Transcriptional Programs and Regeneration Enhancers Underlying Heart Regeneration


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
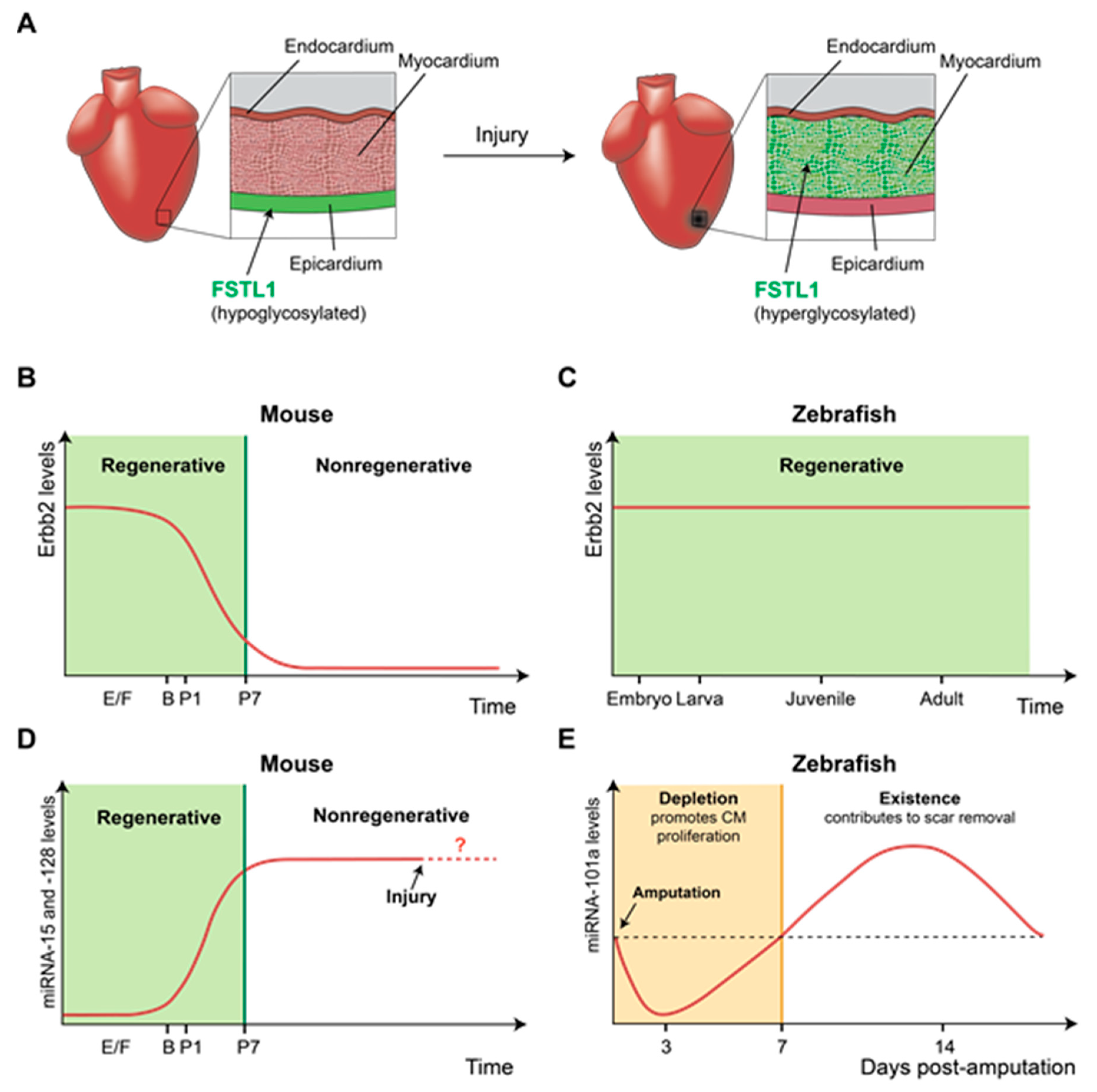
2. Differentially Expressed Genes Underlie Maintenance or Loss of Cardiac Regenerative Capacity
3. Enhancers Are Key Regulatory Elements Controlling Cardiac Gene Expression and Function
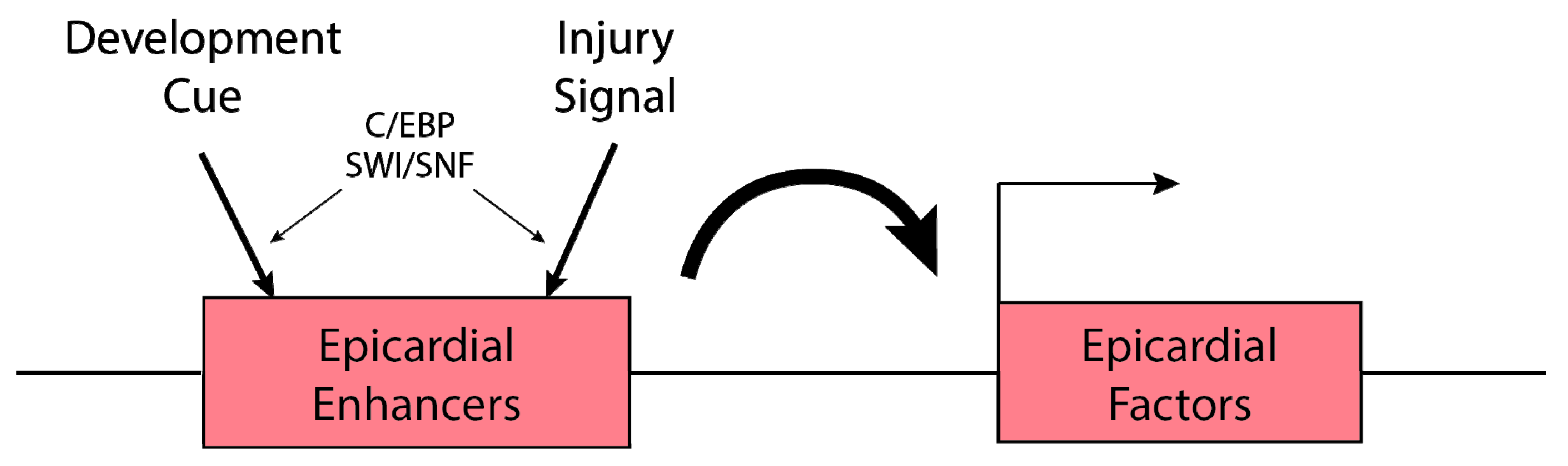
4. Epicardial Enhancers Drive Epicardial Factor Expression in Developing and Injured Hearts
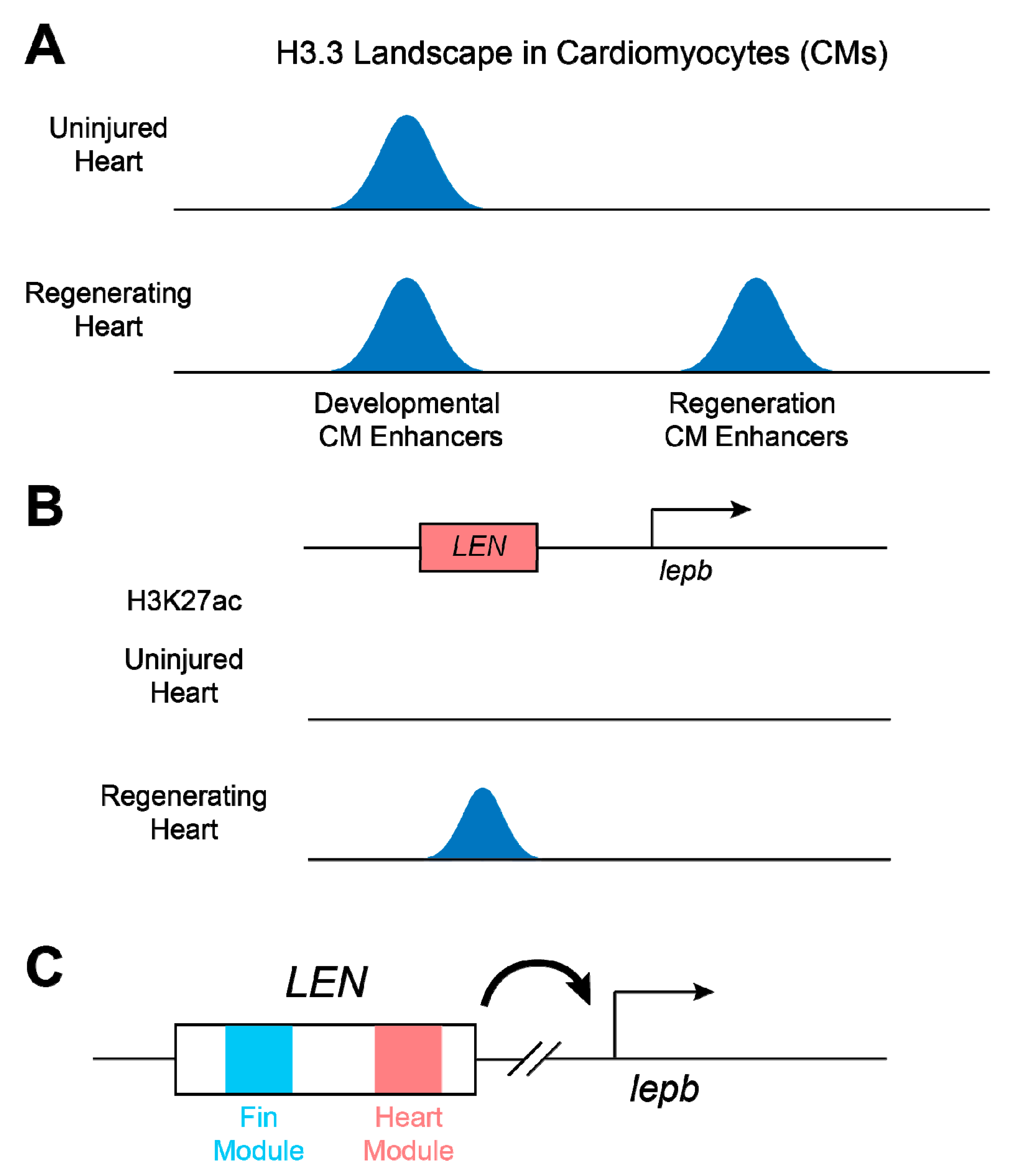
5. Cardiac Tissue Regeneration Enhancers in Zebrafish Ensure Regeneration-Specific Expression
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Richardson, W.J.; Clarke, S.A.; Quinn, T.A.; Holmes, J.W. Physiological Implications of Myocardial Scar Structure. Compr. Physiol. 2015, 5, 1877–1909. [Google Scholar] [CrossRef] [PubMed]
- Poss, K.D.; Wilson, L.G.; Keating, M.T. Heart regeneration in zebrafish. Science 2002, 298, 2188–2190. [Google Scholar] [CrossRef] [PubMed]
- Grivas, J.; Haag, M.; Johnson, A.; Manalo, T.; Roell, J.; Das, T.L.; Brown, E.; Burns, A.R.; Lafontant, P.J. Cardiac repair and regenerative potential in the goldfish (Carassius auratus) heart. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2014, 163, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Lafontant, P.J.; Burns, A.R.; Grivas, J.A.; Lesch, M.A.; Lala, T.D.; Reuter, S.P.; Field, L.J.; Frounfelter, T.D. The giant danio (D. aequipinnatus) as a model of cardiac remodeling and regeneration. Anat. Rec. 2012, 295, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Oberpriller, J.; Oberpriller, J.C. Mitosis in adult newt ventricle. J. Cell Biol. 1971, 49, 560–563. [Google Scholar] [CrossRef]
- Flink, I.L. Cell cycle reentry of ventricular and atrial cardiomyocytes and cells within the epicardium following amputation of the ventricular apex in the axolotl, Amblystoma mexicanum: Confocal microscopic immunofluorescent image analysis of bromodeoxyuridine-labeled nuclei. Anat. Embryol. 2002, 205, 235–244. [Google Scholar] [CrossRef]
- Rumyantsev, P.P. Post-injury DNA synthesis, mitosis and ultrastructural reorganization of adult frog cardiac myocytes. An electron microscopic-autoradiographic study. Z. Zellforsch. Mikrosk. Anat. 1973, 139, 431–450. [Google Scholar] [CrossRef]
- Puente, B.N.; Kimura, W.; Muralidhar, S.A.; Moon, J.; Amatruda, J.F.; Phelps, K.L.; Grinsfelder, D.; Rothermel, B.A.; Chen, R.; Garcia, J.A.; et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell 2014, 157, 565–579. [Google Scholar] [CrossRef]
- Patterson, M.; Barske, L.; Van Handel, B.; Rau, C.D.; Gan, P.; Sharma, A.; Parikh, S.; Denholtz, M.; Huang, Y.; Yamaguchi, Y.; et al. Frequency of mononuclear diploid cardiomyocytes underlies natural variation in heart regeneration. Nat. Genet. 2017, 49, 1346–1353. [Google Scholar] [CrossRef]
- Gonzalez-Rosa, J.M.; Sharpe, M.; Field, D.; Soonpaa, M.H.; Field, L.J.; Burns, C.E.; Burns, C.G. Myocardial Polyploidization Creates a Barrier to Heart Regeneration in Zebrafish. Dev. Cell 2018, 44, 433–446.e437. [Google Scholar] [CrossRef]
- Bassat, E.; Mutlak, Y.E.; Genzelinakh, A.; Shadrin, I.Y.; Baruch Umansky, K.; Yifa, O.; Kain, D.; Rajchman, D.; Leach, J.; Riabov Bassat, D.; et al. The extracellular matrix protein agrin promotes heart regeneration in mice. Nature 2017, 547, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, Y.; Heallen, T.; Leach, J.; Xiao, Y.; Martin, J.F. Dystrophin-glycoprotein complex sequesters Yap to inhibit cardiomyocyte proliferation. Nature 2017, 547, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Quaife-Ryan, G.A.; Sim, C.B.; Ziemann, M.; Kaspi, A.; Rafehi, H.; Ramialison, M.; El-Osta, A.; Hudson, J.E.; Porrello, E.R. Multicellular Transcriptional Analysis of Mammalian Heart Regeneration. Circulation 2017, 136, 1123–1139. [Google Scholar] [CrossRef] [PubMed]
- O’Meara, C.C.; Wamstad, J.A.; Gladstone, R.A.; Fomovsky, G.M.; Butty, V.L.; Shrikumar, A.; Gannon, J.B.; Boyer, L.A.; Lee, R.T. Transcriptional reversion of cardiac myocyte fate during mammalian cardiac regeneration. Circ. Res. 2015, 116, 804–815. [Google Scholar] [CrossRef] [PubMed]
- Wei, K.; Serpooshan, V.; Hurtado, C.; Diez-Cunado, M.; Zhao, M.; Maruyama, S.; Zhu, W.; Fajardo, G.; Noseda, M.; Nakamura, K.; et al. Epicardial FSTL1 reconstitution regenerates the adult mammalian heart. Nature 2015, 525, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Polizzotti, B.D.; Ganapathy, B.; Walsh, S.; Choudhury, S.; Ammanamanchi, N.; Bennett, D.G.; dos Remedios, C.G.; Haubner, B.J.; Penninger, J.M.; Kuhn, B. Neuregulin stimulation of cardiomyocyte regeneration in mice and human myocardium reveals a therapeutic window. Sci. Transl. Med. 2015, 7, 281ra45. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy, B.; Nandhagopal, N.; Polizzotti, B.D.; Bennett, D.; Asan, A.; Wu, Y.; Kuhn, B. Neuregulin-1 Administration Protocols Sufficient for Stimulating Cardiac Regeneration in Young Mice Do Not Induce Somatic, Organ, or Neoplastic Growth. PLoS ONE 2016, 11, e0155456. [Google Scholar] [CrossRef] [PubMed]
- D’Uva, G.; Aharonov, A.; Lauriola, M.; Kain, D.; Yahalom-Ronen, Y.; Carvalho, S.; Weisinger, K.; Bassat, E.; Rajchman, D.; Yifa, O.; et al. ERBB2 triggers mammalian heart regeneration by promoting cardiomyocyte dedifferentiation and proliferation. Nat. Cell Biol. 2015, 17, 627–638. [Google Scholar] [CrossRef]
- Bersell, K.; Arab, S.; Haring, B.; Kuhn, B. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell 2009, 138, 257–270. [Google Scholar] [CrossRef]
- Gemberling, M.; Karra, R.; Dickson, A.L.; Poss, K.D. Nrg1 is an injury-induced cardiomyocyte mitogen for the endogenous heart regeneration program in zebrafish. Elife 2015, 4, e05871. [Google Scholar] [CrossRef]
- Hui, S.P.; Sheng, D.Z.; Sugimoto, K.; Gonzalez-Rajal, A.; Nakagawa, S.; Hesselson, D.; Kikuchi, K. Zebrafish Regulatory T Cells Mediate Organ-Specific Regenerative Programs. Dev. Cell 2017, 43, 659–672.e655. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Johnson, B.A.; Grinsfelder, D.; Canseco, D.; Mammen, P.P.; Rothermel, B.A.; Olson, E.N.; Sadek, H.A. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc. Natl. Acad. Sci. USA 2013, 110, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Feng, Y.; Liang, J.; Yu, H.; Wang, C.; Wang, B.; Wang, M.; Jiang, L.; Meng, W.; Cai, W.; et al. Loss of microRNA-128 promotes cardiomyocyte proliferation and heart regeneration. Nat. Commun. 2018, 9, 700. [Google Scholar] [CrossRef] [PubMed]
- Beauchemin, M.; Smith, A.; Yin, V.P. Dynamic microRNA-101a and Fosab expression controls zebrafish heart regeneration. Development 2015, 142, 4026–4037. [Google Scholar] [CrossRef] [PubMed]
- Yin, V.P.; Lepilina, A.; Smith, A.; Poss, K.D. Regulation of zebrafish heart regeneration by miR-133. Dev. Biol. 2012, 365, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Nie, Y.; Lian, H.; Liu, R.; He, F.; Huang, H.; Hu, S. Acute inflammation stimulates a regenerative response in the neonatal mouse heart. Cell Res. 2015, 25, 1137–1151. [Google Scholar] [CrossRef] [PubMed]
- Aurora, A.B.; Porrello, E.R.; Tan, W.; Mahmoud, A.I.; Hill, J.A.; Bassel-Duby, R.; Sadek, H.A.; Olson, E.N. Macrophages are required for neonatal heart regeneration. J. Clin. Investig. 2014, 124, 1382–1392. [Google Scholar] [CrossRef]
- Lavine, K.J.; Epelman, S.; Uchida, K.; Weber, K.J.; Nichols, C.G.; Schilling, J.D.; Ornitz, D.M.; Randolph, G.J.; Mann, D.L. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc. Natl. Acad. Sci. USA 2014, 111, 16029–16034. [Google Scholar] [CrossRef]
- Zhang, C.; Li, Y.; Wu, Y.; Wang, L.; Wang, X.; Du, J. Interleukin-6/signal transducer and activator of transcription 3 (STAT3) pathway is essential for macrophage infiltration and myoblast proliferation during muscle regeneration. J. Biol. Chem. 2013, 288, 1489–1499. [Google Scholar] [CrossRef]
- Korf-Klingebiel, M.; Reboll, M.R.; Klede, S.; Brod, T.; Pich, A.; Polten, F.; Napp, L.C.; Bauersachs, J.; Ganser, A.; Brinkmann, E.; et al. Myeloid-derived growth factor (C19orf10) mediates cardiac repair following myocardial infarction. Nat. Med. 2015, 21, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Heintzman, N.D.; Hon, G.C.; Hawkins, R.D.; Kheradpour, P.; Stark, A.; Harp, L.F.; Ye, Z.; Lee, L.K.; Stuart, R.K.; Ching, C.W.; et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009, 459, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Nord, A.S.; Blow, M.J.; Attanasio, C.; Akiyama, J.A.; Holt, A.; Hosseini, R.; Phouanenavong, S.; Plajzer-Frick, I.; Shoukry, M.; Afzal, V.; et al. Rapid and pervasive changes in genome-wide enhancer usage during mammalian development. Cell 2013, 155, 1521–1531. [Google Scholar] [CrossRef] [PubMed]
- Shlyueva, D.; Stampfel, G.; Stark, A. Transcriptional enhancers: From properties to genome-wide predictions. Nat. Rev. Genet. 2014, 15, 272–286. [Google Scholar] [CrossRef] [PubMed]
- Rivera, C.M.; Ren, B. Mapping human epigenomes. Cell 2013, 155, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef]
- Bonn, S.; Zinzen, R.P.; Girardot, C.; Gustafson, E.H.; Perez-Gonzalez, A.; Delhomme, N.; Ghavi-Helm, Y.; Wilczynski, B.; Riddell, A.; Furlong, E.E. Tissue-specific analysis of chromatin state identifies temporal signatures of enhancer activity during embryonic development. Nat. Genet. 2012, 44, 148–156. [Google Scholar] [CrossRef]
- Kvon, E.Z. Using transgenic reporter assays to functionally characterize enhancers in animals. Genomics 2015, 106, 185–192. [Google Scholar] [CrossRef]
- Dickel, D.E.; Barozzi, I.; Zhu, Y.; Fukuda-Yuzawa, Y.; Osterwalder, M.; Mannion, B.J.; May, D.; Spurrell, C.H.; Plajzer-Frick, I.; Pickle, C.S.; et al. Genome-wide compendium and functional assessment of in vivo heart enhancers. Nat. Commun. 2016, 7, 12923. [Google Scholar] [CrossRef]
- Osterwalder, M.; Barozzi, I.; Tissieres, V.; Fukuda-Yuzawa, Y.; Mannion, B.J.; Afzal, S.Y.; Lee, E.A.; Zhu, Y.; Plajzer-Frick, I.; Pickle, C.S.; et al. Enhancer redundancy provides phenotypic robustness in mammalian development. Nature 2018, 554, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, K.J.; Katsumura, K.R.; Matson, D.R.; Devadas, P.; Tanimura, N.; Hebert, A.S.; Coon, J.J.; Kim, J.S.; Dewey, C.N.; Keles, S.; et al. GATA Factor-Regulated Samd14 Enhancer Confers Red Blood Cell Regeneration and Survival in Severe Anemia. Dev. Cell 2017, 42, 213–225.e214. [Google Scholar] [CrossRef] [PubMed]
- Narlikar, L.; Sakabe, N.J.; Blanski, A.A.; Arimura, F.E.; Westlund, J.M.; Nobrega, M.A.; Ovcharenko, I. Genome-wide discovery of human heart enhancers. Genome Res. 2010, 20, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Blow, M.J.; McCulley, D.J.; Li, Z.; Zhang, T.; Akiyama, J.A.; Holt, A.; Plajzer-Frick, I.; Shoukry, M.; Wright, C.; Chen, F.; et al. ChIP-Seq identification of weakly conserved heart enhancers. Nat. Genet. 2010, 42, 806–810. [Google Scholar] [CrossRef] [PubMed]
- May, D.; Blow, M.J.; Kaplan, T.; McCulley, D.J.; Jensen, B.C.; Akiyama, J.A.; Holt, A.; Plajzer-Frick, I.; Shoukry, M.; Wright, C.; et al. Large-scale discovery of enhancers from human heart tissue. Nat. Genet. 2011, 44, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Acharya, A.; Baek, S.T.; Huang, G.; Eskiocak, B.; Goetsch, S.; Sung, C.Y.; Banfi, S.; Sauer, M.F.; Olsen, G.S.; Duffield, J.S.; et al. The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development 2012, 139, 2139–2149. [Google Scholar] [CrossRef]
- Grieskamp, T.; Rudat, C.; Ludtke, T.H.; Norden, J.; Kispert, A. Notch signaling regulates smooth muscle differentiation of epicardium-derived cells. Circ. Res. 2011, 108, 813–823. [Google Scholar] [CrossRef]
- Katz, T.C.; Singh, M.K.; Degenhardt, K.; Rivera-Feliciano, J.; Johnson, R.L.; Epstein, J.A.; Tabin, C.J. Distinct compartments of the proepicardial organ give rise to coronary vascular endothelial cells. Dev. Cell 2012, 22, 639–650. [Google Scholar] [CrossRef]
- Kikuchi, K.; Gupta, V.; Wang, J.; Holdway, J.E.; Wills, A.A.; Fang, Y.; Poss, K.D. tcf21+ epicardial cells adopt non-myocardial fates during zebrafish heart development and regeneration. Development 2011, 138, 2895–2902. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Cavallero, S.; Patterson, M.; Shen, H.; Xu, J.; Kumar, S.R.; Sucov, H.M. Adipogenesis and epicardial adipose tissue: A novel fate of the epicardium induced by mesenchymal transformation and PPARgamma activation. Proc. Natl. Acad. Sci. USA 2015, 112, 2070–2075. [Google Scholar] [CrossRef]
- Gonzalez-Rosa, J.M.; Peralta, M.; Mercader, N. Pan-epicardial lineage tracing reveals that epicardium derived cells give rise to myofibroblasts and perivascular cells during zebrafish heart regeneration. Dev. Biol. 2012, 370, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Van Wijk, B.; Gunst, Q.D.; Moorman, A.F.; van den Hoff, M.J. Cardiac regeneration from activated epicardium. PLoS ONE 2012, 7, e44692. [Google Scholar] [CrossRef] [PubMed]
- Dube, K.N.; Thomas, T.M.; Munshaw, S.; Rohling, M.; Riley, P.R.; Smart, N. Recapitulation of developmental mechanisms to revascularize the ischemic heart. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Honor, L.B.; He, H.; Ma, Q.; Oh, J.H.; Butterfield, C.; Lin, R.Z.; Melero-Martin, J.M.; Dolmatova, E.; Duffy, H.S.; et al. Adult mouse epicardium modulates myocardial injury by secreting paracrine factors. J. Clin. Investig. 2011, 121, 1894–1904. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, K.; Holdway, J.E.; Major, R.J.; Blum, N.; Dahn, R.D.; Begemann, G.; Poss, K.D. Retinoic acid production by endocardium and epicardium is an injury response essential for zebrafish heart regeneration. Dev. Cell 2011, 20, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Schnabel, K.; Wu, C.C.; Kurth, T.; Weidinger, G. Regeneration of cryoinjury induced necrotic heart lesions in zebrafish is associated with epicardial activation and cardiomyocyte proliferation. PLoS ONE 2011, 6, e18503. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Poss, K.D. The epicardium as a hub for heart regeneration. Nat. Rev. Cardiol. 2018. [Google Scholar] [CrossRef]
- Huang, G.N.; Thatcher, J.E.; McAnally, J.; Kong, Y.; Qi, X.; Tan, W.; DiMaio, J.M.; Amatruda, J.F.; Gerard, R.D.; Hill, J.A.; et al. C/EBP transcription factors mediate epicardial activation during heart development and injury. Science 2012, 338, 1599–1603. [Google Scholar] [CrossRef]
- Vieira, J.M.; Howard, S.; Villa Del Campo, C.; Bollini, S.; Dube, K.N.; Masters, M.; Barnette, D.N.; Rohling, M.; Sun, X.; Hankins, L.E.; et al. BRG1-SWI/SNF-dependent regulation of the Wt1 transcriptional landscape mediates epicardial activity during heart development and disease. Nat. Commun. 2017, 8, 16034. [Google Scholar] [CrossRef]
- Kapoor, A.; Sekar, R.B.; Hansen, N.F.; Fox-Talbot, K.; Morley, M.; Pihur, V.; Chatterjee, S.; Brandimarto, J.; Moravec, C.S.; Pulit, S.L.; et al. An enhancer polymorphism at the cardiomyocyte intercalated disc protein NOS1AP locus is a major regulator of the QT interval. Am. J. Hum. Genet. 2014, 94, 854–869. [Google Scholar] [CrossRef]
- Smemo, S.; Campos, L.C.; Moskowitz, I.P.; Krieger, J.E.; Pereira, A.C.; Nobrega, M.A. Regulatory variation in a TBX5 enhancer leads to isolated congenital heart disease. Hum. Mol. Genet. 2012, 21, 3255–3263. [Google Scholar] [CrossRef] [PubMed]
- Goldman, J.A.; Kuzu, G.; Lee, N.; Karasik, J.; Gemberling, M.; Foglia, M.J.; Karra, R.; Dickson, A.L.; Sun, F.; Tolstorukov, M.Y.; et al. Resolving Heart Regeneration by Replacement Histone Profiling. Dev. Cell 2017, 40, 392–404.e395. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.D.; Banaszynski, L.A.; Noh, K.M.; Lewis, P.W.; Elsaesser, S.J.; Stadler, S.; Dewell, S.; Law, M.; Guo, X.; Li, X.; et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 2010, 140, 678–691. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Felsenfeld, G. Nucleosome stability mediated by histone variants H3.3 and H2A.Z. Genes Dev. 2007, 21, 1519–1529. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Zang, C.; Wei, G.; Cui, K.; Peng, W.; Zhao, K.; Felsenfeld, G. H3.3/H2A.Z double variant-containing nucleosomes mark ‘nucleosome-free regions’ of active promoters and other regulatory regions. Nat. Genet. 2009, 41, 941–945. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Zhao, J.; Wang, Y.; Wang, M.; Long, H.; Liang, D.; Huang, L.; Wen, Z.; Li, W.; Li, X.; et al. H3.3 actively marks enhancers and primes gene transcription via opening higher-ordered chromatin. Genes Dev. 2013, 27, 2109–2124. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Hu, J.; Karra, R.; Dickson, A.L.; Tornini, V.A.; Nachtrab, G.; Gemberling, M.; Goldman, J.A.; Black, B.L.; Poss, K.D. Modulation of tissue repair by regeneration enhancer elements. Nature 2016, 532, 201–206. [Google Scholar] [CrossRef]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef]
- Long, H.K.; Prescott, S.L.; Wysocka, J. Ever-Changing Landscapes: Transcriptional Enhancers in Development and Evolution. Cell 2016, 167, 1170–1187. [Google Scholar] [CrossRef]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Begeman, I.J.; Kang, J. Transcriptional Programs and Regeneration Enhancers Underlying Heart Regeneration. J. Cardiovasc. Dev. Dis. 2019, 6, 2. https://doi.org/10.3390/jcdd6010002
Begeman IJ, Kang J. Transcriptional Programs and Regeneration Enhancers Underlying Heart Regeneration. Journal of Cardiovascular Development and Disease. 2019; 6(1):2. https://doi.org/10.3390/jcdd6010002
Chicago/Turabian StyleBegeman, Ian J., and Junsu Kang. 2019. "Transcriptional Programs and Regeneration Enhancers Underlying Heart Regeneration" Journal of Cardiovascular Development and Disease 6, no. 1: 2. https://doi.org/10.3390/jcdd6010002
APA StyleBegeman, I. J., & Kang, J. (2019). Transcriptional Programs and Regeneration Enhancers Underlying Heart Regeneration. Journal of Cardiovascular Development and Disease, 6(1), 2. https://doi.org/10.3390/jcdd6010002