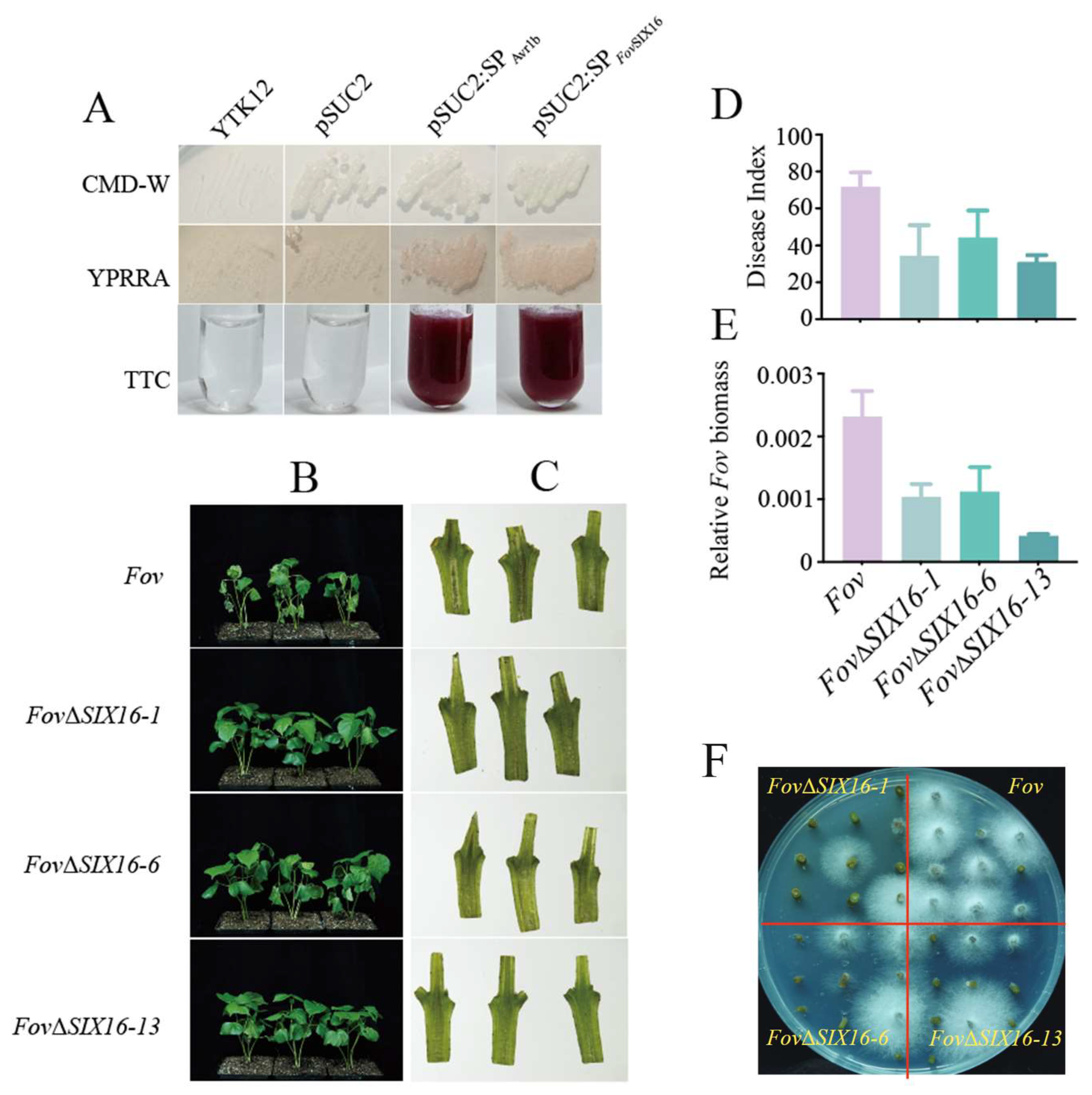
3.5. FovSIX16 Was Required for the Full Virulence of FOV7
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Michielse, C.B.; Rep, M. Pathogen profile update: Fusarium oxysporum. Mol. Plant Pathol. 2009, 10, 311–324. [Google Scholar] [CrossRef]
- Dean, R.; Van Kan, J.A.; Pretorius, Z.A.; Hammond-Kosack, K.E.; Di Pietro, A.; Spanu, P.D.; Rudd, J.J.; Dickman, M.; Kahmann, R.; Ellis, J.; et al. The Top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 2012, 13, 414–430. [Google Scholar] [CrossRef] [PubMed]
- Cox, K.L., Jr.; Babilonia, K.; Wheeler, T.; He, P.; Shan, L. Return of old foes—Recurrence of bacterial blight and Fusarium wilt of cotton. Curr. Opin. Plant Biol. 2019, 50, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Sanogo, S.; Zhang, J. Resistance sources, resistance screening techniques and disease management for Fusarium wilt in cotton. Euphytica 2015, 207, 255–271. [Google Scholar] [CrossRef]
- Cianchetta, A.N.; Davis, R.M. Fusarium wilt of cotton: Management strategies. Crop Prot. 2015, 73, 40–44. [Google Scholar] [CrossRef]
- Davis, R.M.; Colyer, P.D.; Rothrock, C.S.; Kochman, J.K. Fusarium Wilt of Cotton: Population Diversity and Implications for Management. Plant Dis. 2006, 90, 692–703. [Google Scholar] [CrossRef] [PubMed]
- Skovgaard, K.; Nirenberg, H.I.; O’Donnell, K.; Rosendahl, S. Evolution of Fusarium oxysporum f. sp. vasinfectum Races Inferred from Multigene Genealogies. Phytopathology 2001, 91, 1231–1237. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, G.M.; Armstrong, J.K. American Egyptian and Indian Cotton Wilt Fusaria Their Pathogenicity and Relationship to Other Wilt Fusaria; Technical Bulletin; United States Department of Agriculture: Washington, DC, USA, 1960.
- Ulloa, M.; Hutmacher, R.B.; Roberts, P.A.; Wright, S.D.; Nichols, R.L.; Michael Davis, R. Inheritance and QTL mapping of Fusarium wilt race 4 resistance in cotton. Theor. Appl. Genet. 2013, 126, 1405–1418. [Google Scholar] [CrossRef] [PubMed]
- Diaz, J.; Garcia, J.; Lara, C.; Hutmacher, R.B.; Ulloa, M.; Nichols, R.L.; Ellis, M.L. Characterization of Current Fusarium oxysporum f. sp. vasinfectum Isolates from Cotton in the San Joaquin Valley of California and Lower Valley El Paso, Texas. Plant Dis. 2021, 105, 1898–1911. [Google Scholar] [CrossRef]
- Chen, Q.; Ji, X.; Sun, W. Identification of cotton-wilt Fusarium races in China. Sci. Agric. Sin. 1985, 6, 1–6. [Google Scholar]
- Shi, L.Y. Studies on the pathogen of Fusarium vasinfectum and Verticillium dahliae of cotton. Acta Gossypii Sin. 1996, 8, 292–294. [Google Scholar]
- Bell, A.A.; Gu, A.; Olvey, J.; Wagner, T.A.; Tashpulatov, J.J.; Prom, S.; Quintana, J.; Nichols, R.L.; Liu, J. Detection and Characterization of Fusarium oxysporum f. sp. vasinfectum VCG0114 (Race 4) Isolates of Diverse Geographic Origins. Plant Dis. 2019, 103, 1998–2009. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Abdelraheem, A.; Lujan, P.; Idowu, J.; Sullivan, P.; Nichols, R.; Wedegaertner, T.; Zhang, J. Detection and Characterization of Fusarium Wilt (Fusarium oxysporum f. sp. vasinfectum) Race 4 Causing Fusarium Wilt of Cotton Seedlings in New Mexico. Plant Dis. 2021, 105, 3353–3367. [Google Scholar] [CrossRef] [PubMed]
- Houterman, P.M.; Speijer, D.; Dekker, H.L.; CG, D.E.K.; Cornelissen, B.J.; Rep, M. The mixed xylem sap proteome of Fusarium oxysporum-infected tomato plants. Mol. Plant Pathol. 2007, 8, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Houterman, P.M.; Cornelissen, B.J.; Rep, M. Suppression of plant resistance gene-based immunity by a fungal effector. PLoS Pathog. 2008, 4, e1000061. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.M.; Houterman, P.M.; Schreiver, I.; Ma, L.; Amyotte, S.; Chellappan, B.; Boeren, S.; Takken, F.L.W.; Rep, M. MITEs in the promoters of effector genes allow prediction of novel virulence genes in Fusarium oxysporum. BMC Genom. 2013, 14, 119. [Google Scholar] [CrossRef]
- Houterman, P.M.; Ma, L.; van Ooijen, G.; de Vroomen, M.J.; Cornelissen, B.J.; Takken, F.L.; Rep, M. The effector protein Avr2 of the xylem-colonizing fungus Fusarium oxysporum activates the tomato resistance protein I-2 intracellularly. Plant J. 2009, 58, 970–978. [Google Scholar] [CrossRef]
- Rep, M.; van der Does, H.C.; Meijer, M.; van Wijk, R.; Houterman, P.M.; Dekker, H.L.; de Koster, C.G.; Cornelissen, B.J. A small, cysteine-rich protein secreted by Fusarium oxysporum during colonization of xylem vessels is required for I-3-mediated resistance in tomato. Mol. Microbiol. 2004, 53, 1373–1383. [Google Scholar] [CrossRef]
- Ma, L.J.; van der Does, H.C.; Borkovich, K.A.; Coleman, J.J.; Daboussi, M.J.; Di Pietro, A.; Dufresne, M.; Freitag, M.; Grabherr, M.; Henrissat, B.; et al. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 2010, 464, 367–373. [Google Scholar] [CrossRef]
- Ma, L.J.; Geiser, D.M.; Proctor, R.H.; Rooney, A.P.; O’Donnell, K.; Trail, F.; Gardiner, D.M.; Manners, J.M.; Kazan, K. Fusarium pathogenomics. Annu. Rev. Microbiol. 2013, 67, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Rep, M.; Kistler, H.C. The genomic organization of plant pathogenicity in Fusarium species. Curr. Opin. Plant Biol. 2010, 13, 420–426. [Google Scholar] [CrossRef]
- Seo, S.; Pokhrel, A.; Coleman, J.J. The Genome Sequence of Five Genotypes of Fusarium oxysporum f. sp. vasinfectum: A Resource for Studies on Fusarium Wilt of Cotton. Mol. Plant Microbe Interact. 2020, 33, 138–140. [Google Scholar] [CrossRef]
- Srivastava, S.K.; Zeller, K.A.; Sobieraj, J.H.; Nakhla, M.K. Genome Resources of Four Distinct Pathogenic Races Within Fusarium oxysporum f. sp. vasinfectum that Cause Vascular Wilt Disease of Cotton. Phytopathology 2021, 111, 593–596. [Google Scholar] [CrossRef]
- Achari, S.R.; Mann, R.C.; Sharma, M.; Edwards, J. Diagnosis of Fusarium oxysporum f. sp. ciceris causing Fusarium wilt of chickpea using loop-mediated isothermal amplification (LAMP) and conventional end-point PCR. Sci. Rep. 2023, 13, 2640. [Google Scholar] [CrossRef]
- Jobe, T.O.; Ulloa, M.; Ellis, M.L. A high-quality whole-genome sequence, assembly, and gene annotation of Fusarium oxysporum f. sp. vasinfectum (Fov) race 1 from California. Microbiol. Resour. Announc. 2023, 13, e00702-23. [Google Scholar] [CrossRef]
- Jobe, T.O.; Ulloa, M.; Ellis, M.L. Two de novo genome assemblies from pathogenic Fusarium oxysporum f. sp. vasinfectum race 4 (FOV4) isolates from California. Microbiol. Resour. Announc. 2023, 13, e00760-23. [Google Scholar] [CrossRef]
- Xiao, C.L.; Chen, Y.; Xie, S.Q.; Chen, K.N.; Wang, Y.; Han, Y.; Luo, F.; Xie, Z. MECAT: Fast mapping, error correction, and de novo assembly for single-molecule sequencing reads. Nat. Methods 2017, 14, 1072–1074. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2009, 25, 4.10.11–4.10.14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, X.; Tang, H.; Zhang, Q.; Hua, X.; Ma, X.; Zhu, F.; Jones, T.; Zhu, X.; Bowers, J.; et al. Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L. Nat. Genet. 2018, 50, 1565–1573. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015, 16, 157. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. STAG: Species tree inference from all genes. BioRxiv 2018, 267914. [Google Scholar] [CrossRef]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, 1–9. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Yang, J.; Jing, Y.; Xu, L.; Yu, K.; Fang, X. NGenomeSyn: An easy-to-use and flexible tool for publication-ready visualization of syntenic relationships across multiple genomes. Bioinformatics 2023, 39, btad121. [Google Scholar] [CrossRef] [PubMed]
- Li, H. New strategies to improve minimap2 alignment accuracy. Bioinformatics 2021, 37, 4572–4574. [Google Scholar] [CrossRef] [PubMed]
- Abeysekara, N.S.; Bhattacharyya, M.K. Analyses of the xylem sap proteomes identified candidate Fusarium virguliforme proteinacious toxins. PLoS ONE 2014, 9, e93667. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ni, H.; Du, X.; Wang, S.; Ma, X.W.; Nurnberger, T.; Guo, H.S.; Hua, C. The Verticillium-specific protein VdSCP7 localizes to the plant nucleus and modulates immunity to fungal infections. New Phytol. 2017, 215, 368–381. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhang, X.; Xiao, S.; Ma, J.; Shi, W.; Qin, T.; Xi, H.; Nie, X.; You, C.; Xu, Z.; et al. A Single-Nucleotide Mutation in a GLUTAMATE RECEPTOR-LIKE Gene Confers Resistance to Fusarium Wilt in Gossypium hirsutum. Adv. Sci. 2021, 8, 2002723. [Google Scholar] [CrossRef] [PubMed]
- GB/T 22101.4-2009; Technical Specification for Evaluating Resistance of Cotton to Diseases and Insect Pests—Part 4: Fusarium Wilt. China National Standards: Beijing, China, 2009.
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Ortiz, C.S.; Bell, A.A.; Magill, C.W.; Liu, J. Specific PCR Detection of Fusarium oxysporum f. sp. vasinfectum California Race 4 Based on a Unique Tfo1 Insertion Event in the PHO Gene. Plant Dis. 2017, 101, 34–44. [Google Scholar] [CrossRef]
- Sperschneider, J.; Dodds, P.N. EffectorP 3.0: Prediction of Apoplastic and Cytoplasmic Effectors in Fungi and Oomycetes. Mol. Plant Microbe Interact. 2022, 35, 146–156. [Google Scholar] [CrossRef]
- Cianchetta, A.N.; Allen, T.W.; Hutmacher, R.B.; Kemerait, R.C.; Kirkpatrick, T.L.; Lawrence, G.W.; Lawrence, K.S.; Mueller, J.D.; Nichols, R.L.; Olsen, M.W. Survey of Fusarium oxysporum f. sp. vasinfectum in the United States. J. Cotton Sci. 2015, 19, 328–336. [Google Scholar]
- Bell, A.A.; Kemerait, R.C.; Ortiz, C.S.; Prom, S.; Quintana, J.; Nichols, R.L.; Liu, J. Genetic Diversity, Virulence, and Meloidogyne incognita Interactions of Fusarium oxysporum Isolates Causing Cotton Wilt in Georgia. Plant Dis. 2017, 101, 948–956. [Google Scholar] [CrossRef] [PubMed]
- Czislowski, E.; Fraser-Smith, S.; Zander, M.; O’Neill, W.T.; Meldrum, R.A.; Tran-Nguyen, L.T.T.; Batley, J.; Aitken, E.A.B. Investigation of the diversity of effector genes in the banana pathogen, Fusarium oxysporum f. sp. cubense, reveals evidence of horizontal gene transfer. Mol. Plant Pathol. 2018, 19, 1155–1171. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, A.; Rep, M.; Wang, B.; Ashton, A.; Dodds, P.; Ellis, J. Variation in potential effector genes distinguishing Australian and non-Australian isolates of the cotton wilt pathogen Fusarium oxysporum f.sp. vasinfectum. Plant Pathol. 2010, 60, 232–243. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genomic Feature | Fov Race 7 |
|---|---|
| Total length of contigs | 64,259,633 |
| Total length of assemblies | 64,156,821 |
| Percentage of anchoring | 98.60% |
| Number of contigs a | 54 |
| Contig N50 (bp) | 3,564,359 |
| Number of scaffolds b | 30 |
| Scaffold N50 (bp) | 5,083,699 |
| GC content | 47.64% |
| Percentage of repeat sequences | 18.42% |
| Number of genes | 19,633 |
| Number of transcripts | 20,417 |
| BUSCO completeness | 99.20% |
| SIX ID | Gene ID | AA Length | SP a | Induced in planta | Effector Prediction b | SIX Homologs | Core/LS Scaffold |
|---|---|---|---|---|---|---|---|
| FovSIX1 | Fov7_sca01_1094 | 212 | Yes | No | No | ND | Core |
| FovSIX2 | Fov7_sca02_1180 | 567 | Yes | Yes | No | ND | Core |
| FovSIX3 | Fov7_sca04_0306 | 275 | Yes | Yes | Yes | ND | LS |
| FovSIX4 | Fov7_sca04_0935 | 127 | Yes | No | No | ND | LS |
| FovSIX5 | Fov7_sca04_1062 | 107 | Yes | Yes | Yes | ND | LS |
| FovSIX6 | Fov7_sca04_1092 | 118 | Yes | Yes | Yes | SIX9 | LS |
| FovSIX7 | Fov7_sca04_1108 | 87 | Yes | No | Yes | ND | LS |
| FovSIX8 | Fov7_sca05_0196 | 432 | Yes | Yes | No | ND | Core |
| FovSIX9 | Fov7_sca05_0374 | 290 | Yes | Yes | No | ND | Core |
| FovSIX10 | Fov7_sca05_1077 | 106 | No | No | Yes | ND | Core |
| FovSIX11 | Fov7_sca06_0097 | 274 | Yes | No | Yes | ND | Core |
| FovSIX12 | Fov7_sca07_0495 | 154 | No | Yes | Yes | ND | Core |
| FovSIX13 | Fov7_sca07_1331 | 379 | Yes | Yes | NO | ND | Core |
| FovSIX14 | Fov7_sca10_0956 | 419 | Yes | Yes | NO | ND | Core |
| FovSIX15 | Fov7_sca15_0057 | 102 | Yes | Yes | Yes | ND | LS |
| FovSIX16 | Fov7_sca15_0202 | 269 | Yes | Yes | Yes | ND | LS |
| FovSIX17 | Fov7_sca15_0203 | 107 | Yes | Yes | Yes | ND | LS |
| FovSIX18 | Fov7_sca16_0047 | 130 | Yes | Yes | Yes | ND | LS |
| FovSIX19 | Fov7_sca18_0157 | 148 | Yes | Yes | Yes | ND | LS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, D.; Zhang, X.; Ming, Y.; Liu, C.; Zhang, X.; Liu, S.; Zhu, L. Characterization of the High-Quality Genome Sequence and Virulence Factors of Fusarium oxysporum f. sp. vasinfectum Race 7. J. Fungi 2024, 10, 242. https://doi.org/10.3390/jof10040242
Yang D, Zhang X, Ming Y, Liu C, Zhang X, Liu S, Zhu L. Characterization of the High-Quality Genome Sequence and Virulence Factors of Fusarium oxysporum f. sp. vasinfectum Race 7. Journal of Fungi. 2024; 10(4):242. https://doi.org/10.3390/jof10040242
Chicago/Turabian StyleYang, Dingyi, Xiaojun Zhang, Yuqing Ming, Chenglin Liu, Xianlong Zhang, Shiming Liu, and Longfu Zhu. 2024. "Characterization of the High-Quality Genome Sequence and Virulence Factors of Fusarium oxysporum f. sp. vasinfectum Race 7" Journal of Fungi 10, no. 4: 242. https://doi.org/10.3390/jof10040242
APA StyleYang, D., Zhang, X., Ming, Y., Liu, C., Zhang, X., Liu, S., & Zhu, L. (2024). Characterization of the High-Quality Genome Sequence and Virulence Factors of Fusarium oxysporum f. sp. vasinfectum Race 7. Journal of Fungi, 10(4), 242. https://doi.org/10.3390/jof10040242