Abstract
The extracellular matrix (ECM) is a dynamic scaffold within organs and tissues that enables cell morphogenesis and provides structural support. Changes in the composition and organisation of the cardiac ECM are required for normal development. Congenital and age-related cardiac diseases can arise from mis-regulation of structural ECM proteins (Collagen, Laminin) or their receptors (Integrin). Key regulators of ECM turnover include matrix metalloproteinases (MMPs) and their inhibitors, tissue inhibitors of matrix metalloproteinases (TIMPs). MMP expression is increased in mice, pigs, and dogs with cardiomyopathy. The complexity and longevity of vertebrate animals makes a short-lived, genetically tractable model organism, such as Drosophila melanogaster, an attractive candidate for study. We survey ECM macromolecules and their role in heart development and growth, which are conserved between Drosophila and vertebrates, with focus upon the consequences of altered expression or distribution. The Drosophila heart resembles that of vertebrates during early development, and is amenable to in vivo analysis. Experimental manipulation of gene function in a tissue- or temporally-regulated manner can reveal the function of adhesion or ECM genes in the heart. Perturbation of the function of ECM proteins, or of the MMPs that facilitate ECM remodelling, induces cardiomyopathies in Drosophila, including cardiodilation, arrhythmia, and cardia bifida, that provide mechanistic insight into cardiac disease in mammals.
Keywords:
cardiomyopathy; ECM; remodelling; Integrin; Collagen; MMP; TIMP; Drosophila; model organism; genetics 1. Introduction
The heart is a dynamic organ, modifying its morphology and molecular structure in response to physiological changes through embryonic organogenesis, growth, exercise, and aging. Much of what is known of the genetic basis of heart formation and function has emerged from genetic models of heart function such as the mouse and zebrafish. There is but a single invertebrate genetic model for heart development and aging; the fruit fly Drosophila. Upon first glance, the fruit fly would seem an unlikely candidate for cardiac research; its heart is a dorsally-positioned, linear tube that is not essential for the delivery of oxygen to tissues and has no stem cells [1,2,3]. Therefore, why then have dozens of laboratories published over 1500 papers on this arcane model? The answer lies in the remarkable conservation of gene identity, expression and function between the fly heart and the hearts of mammals of medical or veterinary significance. Insights into gene function in the fly translate rapidly to vertebrates. Here we shall briefly review the fly model and the extent of cardiac gene conservation, with focus on one family of genes required to establish and maintain the extra cellular matrix (ECM); the matrix metalloproteinases (MMPs). Genes of the ECM underlie morphological adaptations in the heart that are linked to numerous cardiac diseases present in mice, dogs, pigs, and humans [4,5,6,7,8]. In this review, we shall illustrate how insights into gene function from the Drosophila model are relevant to our understanding of the basis of mammalian disorders.
2. ECM Regulation and Cardiac Dysfunction
The ECM is a dynamic three-dimensional network of proteoglycans, glycoproteins, and fibrous proteins linking and protecting the intercellular regions within organs and tissues, including the heart (reviewed in [9,10]). It serves as a scaffold, providing structural support to organise cells, transmit tension through tissues, and mitigate damage from mechanical stressors [11]. ECM remodelling is key to cardiac dysfunction and repair; the disruption of structural and regulatory ECM proteins is linked to the progression of myriad heart and vascular diseases. Increased ECM deposition is a hallmark of dilated cardiomyopathy (DCM), hypertrophy, and heart failure in humans (reviewed in [12,13,14,15]).
Congenital, pathological, and age-related cardiac disorders are a leading cause of death amongst many mammals, including humans and canines [16,17,18,19]. Heart diseases affecting the ECM, such as congestive heart failure and cardiomyopathy, are common to many dog breeds [20]. Amongst smaller dogs, mitral valve disease accounts for the highest number of cases of non-congenital cardiac failure, whereas cardiomyopathy is prevalent amongst larger dogs [21,22]. DCM has been characterised in Doberman Pinschers [23,24], Great Danes [25], Boxers [26,27], and Irish Wolfhounds [28], while sub-aortic stenosis has been examined in Newfoundlands [29,30] and Golden Retrievers [31], and ventricular arrhythmia is found in many breeds (reviewed in [20,21]).
Many cardiac disorders are difficult to elucidate due to the existence of complex gene and protein interactions, and functional redundancy. These disorders might be examined within simpler systems more amenable to manipulation, such as the fruit fly Drosophila melanogaster. Owing to the functional and compositional similarities between fly and vertebrate hearts, the study of cardiac disease gene candidates in Drosophila has the potential to illuminate genetic interactions underlying polygenic syndromes, which may not be readily determined in other organisms given the limitations imposed by life history traits and the availability of genetic tools (reviewed in [32,33,34]).
3. The Drosophila Model
Drosophila is a proven and powerful model for the study of cardiogenesis, cardiac aging, and heart disease [33,35]. This system boasts numerous advantages over vertebrate models owing to its relative simplicity, short generation time, and the availability of genetic tools. The diversity of genetic approaches is reviewed more comprehensively in [36]. Moreover, a sizeable library of mutant and transgenic strains is available through various stock centres [37,38,39].
3.1. Simplicity and Homology
Compared to vertebrates, Drosophila possesses a smaller genome encoding fewer protein variants (reviewed in [40,41,42]); Drosophila has fewer than 15,000 predicted protein-coding genes [43,44] compared to the (approximately) 19,000 in humans and mice [45,46]. As a result, a genetic approach may be utilised to manipulate the expression or structure of entire gene families simply by generating mutant alleles or inserting transgenes. This has enabled the inducible switching of, for example, all Integrin function or all endocytosis within intact organisms [47,48]. Furthermore, the expression of mutant isoforms of human genes in Drosophila can recapitulate human cardiac disease [49,50].
The Drosophila heart, or dorsal vessel, is a linear valved tube, and thus structurally simpler than the looped and multi-chambered vertebrate heart. Both hearts are derived from developmentally homologous lateral mesodermal precursors and share the same pattern of specification and medial migration to the midline to form a cardiac tube (reviewed in [32]). Cardiogenesis in vertebrates and invertebrates involves many conserved molecular mechanisms [51,52]. For example, the transcription factors and signalling cascade specifying cardiogenesis are highly conserved; homeobox transcription factors activate a transcriptional cascade in the pre-cardial mesoderm that promotes cardioblast (CB) specification and later differentiation [53,54]. The homeobox transcription factor Tinman (Tin), which specifies the visceral and cardiac mesoderm, is homologous with mammalian Nkx2-5, though these differ in their deployment and may have adapted unique cardiogenic roles [55,56]. Tin is regulated by the TGF-β growth factor Decapentaplegic (Dpp), a BMP2/4-homologue [57,58]. Pannier, involved in CB specification in Drosophila, is homologous with GATA4 [59]. Drosophila possesses a single Mef2 gene, Dmef2, which is expressed in myocardial precursors [60,61]. Its Hand gene, encoding a basic helix-loop-helix (bHLH) protein expressed in cardiac precursors, has orthologues in vertebrates, including chicks and mice [51,62,63,64]. Vertebrate COUP-TF/NR2F is homologous with Drosophila’s Seven-up (Svp) [65], and Islet-1 (Isl1), which is involved in cardiac precursor specification, has a Drosophila counterpart in Tailup (Tup) [66].
Proteomic analysis reveals a general conservation of function between Drosophila and vertebrate cardiac proteins, which retain critical domain and structural similarities despite lower levels of over-all sequence identity [67]. This includes significant conservation of structural ECM proteins and receptors between Drosophila and mammalian species; for instance, Collagens, Laminins, Perlecan, and Integrins are all present in Drosophila (reviewed in [67]). Conserved proteins include those mapped to known vertebrate mutations resulting in cardiac disease and dysfunction [68]. For example, proteomics has identified Troponin-T as a conserved protein, which can trigger cardiomyopathy in humans if mutated in the Tropomyosin binding region. Similar mutations in the Drosophila homologue causes restrictive cardiomyopathy [69].
3.2. Life History
Unlike mammals, which may survive for years or decades and may require months or years to become reproductively competent, Drosophila species are short-lived, with an average lifespan of less than 100 days under normal laboratory conditions, and a generation time of only ten days. This facilitates longevity studies examining the effects of heart-related genes on aging [1,70,71]. Although heart function is necessary for adult survivorship, a poorly functioning heart is sufficient throughout embryonic and early larval stages of Drosophila growth, since the oxygenation of tissues occurs through the tracheal system (reviewed in [72]). This allows for the study of severe mutant or mis-expression phenotypes that, in other organisms, would prove lethal in early development.
3.3. Generating Genetic Mosaics
Although CRISPR-mediated approaches to genetic modification are becoming increasingly prevalent (e.g., [73,74]), the historical advantage of Drosophila over other genetic models lies in the ability to insert novel genetic sequences (such as gene enhancers, inverted sequences for RNAi, exogenous genes, or fluorescently tagged sequences), or to interrupt endogenous genes, using modified mobile genetic elements (e.g., P-element transposons) [75,76,77,78]. Temporal or spatial genetic mosaics can be created through site-directed DNA recombination with transposons containing yeast recombinase [79,80,81]. Mitotic recombination can be induced by tissue-specific enhancers, temperature shifts, or drugs to generate gain-of-function mutants or loss-of-function/genetic null mutants [79].
The Gal4/UAS system, originally adapted from the yeast Saccharomyces cerevisiae, has found common usage in Drosophila, enabling the precise spatial regulation of specific gene transcription [82,83]. This system can be exploited to drive the expression of transgenes (such as fluorescent reporters), or induce mis-expression, either through over-expression or RNA interference (RNAi)-mediated knock-down [84]. Temporal control can be exerted with the expression of a temperature sensitive inhibitor of Gal4 [85].
3.4. In Vivo Imaging
A key advantage of Drosophila in the study of the heart is the ease with which live imaging can be performed. Transgenic Drosophila can express fluorescent reporters in vivo by way of enhancer or protein traps. Enhancer traps are fluorescent reporter proteins (e.g., green or red fluorescent protein; GFP/RFP) expressed under the control of the endogenous enhancer [86,87]. Protein traps result from the insertion of a fluorescent gene transposon into a native gene, adding the GFP/RFP coding sequence as a new exon within the functioning endogenous protein [86]. Fosmid constructs may be used to insert transgenes encoding C-terminally tagged proteins [88]. The levels and localisation of these proteins can then be assessed within a living system [89,90,91].
The Drosophila embryonic and larval cuticle is translucent, and many internal structures remain visible in the live organism. Cardiogenesis may be observed in embryos using fluorescence microscopy [1,89]. The internal structures of the larger and more opaque larvae may be imaged using optical coherence tomography (OCT) [49]. OCT is non-invasive and high-speed, providing a rapid, high-throughput means of quantifying or qualifying contractility, rhythmicity, and heart chamber size and topography [1,92,93].
4. The Drosophila Heart
4.1. Early Morphogenesis
In contrast to vertebrates, Drosophila possesses an open circulatory system of low hydrostatic pressure. The insect circulatory system contains hemolymph, a nutrient- and hemocyte-rich interstitial fluid that directly bathes tissues and organs, but which is not essential for oxygen transport (reviewed in [94]). The dorsal vessel is a linear tube formed during embryogenesis. Drosophila cardiac precursors arise in a manner analogous to their vertebrate counterparts [95,96]. As the mesodermal precursors enter through the ventral gastrulation furrow, they flatten to associate with the interior (basal) side of the ectoderm. The lateral-most mesodermal cells receive the strongest ectodermal signal from the Dpp (BMP) and Wingless (Wnt) ligands, whose activities converge to activate heart-specific transcription factors, beginning with Tin (Nkx2-5) (Figure 1A). Subsequently, the lateral ectoderm migrates to the dorsal surface of the embryo to displace the amnioserosa, a transient tissue overlaying the yolk (Figure 1B). Tin-expressing heart precursors maintain an association with the dorsal ectoderm and accompany its migration. As the ectodermal cells approach their contralateral partners upon dorsal closure, the post-mitotic heart cells (CBs) migrate independently to meet their partners also [89,97]. The CBs assume a characteristic teardrop shape, whereupon the ectodermally-exposed edge of the apical surface extends motile processes medially, while the interior edge of the apical surface remains quiescent (Figure 1B’). This motile domain forms a Leading Edge, and exhibits guidance behaviour typical of Collective Cell Migration [89]. The Leading Edge is the first region to make contralateral contact, and matures into the Cadherin-based dorsal midline cell junction of the heart tube (Figure 1C’). Concurrently, the CB soma curves towards the midline to form the ventral seam of the heart tube (reviewed in [72,98]). During this process, the pre-luminal domain is constrained within the heart tube (Figure 1C”). The cardiac cells, cardiomyocytes, enclose the heart lumen. These are flanked by non-contractile pericardial cells (PCs) that function as detoxifying nephrocytes [99,100]. The heart tube is suspended from the epidermis by seven pairs of alary muscles, performing the role of a diaphragm [32,101].
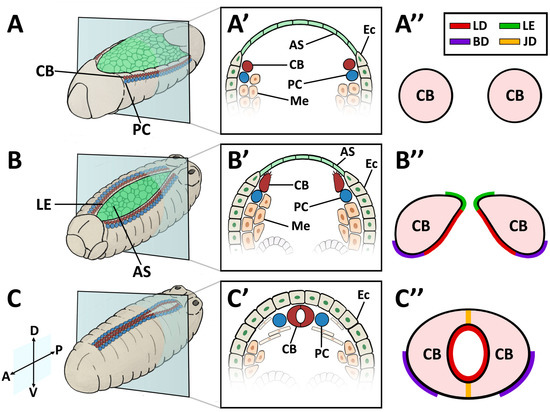
Figure 1.
Cardioblast (CB) migration during Drosophila embryogenesis. (A–A”) Stage 12–13 embryo. (A) Perspective view of a whole embryo showing cardiac precursor cells associated with the dorsal ectoderm prior to dorsal closure; (A’) Cross-sectional impression showing the lateral-most mesodermal cells, which have become specified as CB precursors; (A”) CB precursors prior to the extension of the motile apical domain. (B–B”) Stage 14–15 embryo. (B) Lateral ectoderm and CBs migrate dorso-medially and displace the amnioserosa; (B’) Cross-sectional impression showing Leading Edge activity. CBs extend a motile apical domain towards the midline; (B”) CBs assume a teardrop shape with the Cadherin-rich apical domain forming a Leading Edge. The basal domain is quiescent. The pre-luminal domain localises Dystroglycan, βPS-Integrin, Multiplexin, and the Slit-Robo complex. (C–C”) Stage 17 embryo. (C) CBs make contralateral contact at the midline upon dorsal closure. (C’) Cross-sectional impression showing paired CBs at the dorsal midline, flanked by PCs; (C”) Formation of the heart tube. CBs form a dorsal and ventral seam at the Cadherin-based junctional domain to enclose a lumen. Dorsal at top. A: anterior; AS: amnioserosa; BD: basal domain; CB: cardioblast; D: dorsal; Ec: ectoderm; JD: junctional domain; LE: Leading Edge; LD: luminal domain; Me: mesoderm; P: posterior; PC: pericardial cell; V: ventral.
4.2. Embryonic and Larval Heart
The embryonic and larval dorsal vessel exhibits anterior-posterior polarisation, and is bisected by intercardiac valve cells to form two distinct regions; the posterior heart, and the narrower anterior aorta (reviewed in [32,94]). The entire heart is comprised initially of 104 CBs, which differentiate into contractile cardiomyocytes, ostial (inlet) cells, and intercardiac valve cells [3,52]. Despite lengthening nearly five-fold by the time of adulthood, the Drosophila cardiac system contains no stem cells; there is no CB proliferation or migration post-embryogenesis, nor is there replacement in response to tissue loss or damage [1,11,102].
The embryonic dorsal vessel extends from segment T2 (Thoracic segment 2) to A8 (Abdominal segment 8), with the contractile heart chamber encompassing segments A5 through A8 [62,98,102]. Heart chamber identity is specified by the Hox gene abdominal-A (abd-A), whereas Ultrabithorax (Ubx) specifies the posterior aorta [103]. The dorsal vessel exhibits segmental patterning, with a majority of segments formed from six pairs of cardiomyocytes; the two anterior-most pairs express seven-up (NR2F) and are destined to become ostial cells, whereas the four larger posterior pairs express tinman (NKX2-5) and will develop as contractile cells (reviewed in [98]). All of these cells are CB derivatives. Throughout the larval phase (first to third instar), thin and thick myofilaments accrue in a circular pattern within cardiac cells, concomitant with lumen expansion [3]. PCs enlarge and decrease in number through apoptosis [62,94,104].
4.3. Adult Heart
During metamorphosis, the dorsal vessel experiences extensive remodelling. Twenty of the original 104 CBs undergo apoptosis, resulting in the loss of all cardiac cells in segments A6 through A8, such that segment A5 becomes the terminal heart chamber [3,62,98]. The diameter of the lumen increases as cardiomyocytes enlarge. Aortal myocytes produce additional myofibrils and become contractile [3]. Further differentiation occurs, resulting in the formation of additional ostial cells (from three pairs in larvae to five in adults) and valve cells (from one intercardial valve to three) [94,102]. Lymph gland-like cells associated with the anterior dorsal vessel migrate and differentiate into myoblastic longitudinal cardiac fibres [105].
5. The Drosophila Heart ECM
5.1. Form and Function
ECM and its receptors play a critical role in Drosophila cardiogenesis and morphogenesis through the formation of migration corridor cell anchoring points, and through the regulation of signalling and guidance cues that specify the cardiac luminal domain [106]. In the developed larval and adult heart, the ECM maintains connections between cardiomyocytes and adjacent supporting cells such as alary muscles, restores diastolic heart diameter, and helps synchronise myocyte contraction (reviewed in [107,108]).
The ECM is comprised principally of fibrous proteins (e.g., Collagens, Elastin), integrated glycoproteins (e.g., Fibronectin, Perlecan, Nidogen), and growth factors (reviewed in [10]). Key transmembrane cell-adhesion receptors include Integrins, Syndecans, and Dystroglycan [109,110,111,112,113]. The molecular composition of the ECM changes with function and location (reviewed in [10,114]). For the purpose of this review, we shall focus on the Collagen-IV-dominated basement membrane (basal lamina), but not the interstitial ECM, which assists in positioning the heart.
The sheet-like BM forms a compact and ordered proteoglycan matrix abutting endothelial and epithelial cell monolayers (Figure 2). It is dominated by Collagen and Laminin networks, which are further stabilised by Nidogen and Perlecans (reviewed in [10,114]). In most polarised cells, BM proteins are laid down at the basal but not apical surfaces; thus the BM is involved in defining cell apical-basal polarity (reviewed in [115,116]). Migrating cells such as CBs display asymmetric localisation of BM proteins; certain BM constituents and their receptors aggregate to both the basal and luminal domains, and some, such as Multiplexin or Pericardin, are restricted to one domain (Figure 3 and Figure 4) [101,110,117,118,119,120].
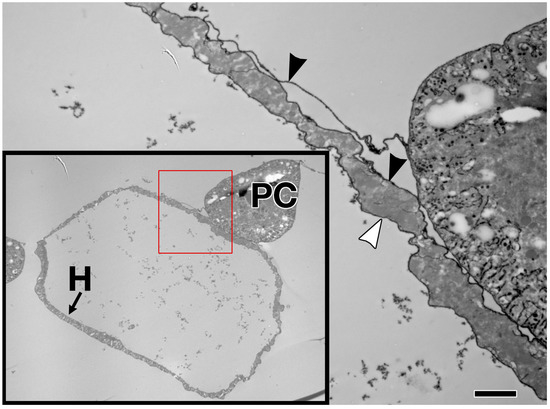
Figure 2.
Extracellular matrix in the larval Drosophila heart. Enlargement of the cross section shown within the red box (lower left) reveals Ruthenium Red-labelled ECM proteoglycans on both the external (black arrowhead) and luminal (white arrowhead) surfaces of the heart muscle cell, with no endothelium. H: heart muscle; PC: pericardial cell. Scale bar is 2.0 microns.
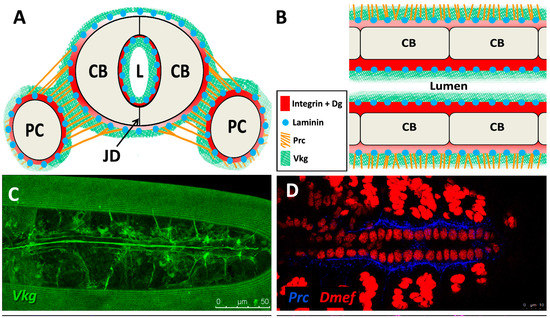
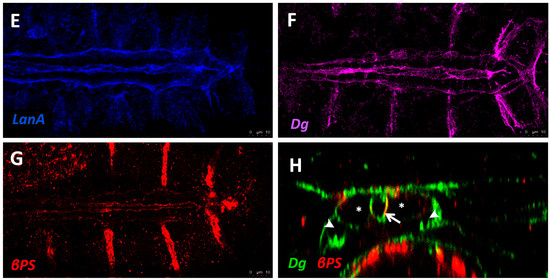
Figure 3.
Localisation of ECM proteins and receptors in the embryonic Drosophila heart. (A) Cross-sectional impression of a stage 17 embryonic dorsal vessel with flanking PCs. The transmembrane receptors βPS-Integrin (βPS) and Dystroglycan (Dg) are present along the cardioblast surface, but are concentrated at the luminal domain. Dg is excluded from the junctional domain. Laminin A (LanA) is present ubiquitously. Viking Collagen-IV (Vkg) localises to the basal lamina and is expressed within the heart lumen. Pericardin (Prc) is absent from the lumen. Dorsal at top. CB: cardioblast; JD: junctional domain; L: lumen; PC: pericardial cell; (B) A portion of the embryonic dorsal vessel in coronal view (frontal plane), illustrating CBs but not PCs. Alary muscles have not been included for simplicity; (C–G) Distribution of ECM proteins and receptors in stage 17 embryos. Anterior on left. (C) Vkg is expressed along the basal lamina of cardiac cells, localised to the luminal and abluminal surfaces. The striated lines running parallel the heart tube is an eggshell artefact; (D) Prc forms bundles localised to the abluminal surface of the heart tube, and envelopes the PCs; (E) LanA is expressed along the luminal and abluminal cardiac cell surface and within muscle costameres; (F) Dg localises to the apical CB surfaces, and to costameres; (G) βPS-Integrin is expressed primarily along the CB apical and luminal surfaces, and to a lesser extent along the basal surface; (H) Cross-sectional distribution of Dg and βPS-Integrin in a stage 17 embryo. Dg is strongly expressed along the CB luminal surface, and is also present on the abluminal surface, with weak ventral expression. Integrin is strongly expressed along the CB luminal surface, and weakly expressed along the abluminal surface. Dorsal at top. Asterisks label CBs; arrows label the heart lumen; arrowheads label PCs.
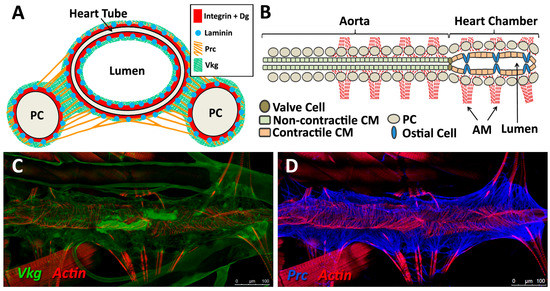
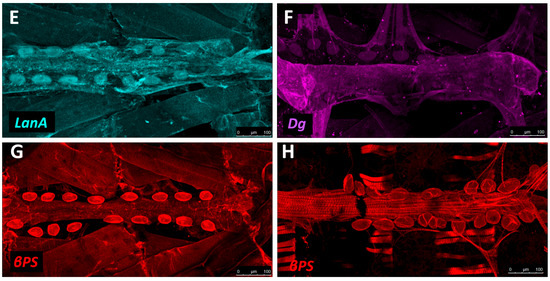
Figure 4.
Localisation of ECM proteins and receptors in the larval Drosophila heart. (A) Cross-sectional impression of a third instar larval dorsal vessel with flanking PCs. The transmembrane receptors βPS-Integrin and Dg are present along the luminal domain and at the abluminal surfaces of the heart tube. LanA is ubiquitous. Vkg localises to the basal lamina and, at lower levels, within the heart lumen. Prc is absent from the lumen. Dorsal at top; (B) The larval dorsal vessel in coronal view (frontal plane), illustrating the aorta and the heart chamber, divided by valve cells. The aorta is comprised of non-contractile cardiomyocytes. The heart chamber is wider and formed from contractile cardiomyocytes and non-contractile ostial cells. Anterior on left. AM: alary muscle; CM: cardiomyocyte; (C–G) Distribution of ECM proteins and receptors in late third instar larvae. Anterior on left; (C) Vkg is expressed along the basal lamina of cardiac cells, as well as in hemocytes and along the alary muscles and trachea. Actin forms parallel myofibrils that envelop the heart tube, and localises to alary muscles; (D) Prc networks localise to the abluminal surface of the heart tube, and envelope the PCs; (E) LanA is expressed along the luminal and abluminal cardiac cell surface, along the pericardial cell surface, and within muscle costameres (banded pattern); (F) Dg is expressed along the heart tube and pericardial cell surface, and within the costameres. Alary muscle tissue is also labelled; (G) βPS-Integrin is expressed along the heart tube and pericardial cell surface. Integrin is expressed along costameres (banded pattern); (H) Five-day-old adult stained for βPS-Integrin. Integrin is expressed by new longitudinal muscles ventral to the heart tube, and by PC and costameres.
5.2. Basement Membrane Constituents
Major ECM proteins identified in the Drosophila heart and their vertebrate homologues are summarised in Table 1, Table 2 and Table 3. Major components are discussed below.

Table 1.
Structural proteins of the BM.
Table 2.
Receptors and signalling cues of the BM.
Table 3.
Remodelling proteases of the BM.
5.3. Structural Proteins
Collagens are the core structural proteins of the ECM, and contribute to the stability and tensile strength of the cardiac tissue (reviewed in [10]). Three conserved genes in Drosophila encode BM Collagens; multiplexin (mp), Cg25C, and viking (vkg), while a fourth, pericardin (prc), encodes a unique Collagen-like protein (reviewed in [170]). Vkg and Cg25C together form the heterotrimeric Drosophila Collagen-IV, which assembles at the basal lamina to provide structural support through linkage with a Laminin-Nidogen complex [119,132]. Prc is a domain-specific Collagen-IV-like cardiac ECM protein required for the adhesion of nephrocytes (PCs) to heart muscle cells [11,101]. Prc is secreted by embryonic PCs and by the larval fat body [101,119]. Prc is recruited to the abluminal domain of cardiac cells by Lonely Heart (Loh), a secreted disintegrin and metalloproteinase with Thrombospondin repeats (ADAMTS)-like protein [11,171]. Multiplexin (Mp) is orthologous with mammalian Collagen-XV/XVIII [172]. It is apically secreted by embryonic cardiac cells, where it localises to the CB luminal domain during the closure of the dorsal vessel [118]. Mp enhances luminal expansion, and its localisation to the posterior dorsal vessel is responsible for the larger heart lumen relative to that of the aorta [118].
Laminin glycoproteins are thought to be the first component of the ECM recruited to the BM [119,128]. They bind the BM cell-surface receptors Integrin and Dystroglycan (Dg), and are necessary for embryonic cell adhesion, migration, and differentiation [128,173,174]. Two heterotrimers are produced in invertebrates compared to 15 or more in mammals [119,175].
Matricellular proteins such as secreted protein acidic and rich in cysteine (SPARC), Thrombospondins (TSPs), and Loh are matrix components serving to link cell surface receptors, proteases, and structural proteins (reviewed in [5,171]). SPARC is an evolutionarily conserved glycoprotein involved in tissue remodelling through the modulation of Collagen deposition and fusion [5,133]. SPARC is secreted by hemocytes to form the basal lamina, where it is implicated in both angiogenesis and angiostasis [132]. TSPs are multi-domain glycoproteins that bind numerous ECM proteins, including Collagens, as well as cell surface receptors, such as Integrins [136,176]. Their inhibitory effect on proteases contributes to matrix stabilisation (reviewed in [5]).
5.4. Receptors
Integrins are transmembrane receptors for Collagens and Laminins. They are essential for the formation of cell-matrix linkages, and serve to connect the ECM to the Actin cytoskeleton (reviewed in [109,177]). Integrin signalling is moderated by numerous intercellular linkers, including focal adhesion kinase (Fak), Integrin-linked kinase (Ilk), and Talin [1,141]. During early cardiogenesis, Integrins are necessary to establish the apical ECM, and for the assembly of Collagen, Laminin, and Dystroglycan to form the luminal ECM [110]. The luminal ECM assembles while the CBs are migrating medially (Figure 1B”). Initially, the distribution of Integrin, Laminin, and Collagen is diffuse along the apical and basal CB surface. During the last hours of migration, the activity of the apical Cadherin-rich Leading Edge process increases, and the adjacent apical pre-luminal domain accumulates higher levels of Integrin [89,110]. Apical domain definition and the later emergence of the Leading Edge require Integrin function, as well as that of its cytoskeletal linker protein, Talin [90,110].
Integrin targeting and turnover at the apical ECM is required for the later targeting and retention of the morphogen Slit and its receptor, Robo, to the luminal domain [106,154]. Robo signalling destabilises Cadherin adhesions and acts to delineate the junctional and luminal domains of the heart tube [106]. Loss of function of any of the luminal receptors and ECM components results in the expansion of the Cadherin domain over the apical surface, and a reduced or absent lumen. Other key apical surface receptors are dependent upon Integrin for targeting and stabilisation [110]. These receptors further stabilise and define cardial cell luminal and junctional domains. Syndecan (Sdc), a transmembrane heparin sulfate proteoglycan (HSPG), binds the guidance cue Slit and acts to apicalise the Slit/Roundabout (Robo) complex to promote localised ECM assembly [113,150,152,154]. The Netrin receptors Uncoordinated-5 (Unc5) and Frazzled (homologous with Deleted in Colorectal Cancer) play complementary roles in the regulation of lumen size [157].
Shortly before hatching, the CBs differentiate into cardiomyocytes and re-organise their ECM. Levels of Slit and Robo decline and the Integrin adhesion complex relocates to cardiomyocyte adhesions and muscle costameres. This process is poorly understood, since the development of the cuticle renders the heart largely inaccessible at this stage. During post-embryonic heart growth and morphogenesis, continuous Integrin turnover is necessary for the remodelling of cardiac muscle insertions assoicated with growth and metamorphosis [1].
The Dystrophin-glycoprotein complex (DGC) is involved in signalling and stabilisation of the ECM (reviewed in [178,179]). Dystrophin (Dys) links transmembrane Dg to Actin, connecting the ECM to the cytoskeleton [35,180]. Dg serves as a transmembrane receptor for Laminin [5,115,181,182]. Dg localises to the CB basal and luminal surfaces where it stabilises apical-basal polarity [90,112,183].
6. ECM Regulation and Turnover
6.1. MMPs and TIMPs
The chemical and biophysical properties of tissues change over time to reflect alterations in cell function, physiology, tension, elasticity, and shape. Constant turnover of the ECM allows for the renewal of proteins, the introduction and post-translational modification of different proteins or carbohydrates, and the remodelling of the density and geometry of protein cross-linking (reviewed in [9]). Integral to this process is the regulated cleavage of ECM proteins by a family of conserved zinc-dependent endopeptidases termed matrix metalloproteinases (MMPs) (reviewed in [42,184]). MMP expression is altered in mammalian heart diseases and during myocardial infarction [185,186,187,188,189,190]. MMP function in the Drosophila heart is under study, revealing important roles in lumen establishment and in defining compartment-specific ECM composition [159].
MMPs fall broadly into two classes, transmembrane MMPs and secreted MMP; however the majority of vertebrate MMPs are secreted (reviewed in [41]). MMPs are synthesised as latent zymogens, and are activated by proteolysis or conformational changes via a cysteine-switch mechanism [191]. They are regulated at the post-translational level by endogenous tissue inhibitors of matrix metalloproteinases (TIMPs) (reviewed in [192]). Most vertebrates and invertebrates express multiple MMPs with partially overlapping substrates, which, taken together, are capable of cleaving nearly every kind of ECM protein (reviewed in [184,193]). MMPs are also capable of cleaving non-ECM proteins, including cytokines, chemokines, clotting factors, pericellular proteins, and cell-surface receptors, and can activate other proteinases [40,41,194,195,196]. This substrate versatility sees MMPs involved in myriad developmental and homeostatic processes such as angiogenesis, coagulation and wound healing, Collective Cell Migration, and bone modelling, as well as pathologies such as cancer and heart disease [89,193,194].
6.2. ECM Remodelling in Vertebrates
Numerous MMP and TIMP variants are encoded by vertebrates; for instance, humans possess 23 MMPs and four TIMPs, which collectively inhibit all MMPs (reviewed in [40]). Vertebrate MMPs fall into six categories on the basis of substrate affinity or localisation; collagenases, gelatinases, matrilysins, stromelysins, membrane-type (MT)-MMPs, and other MMPs (reviewed in [184]). Collagenase1/2/3 (MMP1/8/13, respectively), secreted GelatinaseA/B (MMP2/9, respectively), Stromalysin1 (MMP3), and Matrilysin1 (MMP7) target substrates within the vertebrate myocardium (reviewed in [169]). All four TIMPS (TIMP1/2/3/4) are expressed within the murine myocardium, with TIMP4 being largely restricted to the heart [197,198,199].
MMPs therefore exhibit some degree of redundancy, and null mutants for a single MMP do not always reveal prominent embryonic phenotypes. In mice, many MMPs appear dispensable for embryogenesis (reviewed in [40]). MMPs are nonetheless required for vertebrate heart development through their role as agents of ECM remodelling and turnover, and careful regulation of MMPs is necessary to maintain cardiac homeostasis post-embryogenesis [161,200,201].
6.3. ECM Remodelling in Drosophila
Drosophila encodes only two MMPs; a secreted MMP (MMP1), and the trans-membrane MMP (MMP2), both of which are regulated by a singular TIMP [2,202,203,204]. Phylogenetic analysis indicates that the Drosophila MMPs are each more similar to their vertebrate homologues than they are to one another [2]. However, MMP1 and MMP2 have partially overlapping substrates; for instance, both are capable of hydrolysing Fibronectin and non-fibrillar Collagens, though MMP1 is incapable of cleaving Laminin [203]. Temporal expression patterns also differ; MMP1 is expressed throughout embryogenesis and larval growth, but not adulthood, whereas MMP2 is expressed from late embryogenesis through adulthood [2,203,204].
In Drosophila, MMPs are dispensable for embryonic survival but are critical for tissue remodelling at later developmental stages; embryonic expression of one or both MMPs is required for axonal fasciculation [205], cardiogenesis [159], and tracheal expansion [2].
MMP1 and MMP2 are essential for cardiac development, operating co-operatively to enhance Leading Edge motility during embryonic CB precursor migration, and then in a complementary manner to establish lumen expansion [159]. MMP1 localises to the (pre)luminal domain and constrains the size of the presumptive midline adhesive (junctional) domain. MMP2 localises to the Leading Edge, where it facilitates Collective Cell Migration and CB polarisation by regulating the apical domain of the ECM. MMP2 is required to restrict lumen-specific ECM proteins (e.g., Slit, Dystroglycan, and Collagen) to the luminal domain.
In both Drosophila and vertebrates, MMPs play analogous roles in heart morphogenesis and cardiomyopathy. Future studies on this and other genetic models should reveal more about how cardiac ECM remodelling contributes to stress and aging, and identify therapeutic targets for diagnosis or therapy.
7. Cardiac Aging and ECM Disruption
Cardiac performance declines progressively in aging Drosophila and vertebrates (reviewed in [35,206]). Mis-regulation of ECM components in the form of altered expression or accumulation is characteristic of cardiac aging and dysfunction. Changes in the synthesis, deposition, and degradation of cardiac ECM are correlated with fibrosis and hypertrophy that mark the aging or diseased vertebrate heart (reviewed in [5,207,208]). Fibrosis results from an accretion of ECM proteins (loss of homeostasis) following accumulation and differentiation of cardiac fibroblasts into pro-fibrotic myofibroblasts (largely responsible for fibrillar Collagen synthesis [209]) that renders the heart less elastic, and is correlated with reduced or non-heterogeneous electrical conduction [210,211], elevated incidence of arrhythmia [212,213], and heart failure [214,215]. Hypertrophy emerges in the backdrop of progressive myocyte loss and vascular stiffening in aging vertebrates [216]. Cardiac dilation similarly arises from alterations in ECM regulation and involves a weakening of Collagen-cytoskeletal linkages, though the resultant increase in luminal volume occurs without a compensatory increase in chamber wall thickness, as is the case in hypertrophy, resulting in impaired muscle integrity throughout development and growth [217]. Dilation is correlated with interstitial fibrosis [7,218] and reduced contractility that may result in diminished cardiac output and decreased life expectancy [1,4]. These phenomena impair myocardial compliance and contribute to contractile dysfunction, impeding relaxation and increasing diastolic pressure (for a detailed review, see [206,215,219]).
Unfortunately, many mutations of structural ECM genes prove embryonic-lethal for both vertebrates and Drosophila, hampering the examination of adult phenotypes. However, temporal activation of gene expression in Drosophila (e.g., by employing the temperature-sensitive Gal80TS inhibitor of Gal4) can circumvent this constraint.
7.1. Altered Expression or Deposition of Structural Proteins
Collagens accumulate naturally in the aging vertebrate heart [108,220,221], and changes in Collagen levels are noted during heart failure [222]. Concomitant over-expression of myocardial Collagen-VI α2 and the guidance receptor Down syndrome cell adhesion molecule (Dscam) results in cardiac defects in both flies and mammals; adult Drosophila show a reduction in heart rate and arrhythmic contractions due to altered cell-substrate adhesion, and mice develop cardiac hypertrophy and atrial-septal defects [146]. Drosophila with reduced Mp (Collagen-XV/XVIII) exhibit embryonic luminal defects and impaired contractility (reduced fractional shortening) as adults [118], while mice deficient in Collagen-XV develop cardiomyopathies and exhibit myofibrillar disorganisation and cardiac stiffening [125]. Drosophila heterozygous for Cg25C (Collagen-IV α1) manifest sarcomeric defects along the musculature of the adult oviduct, and embryos have abnormal Perlecan localisation along the cardiac BM and accumulation within hemocytes [119]. A reduction of either Collagen-IV (Vkg or Prc) or Laminin expression within PCs and cardiomyocytes with RNA interference mitigates age-related defects; the thickness of the cardiac BM is reduced and lifespan increases [121]. Compared to control adults, individuals with reduced Collagen-IV or Laminin experience a less severe reduction in systolic and diastolic diameter with age, and show increased fractional shortening.
Loh (ADAMTSL-6) and Prc (Collagen-IV-like protein) mutants reveal a different class of cardiac phenotypes, including PC detachment from the heart tube, compromised circulation, and reduced lifespan [11]. Drosophila reared on a high sucrose diet accumulate Prc within cardiac tissues and show myofibrillar disorganisation; these develop cardiac arrhythmia, asystoli, and fibrillation as adults, and exhibit impaired contractility similar to the dilated cardiomyopathy observed in diabetic mammals [223].
7.2. Mis-Regulation of Receptors and Linker Proteins
Integrin levels increase in the aging Drosophila heart [141] and mammalian vascular smooth muscle [142], but are reduced in older murine hearts [220]. In Drosophila, a mild reduction of β1-Integrin or Integrin-linked kinase (Ilk) expression in cardiomyocytes appears cardioprotective and may mitigate the effects of age-dependent Integrin accumulation, resulting in reduced incidence of arrhythmia with age, as well as decreased myocardial stiffness and increased lifespan. In rat cardiac fibroblasts, reduction of Ilk is correlated with a decline in the markers of cellular senescence [224]. However, the β1-Integrin/Ilk complex is necessary for normal cardiac development and contractile activity, and inhibition within cardiomyocytes and PCs beyond a certain threshold results in cardioblast adhesion defects and arrhythmia in Drosophila [141], while β1-Integrin/Ilk knock-out mice develop myocardial fibrosis, cardiomyopathy, and heart failure [143,225]. Over-expression of β1-Integrin within young adult Drosophila hearts increases myocardial stiffness and accelerates heart aging, leading to arrhythmia, decreased diastolic diameter, and decreased fractional shortening common in older hearts [141]. Though cardiac stiffening increases with age and is correlated with failing hearts, inelasticity in and of itself is not necessarily indicative of dysfunction [226]. For instance, up-regulation of the focal adhesion protein Vinculin reinforces the Drosophila cardiac cytoskeleton, resulting in reduced cardiac diameter but increased shortening and lengthening velocities, thus mitigating age-related decline in contractility and increasing longevity [227]. Careful regulation of ECM proteins is therefore necessary to ensure cardiac health.
Inhibition of Dystrophin (Dys) expression in Drosophila results in cardiac dilation and reduced fractional shortening, reminiscent of dilated cardiomyopathy in mammals [71]. Individuals further experience a more rapid onset of age-dependent myofibrillar disorganisation and decreased lifespan. In mammals, mutation of the dys gene is a cause of muscular dystrophy (MD), in which cardiomyopathies and heart failure are symptomatic, and cardiac dilation and arrhythmia are observed (reviewed in [228]).
Drosophila, unlike vertebrates, possesses but a single Talin gene (rhea) [47,229]. Ubiquitous loss of Talin function during Drosophila embryogensis compromises cardioblast polarity and promotes the formation of ectopic cardiac lumens [90], while localised inhibition of Talin within the myocardium causes loss of contact between neighbouring myocytes, as well as accumulation of Prc (Collagen-IV) and reduced longevity [1]. Myocardial Talin reduction results in compromised contractility (reduced cardiac output) and cardiac dilation that resembles dilated cardiomyopathy in vertebrates (Figure 5B).
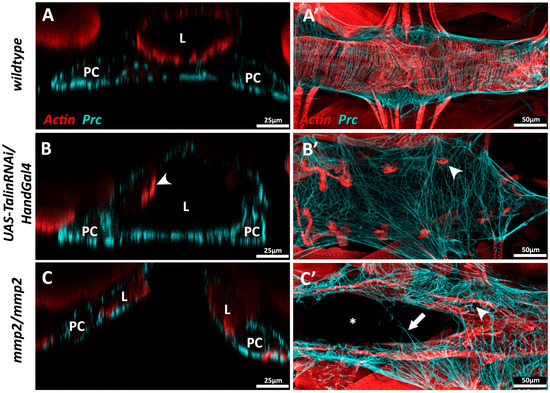
Figure 5.
Mis-regulation of ECM proteins in Drosophila results in cardiac dilation or cardia bifida. (A–A’) Wildtype third instar dorsal vessel in cross-sectional (A) and coronal view (A’). Actin (red) forms regular transverse myofibrils along the length of the heart tube. Abluminal Prc networks (cyan) envelop the heart tube and pericardial cells (PCs); (B–B’) Dorsal vessel of a third instar UAS-TalinRNAi/HandGal4 larva in which Talin expression has been inhibited via RNA interference in cardiomyocytes and PCs during late embryogenesis and throughout larval growth. (B) Cross-sectional view illustrating dilation of the cardiac lumen. Actin myofibrils do not fully envelop the heart tube, and appear constrained to the lateral regions of the vessel (arrowhead) (B’) Actin myofibrils are fewer in number and reduced in length (arrowhead), and Prc bundles are more broadly distributed; (C–C’) Homozygous third instar mmp2w307 phenotypic null mutants show cardia bifida. (C) Cross-sectional view reveals two discrete heart vessels. The Prc network envelops the abluminal domains of both vessels. (C’) Portions of the mmp2 mutant heart chamber are split, while other regions show successful contralateral contact between cardioblasts to enclose a singular lumen. A small number of Prc bundles connect the two discrete heart vessels (arrow). The Actin cytoskeleton appears disorganised; myofibrils envelop the heart vessels but gaps are visible between parallel fibres (arrowhead). L: heart lumen; PC: pericardial cell.
7.3. Mis-Expression of MMPs
Both Drosophila MMPs (MMP1 and MMP2) are required for normal cardiac development. In embryonic MMP1 mutants, the junctional domain of the Drosophila cardioblast (CB) expands at the expense of the luminal domain; these individuals exhibit less organised cardiomyocyte migration and possess a small heart lumen [159]. In MMP2 mutants, luminal proteins are broadly expressed along the entire surface of the CB, and no junctional domain is formed. MMP2 mutants that survive embryogenesis may display luminal defects such as cardia bifida, wherein portions of the heart are split into bilateral tubes (Figure 5C) [159,160].
MMP expression is altered in aged and diseased vertebrate hearts. In older humans, for example, levels of myocardial MMP2/7 and TIMP1/2/4 (from circulating plasma) are increased, whereas those of MMP9 are lowered, resulting in the net reduction of cardiac MMP expression relative to that of TIMP, representing a diminished remodelling capability [230]. In rats and mice experiencing myocardial fibrosis during systolic heart failure or dilated cardiomyopathy, MMP1/2/3/7/8/9/12/13/14 and TIMP1/2 mRNA expression is shown to increase, while TIMP3/4 protein levels drop (reviewed in [4]). Post-infarction cardiac remodelling in vertebrates is dictated in large part by the activities of specific MMPs. During heart failure, MMPs act to promote tissue remodelling and so increase susceptibility to fibrillation [231,232]. For example, MMP2/9, which promote the migration of cardiac progenitor cells during embryogenesis, are up-regulated in the adult pig left ventricle after myocardial infarction [164,201]. Healthy canine hearts constitutively express TIMP3/4 mRNAs, but not MMP mRNAs or those of TIMP1/2 [161]. However, in those manifesting abnormalities, MMP1/2/3/9/13 and TIMP1/2/3/4 mRNAs are up-regulated in myocardial tissue. Moreover, diverse cardiac disorders in canines reveal non-identical mRNA expression profiles for these MMPs and TIMPs within different cardiac tissues, suggesting that specific MMPs and TIMPs play specific roles in certain diseases [161].
8. Conclusions
Cardiac disorders are prevalent in many species of veterinary significance. Since the remodelling of ECM is a common thread between cardiac disorders and diseases, the importance of elucidating the mechanism of ECM regulation cannot be understated. The homology of developmental pathways and molecular mechanisms, relative simplicity, short generation time, and availability of genetic tools make Drosophila an indispensible model for the study of cardiogenesis and heart remodelling. A number of vertebrate disorders, such as cardiac dilation and ECM weakness, have complementary morphology in Drosophila, and are triggered by the loss or over-expression of the same genes. The studies reviewed here reveal that several ECM proteins and receptors, such as Collagens and the Integrin adhesion proteome, are required continuously to permit the heart to form, grow, and adapt to changes in activity or load. Other ECM proteins reveal their function with age, when reduced elasticity correlates with reduced lifespan. Further understanding of the physiological and biomechanical response to disorders of the ECM in Drosophila may reveal future insights into mammalian cardiovascular diseases.
Note Added in Proof
Since submission of this manuscript, it has been demonstrated that both Drosophila MMPs (MMP1 and MMP2) can be secreted or remain membrane-bound (LaFever, K. S.; Wang, X.; Page-McCaw, P.; Bhave, G.; Page-McCaw, A. Both Drosophila matrix metalloproteinases have released and membrane-tethered forms but have different substrates. Sci. Rep. 2017, 7, 44560) [233].
Acknowledgments
We thank Jessica Vanderploeg, Meryl Acker, and Qanber Raza for providing confocal micrographs. Research reported here was supported by the Natural Sciences and Engineering Research Council of Canada.
Author Contributions
Chris J. R. Hughes composed the review with input from J. Roger Jacobs.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bogatan, S.; Cevik, D.; Demidov, V.; Vanderploeg, J.; Panchbhaya, A.; Vitkin, A.; Jacobs, J.R. Talin is required continuously for cardiomyocyte remodeling during heart growth in Drosophila. PLoS ONE 2015, 10, e0131238. [Google Scholar] [CrossRef] [PubMed]
- Page-McCaw, A.; Serano, J.; Sante, J.M.; Rubin, G.M. Drosophila matrix metalloproteinases are required for tissue remodeling, but not embryonic development. Dev. Cell 2003, 4, 95–106. [Google Scholar] [CrossRef]
- Lehmacher, C.; Abeln, B.; Paululat, A. The ultrastructure of Drosophila heart cells. Arthropod Struct. Dev. 2012, 41, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Louzao-Martinez, L.; Vink, A.; Harakalova, M.; Asselbergs, F.W.; Verhaar, M.C.; Cheng, C. Characteristic adaptations of the extracellular matrix in dilated cardiomyopathy. Int. J. Cardiol. 2016, 220, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Matricellular proteins in cardiac inflammation and fibrosis. Physiol. Rev. 2012, 92, 635–688. [Google Scholar] [CrossRef] [PubMed]
- Łój, M.; Garncarz, M.; Jank, M. Genomic and genetic aspects of heart failure in dogs—A review. Acta Vet. Hung. 2012, 60, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Schenke-Layland, K.; Stock, U.A.; Nsair, A.; Xie, J.; Angelis, E.; Fonseca, C.G.; Larbig, R.; Mahajan, A.; Shivkumar, K.; Fishbein, M.C.; et al. Cardiomyopathy is associated with structural remodelling of heart valve extracellular matrix. Eur. Heart J. 2009, 30, 2254–2265. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.T.; Liu, S.K.; Liu, M.; Chen, S.P.; Lin, Y.H.; Mao, S.J.T.; Chu, R. Characterization and quantitation of extracellular collagen matrix in myocardium of pigs with spontaneously occurring hypertrophic cardiomyopathy. Cardiovasc. Pathol. 1999, 8, 169–175. [Google Scholar] [CrossRef]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [PubMed]
- Kular, J.K.; Basu, S.; Sharma, R.I. The extracellular matrix: Structure, composition, age-related differences, tools for analysis and applications for tissue engineering. J. Tissue Eng. 2014, 5, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Drechsler, M.; Schmidt, A.C.; Meyer, H.; Paululat, A. The Conserved ADAMTS-like protein lonely heart mediates matrix formation and cardiac tissue integrity. PLoS Genet. 2013, 9, e1003616. [Google Scholar] [CrossRef] [PubMed]
- Berk, B.C.; Fujiwara, K.; Lehoux, S. ECM remodeling in hypertensive heart disease. J. Clin. Investig. 2007, 117, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Leonard, B.L.; Smaill, B.H.; LeGrice, I.J. Structural remodeling and mechanical function in heart failure. Microsc. Microanal. 2012, 18, 50–67. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.; Takawale, A.; Lee, J.; Kassiri, Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenes. Tissue Repair 2012, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Bayomy, A.F.; Bauer, M.; Qiu, Y.; Liao, R. Regeneration in heart disease—Is ECM the key? Life Sci. 2012, 91, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Tidholm, A. Retrospective study of congenital heart defects in 151 dogs. J. Small Anim. Pract. 1997, 38, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Van Der Linde, D.; Konings, E.E.M.; Slager, M.A.; Witsenburg, M.; Helbing, W.A.; Takkenberg, J.J.M.; Roos-Hesselink, J.W. Birth prevalence of congenital heart disease worldwide: A systematic review and meta-analysis. J. Am. Coll. Cardiol. 2011, 58, 2241–2247. [Google Scholar] [CrossRef] [PubMed]
- Pierpont, M.E.; Basson, C.T.; Benson, D.W.; Gelb, B.D.; Giglia, T.M.; Goldmuntz, E.; McGee, G.; Sable, C.A.; Srivastava, D.; Webb, C.L. Genetic basis for congenital heart defects: Current knowledge. Circulation 2007, 115, 3015–3038. [Google Scholar] [CrossRef] [PubMed]
- Hamlin, R.L. Geriatric heart diseases in dogs. Vet. Clin. Small Anim. Pract. 2005, 35, 597–615. [Google Scholar] [CrossRef] [PubMed]
- Parker, H.G.; Meurs, K.M.; Ostrander, E.A. Finding cardiovascular disease genes in the dog. J. Vet. Cardiol. 2006, 8, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Schoenebeck, J.J.; Ostrander, E.A. Insights into morphology and disease from the dog genome project. Annu. Rev. Cell Dev. Biol. 2014, 30, 535–560. [Google Scholar]
- Falk, V.; Garbade, J.; Walther, T. Experimental Models of Heart Failure. In Practical Methods in Cardiovascular Research; Dhein, S., Mohr, F.W., Delmar, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2005; pp. 83–110. [Google Scholar]
- Steudemann, C.; Bauersachs, S.; Weber, K.; Wess, G. Detection and comparison of microRNA expression in the serum of Doberman Pinschers with dilated cardiomyopathy and healthy controls. BMC Vet. Res. 2013, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Meurs, K.M.; Fox, P.R.; Norgard, M.; Spier, A.W.; Lamb, A.; Koplitz, S.L.; Baumwart, R.D. A prospective genetic evaluation of familial dilated cardiomyopathy in the Doberman pinscher. J. Vet. Intern. Med. 2007, 21, 1016–1020. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, H.M.; Fonfara, S.; López-Alvarez, J.; Cripps, P.; Dukes-McEwan, J. Screening for Dilated Cardiomyopathy in Great Danes in the United Kingdom. J. Vet. Intern. Med. 2012, 26, 1140–1147. [Google Scholar] [CrossRef] [PubMed]
- Meurs, K.M.; Stern, J.A.; Sisson, D.D.; Kittleson, M.D.; Cunningham, S.M.; Ames, M.K.; Atkins, C.E.; Defrancesco, T.; Hodge, T.E.; Keene, B.W.; et al. Association of Dilated Cardiomyopathy with the Striatin Mutation Genotype in Boxer Dogs. J. Vet. Intern. Med. 2013, 27, 1437–1440. [Google Scholar] [CrossRef] [PubMed]
- Palermo, V.; Stafford Johnson, M.J.; Sala, E.; Brambilla, P.G.; Martin, M.W.S. Cardiomyopathy in Boxer dogs: A retrospective study of the clinical presentation, diagnostic findings and survival. J. Vet. Cardiol. 2011, 13, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Vollmar, A. The prevalence of cardiomyopathy in the Irish wolfhound: A clinical study of 500 dogs. J. Am. Anim. Hosp. Assoc. 2000, 36, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Reist-Marti, S.B.; Dolf, G.; Leeb, T.; Kottmann, S.; Kietzmann, S.; Butenhoff, K.; Rieder, S. Genetic evidence of subaortic stenosis in the Newfoundland dog. Vet. Rec. 2012, 170, 597. [Google Scholar] [CrossRef] [PubMed]
- Pyle, R.L.; Patterson, D.F.; Chacko, S. The genetics and pathology of discrete subaortic stenosis in the Newfoundland dog. Am. Heart J. 1976, 92, 324–334. [Google Scholar] [CrossRef]
- Stern, J.A.; Meurs, K.M.; Nelson, O.L.; Lahmers, S.M.; Lehmkuhl, L.B. Familial subvalvular aortic stenosis in golden retrievers: Inheritance and echocardiographic findings. J. Small Anim. Pract. 2012, 53, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Bodmer, R. Heart Development and Its Relationship in Drosophila to Vertebrates. Trends Cardiovasc. Med. 1995, 5, 21–28. [Google Scholar] [CrossRef]
- Bier, E.; Bodmer, R. Drosophila, an emerging model for cardiac disease. Gene 2004, 342, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bier, E. Drosophila, the golden bug, emerges as a tool for human genetics. Nat. Rev. Genet. 2005, 6, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, M.; Ocorr, K.; Bodmer, R.; Cartry, J. Drosophila as a model to study cardiac aging. Exp. Gerontol. 2011, 46, 326–330. [Google Scholar] [CrossRef] [PubMed]
- del Valle Rodríguez, A.; Didiano, D.; Desplan, C. Power tools for gene expression and clonal analysis in Drosophila. Nat. Methods 2012, 9, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Bellen, H.J.; Levis, R.W.; He, Y.; Carlson, J.W.; Evans-Holm, M.; Bae, E.; Kim, J.; Metaxakis, A.; Savakis, C.; Schulze, K.L.; et al. The Drosophila gene disruption project: Progress using transposons with distinctive site specificities. Genetics 2011, 188, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Dietzl, G.; Chen, D.; Schnorrer, F.; Su, K.-C.; Barinova, Y.; Fellner, M.; Gasser, B.; Kinsey, K.; Oppel, S.; Scheiblauer, S.; et al. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 2007, 448, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Zhou, R.; Czech, B.; Liu, L.-P.; Holderbaum, L.; Yang-Zhou, D.; Shim, S.; Handler, D.; Karpowicz, P.; Binari, R.; et al. A genome-scale shRNA resource for transgenic RNAi in Drosophila. Nat. Methods 2011, 8, 405–407. [Google Scholar] [CrossRef] [PubMed]
- Page-McCaw, A.; Ewald, A.J.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Sternlicht, M.; Werb, Z. How Matrix metalloproteinases regulate cell behavior. Annu. Rev. Cell Biol. 2001, 17, 463–516. [Google Scholar] [CrossRef] [PubMed]
- Page-McCaw, A. Remodeling the model organism: Matrix metalloproteinase functions in invertebrates. Semin. Cell Dev. Biol. 2008, 19, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.D.; Celniker, S.E.; Holt, R.A.; Evans, C.A.; Gocayne, J.D.; Amanatides, P.G.; Scherer, S.E.; Li, P.W.; Hoskins, R.A.; Galle, R.F.; et al. The genome sequence of Drosophila melanogaster. Science 2000, 287, 2185–2195. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.B.; Boley, N.; Eisman, R.; May, G.E.; Stoiber, M.H.; Duff, M.O.; Booth, B.W.; Wen, J.; Park, S.; Suzuki, A.M.; et al. Diversity and dynamics of the Drosophila transcriptome. Nature 2014, 512, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Guenet, J.L. The mouse genome. Genome Res. 2005, 15, 1729–1740. [Google Scholar] [CrossRef] [PubMed]
- Ezkurdia, I.; Juan, D.; Rodriguez, J.M.; Frankish, A.; Diekhans, M.; Harrow, J.; Vazquez, J.; Valencia, A.; Tress, M.L. Multiple evidence strands suggest that theremay be as few as 19,000 human protein-coding genes. Hum. Mol. Genet. 2014, 23, 5866–5878. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.H.; Gregory, S.L.; Rickoll, W.L.; Fessler, L.I.; Prout, M.; White, R.A.H.; Fristrom, J.W. Talin is essential for integrin function in Drosophila. Dev. Cell 2002, 3, 569–579. [Google Scholar] [CrossRef]
- Chang, H.C.; Newmyer, S.L.; Hull, M.J.; Ebersold, M.; Schmid, S.L.; Mellman, I. Hsc70 is required for endocytosis and clathrin function in Drosophila. J. Cell Biol. 2002, 159, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.J.; Amrein, H.; Izatt, J.A.; Choma, M.A.; Reedy, M.C.; Rockman, H.A. Drosophila as a model for the identification of genes causing adult human heart disease. PNAS 2006, 103, 1394–1399. [Google Scholar] [CrossRef] [PubMed]
- Cammarato, A.; Dambacher, C.M.; Knowles, A.F.; Kronert, W.A.; Bodmer, R.; Ocorr, K.; Bernstein, S.I. Myosin Transducer Mutations Differentially Affect Motor Function, Myofibril Structure, and the Performance of Skeletal and Cardiac Muscles. Mol. Biol. Cell 2008, 19, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Bodmer, R.; Venkatesh, T.V. Heart development in Drosophila and vertebrates: Conservation of molecular mechanisms. Dev. Genet. 1998, 22, 181–186. [Google Scholar] [CrossRef]
- Zeitouni, B.; Sénatore, S.; Séverac, D.; Aknin, C.; Sémériva, M.; Perrin, L. Signalling pathways involved in adult heart formation revealed by gene expression profiling in Drosophila. PLoS Genet. 2007, 3, 1907–1921. [Google Scholar] [CrossRef] [PubMed]
- Vogler, G.; Bodmer, R. Cellular Mechanisms of Drosophila Heart Morphogenesis. J. Cardiovasc. Dev. Dis. 2015, 2, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, K.; Fossett, N.; Molkentin, J.D.; Schulz, R.A. The zinc finger proteins Pannier and GATA4 function as cardiogenic factors in Drosophila. Development 1999, 126, 5679–5688. [Google Scholar] [PubMed]
- Park, M.; Lewis, C.; Turbay, D.; Chung, A.; Chen, J.N.; Evans, S.; Breitbart, R.E.; Fishman, M.C.; Izumo, S.; Bodmer, R. Differential rescue of visceral and cardiac defects in Drosophila by vertebrate tinman-related genes. Proc. Natl. Acad. Sci. USA 1998, 95, 9366–9371. [Google Scholar] [CrossRef] [PubMed]
- Ranganayakulu, G.; Elliott, D.A.; Harvey, R.P.; Olson, E.N. Divergent roles for NK-2 class homeobox genes in cardiogenesis in flies and mice. Development 1998, 125, 3037–3048. [Google Scholar] [PubMed]
- Harvey, R.P. NK-2 homeobox genes and heart development. Dev. Biol. 1996, 178, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Azpiazu, N.; Frasch, M. Tinman and bagpipe: Two homeo box genes that determine cell fates in the dorsal mesoderm of Drosophila. Genes Dev. 1993, 7, 1325–1340. [Google Scholar] [CrossRef] [PubMed]
- Klinedinst, S.L.; Bodmer, R. Gata factor Pannier is required to establish competence for heart progenitor formation. Development 2003, 130, 3027–3038. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.T.; Bodmer, R.; Abmayr, S.M.; McDermott, J.C.; Spoerel, N.A. D-mef2: A Drosophila mesoderm-specific MADS box-containing gene with a biphasic expression profile during embryogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 7520–7524. [Google Scholar] [CrossRef] [PubMed]
- Bour, B.A.; O’Brien, M.A.; Lockwood, W.L.; Goldstein, E.S.; Bodmer, R.; Taghert, P.H.; Abmayr, S.M.; Nguyen, H.T. Drosophila MEF2, a transcription factor that is essential for myogenesis. Genes Dev. 1995, 9, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Sellin, J.; Albrecht, S.; Kölsch, V.; Paululat, A. Dynamics of heart differentiation, visualized utilizing heart enhancer elements of the Drosophila melanogaster bHLH transcription factor Hand. Gene Expr. Patterns 2006, 6, 360–375. [Google Scholar] [CrossRef] [PubMed]
- Hallier, B.; Hoffmann, J.; Roeder, T.; Tögel, M.; Meyer, H.; Paululat, A. The bHLH Transcription Factor Hand Regulates the Expression of Genes Critical to Heart and Muscle Function in Drosophila melanogaster. PLoS ONE 2015, 10, e0134204. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Yi, P.; Li, X.; Olson, E.N. Hand, an evolutionarily conserved bHLH transcription factor required for Drosophila cardiogenesis and hematopoiesis. Development 2006, 133, 1175–1182. [Google Scholar] [CrossRef] [PubMed]
- Lo, P.C.H.; Frasch, M. A role for the COUP-TF-related gene seven-up in the diversification of cardioblast identities in the dorsal vessel of Drosophila. Mech. Dev. 2001, 104, 49–60. [Google Scholar] [CrossRef]
- Mann, T.; Bodmer, R.; Pandur, P. The Drosophila homolog of vertebrate Islet1 is a key component in early cardiogenesis. Development 2009, 136, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Kooij, V.; Venkatraman, V.; Tra, J.; Kirk, J.A.; Rowell, J.; Blice-Baum, A.; Cammarato, A.; Van Eyk, J.E. Sizing up models of heart failure: Proteomics from flies to humans. Proteom. Clin. Appl. 2014, 8, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Cammarato, A.; Ahrens, C.H.; Alayari, N.N.; Qeli, E.; Rucker, J.; Reedy, M.C.; Zmasek, C.M.; Gucek, M.; Cole, R.N.; Van Eyk, J.E.; et al. A mighty small heart: The cardiac proteome of adult Drosophila melanogaster. PLoS ONE 2011, 6, e18497. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, M.C.; Kaushik, G.; Engler, A.J.; Lehman, W.; Cammarato, A. A Drosophila melanogaster model of diastolic dysfunction and cardiomyopathy based on impaired troponin-T function. Circ. Res. 2014, 114, e6–e17. [Google Scholar] [CrossRef] [PubMed]
- Ocorr, K.; Perrin, L.; Lim, H.-Y.Y.; Qian, L.; Wu, X.; Bodmer, R. Genetic control of heart function and aging in Drosophila. Trends Cardiovasc. Med. 2007, 17, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Taghli-Lamallem, O.; Akasaka, T.; Hogg, G.; Nudel, U.; Yaffe, D.; Chamberlain, J.S.; Ocorr, K.; Bodmer, R. Dystrophin deficiency in Drosophila reduces lifespan and causes a dilated cardiomyopathy phenotype. Aging Cell 2008, 7, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Medioni, C.; Sénatore, S.; Salmand, P.A.; Lalevée, N.; Perrin, L.; Sémériva, M. The fabulous destiny of the Drosophila heart. Curr. Opin. Genet. Dev. 2009, 19, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Bassett, A.R.; Tibbit, C.; Ponting, C.P.; Liu, J.L. Highly Efficient Targeted Mutagenesis of Drosophila with the CRISPR/Cas9 System. Cell Rep. 2013, 4, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Bassett, A.R.; Liu, J.-L. CRISPR/Cas9 and genome editing in Drosophila. J. Genet. Genom. 2014, 41, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Bellen, H.J.; Kane, C.J.O.; Wilson, C.; Grossniklaus, U.; Pearson, R.K.; Gehring, W.J. P-element-mediated enhancer detection: A versatile method to study development in Drosophila. Genes Dev. 1989, 3, 1288–1300. [Google Scholar] [CrossRef] [PubMed]
- Perrimon, N.; Noll, E.; McCall, K.; Brand, A. Generating lineage-specific markers to study Drosophila development. Dev. Genet. 1991, 12, 238–252. [Google Scholar] [CrossRef] [PubMed]
- Venken, K.J.T.; Sarrion-Perdigones, A.; Vandeventer, P.J.; Abel, N.S.; Christiansen, A.E.; Hoffman, K.L. Genome engineering: Drosophila melanogaster and beyond. WIREs Dev. Biol. 2015, 5, 233–267. [Google Scholar] [CrossRef] [PubMed]
- Venken, K.J.T.; Bellen, H.J. Emerging technologies for gene manipulation in Drosophila melanogaster. Nat. Rev. Genet. 2005, 6, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Golic, K.G.; Lindquist, S. The FLP recombinase of yeast catalyzes site-specific recombination in the drosophila genome. Cell 1989, 59, 499–509. [Google Scholar] [CrossRef]
- Blair, S.S. Genetic mosaic techniques for studying Drosophila development. Development 2003, 130, 5065–5072. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.J.; Rockman, H.A. Drosophila, Genetic Screens, and Cardiac Function. Circ. Res. 2011, 109, 794–806. [Google Scholar] [CrossRef] [PubMed]
- Brand, A.H.; Perrimon, N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 1993, 118, 401–415. [Google Scholar] [PubMed]
- Duffy, J.B. GAL4 system in Drosophila: A fly geneticist’s Swiss army knife. Genesis 2002, 34, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Yeh, E.; Gustafson, K.; Boulianne, G.L. Green fluorescent protein as a vital marker and reporter of gene expression in Drosophila. Proc. Natl. Acad. Sci. USA 1995, 92, 7036–7040. [Google Scholar] [CrossRef] [PubMed]
- Suster, M.L.; Seugnet, L.; Bate, M.; Sokolowski, M.B. Refining GAL4-driven transgene expression in Drosophila with a GAL80 enhancer-trap. Genesis 2004, 39, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Morin, X.; Daneman, R.; Zavortink, M.; Chia, W. A protein trap strategy to detect GFP-tagged proteins expressed from their endogenous loci in Drosophila. PNAS 2001, 98, 15050–15055. [Google Scholar] [CrossRef] [PubMed]
- Clyne, P.J.; Brotman, J.S.; Sweeney, S.T.; Davis, G. Green Fluorescent Protein Tagging Drosophila Proteins at Their Native Genomic Loci with Small P Elements. Genetics 2003, 165, 1433–1441. [Google Scholar] [PubMed]
- Sarov, M.; Barz, C.; Jambor, H.; Hein, M.Y.; Schmied, C.; Suchold, D.; Stender, B.; Janosch, S.; Vinay Vikas, K.J.; Krishnan, R.T.; et al. A genome-wide resource for the analysis of protein localisation in Drosophila. Elife 2016, 5, 1–38. [Google Scholar] [CrossRef] [PubMed]
- Raza, Q.; Jacobs, J.R. Guidance signalling regulates Leading Edge behaviour during collective cell migration of cardiac cells in Drosophila. Dev. Biol. 2016, 419, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Vanderploeg, J.; Jacobs, J.R. Talin is required to position and expand the luminal domain of the Drosophila heart tube. Dev. Biol. 2015, 405, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Vanderploeg, J.; Jacobs, J.R. Mapping heart development in flies: Src42A acts non-autonomously to promote heart tube formation in Drosophila. Vet. Sci. 2017, 4. [Google Scholar] [CrossRef]
- Ocorr, K.; Vogler, G.; Bodmer, R. Methods to assess Drosophila heart development, function and aging. Methods 2014, 68, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Bradu, A.; Ma, L.; Bloor, J.W.; Podoleanu, A. Dual optical coherence tomography/fluorescence microscopy for monitoring of Drosophila melanogaster larval heart. J. Biophotonics 2009, 2, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Rotstein, B.; Paululat, A. On the Morphology of the Drosophila Heart. J. Cardiovasc. Dev. Dis. 2016, 3, 15. [Google Scholar] [CrossRef]
- Reim, I.; Frasch, M. The Dorsocross T-box genes are key components of the regulatory network controlling early cardiogenesis in Drosophila. Development 2005, 132, 4911–4925. [Google Scholar] [CrossRef] [PubMed]
- Bryantsev, A.L.; Cripps, R.M. Cardiac gene regulatory networks in Drosophila. Biochim. Biophys. Acta Gene Regul. Mech. 2009, 1789, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Haack, T.; Schneider, M.; Schwendele, B.; Renault, A.D. Drosophila heart cell movement to the midline occurs through both cell autonomous migration and dorsal closure. Dev. Biol. 2014, 396, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Schulz, R.A. Heart development in Drosophila. Semin. Cell Dev. Biol. 2007, 18, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Denholm, B.; Skaer, H. Bringing together components of the fly renal system. Curr. Opin. Genet. Dev. 2009, 19, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Weavers, H.; Prieto-Sánchez, S.; Grawe, F.; Garcia-López, A.; Artero, R.; Wilsch-Bräuninger, M.; Ruiz-Gómez, M.; Skaer, H.; Denholm, B. The insect nephrocyte is a podocyte-like cell with a filtration slit diaphragm. Nature 2009, 457, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Chartier, A.; Zaffran, S.; Astier, M.; Sémériva, M.; Gratecos, D. Pericardin, a Drosophila type IV collagen-like protein is involved in the morphogenesis and maintenance of the heart epithelium during dorsal ectoderm closure. Development 2002, 129, 3241–3253. [Google Scholar] [PubMed]
- Molina, M.R.; Cripps, R.M. Ostia, the inflow tracts of the Drosophila heart, develop from a genetically distinct subset of cardial cells. Mech. Dev. 2001, 109, 51–59. [Google Scholar] [CrossRef]
- Lo, P.C.H.; Frasch, M. Establishing A-P polarity in the embryonic heart tube: A conserved function of Hox genes in Drosophila and vertebrates? Trends Cardiovasc. Med. 2003, 13, 182–187. [Google Scholar] [CrossRef]
- Ivy, J.R.; Drechsler, M.; Catterson, J.H.; Bodmer, R.; Ocorr, K.; Paululat, A.; Hartley, P.S. Klf15 is critical for the development and differentiation of drosophila nephrocytes. PLoS ONE 2015, 10, e0134620. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.P.; Nongthomba, U.; Kelly Tanaka, K.K.; Denton, M.L.B.; Meadows, S.M.; Bancroft, N.; Molina, M.R.; Cripps, R.M. Cardiac remodeling in Drosophila arises from changes in actin gene expression and from a contribution of lymph gland-like cells to the heart musculature. Mech. Dev. 2011, 128, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Martínez, E.; Soplop, N.H.; Patel, R.; Kramer, S.G. Repulsion by Slit and Roundabout prevents Shotgun/E-cadherin-mediated cell adhesion during Drosophila heart tube lumen formation. J. Cell Biol. 2008, 182, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Heeneman, S.; Cleutjens, J.P.; Faber, B.C.; Creemers, E.E.; Van Suylen, R.; Lutgens, E.; Cleutjens, K.B.; Daemen, M.J. The dynamic extracellular matrix: Intervention strategies during heart failure and atherosclerosis. J. Pathol. 2003, 200, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Horn, M.A.; Trafford, A.W. Aging and the cardiac collagen matrix: Novel mediators of fibrotic remodelling. J. Mol. Cell. Cardiol. 2016, 93, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Integrins: A family of cell surface receptors. Cell 1987, 48, 549–554. [Google Scholar] [CrossRef]
- Vanderploeg, J.; Vazquez Paz, L.L.; MacMullin, A.; Jacobs, J.R. Integrins are required for cardioblast polarisation in Drosophila. BMC Dev. Biol. 2012, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Williamson, R.A.; Henry, M.D.; Daniels, K.J.; Hrstka, R.F.; Lee, J.C.; Sunada, Y.; Ibraghimov-Beskrovnaya, O.; Campbell, K.P. Dystroglycan is essential for early embryonic development: Disruption of Reichert’s membrane in Dag1-null mice. Hum. Mol. Genet. 1997, 6, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.-M.; Schneider, M.; Frock, R.; Castillejo-Lopez, C.; Gaman, E.A.; Baumgartner, S.; Ruohola-Baker, H. Dystroglycan is required for polarizing the epithelial cells and the oocyte in Drosophila. Development 2003, 130, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Xian, X.; Gopal, S.; Couchman, J.R. Syndecans as receptors and organizers of the extracellular matrix. Cell Tissue Res. 2010, 339, 31–46. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Macdonald, B.; Kalluri, R. Structure and function of basement membranes. Exp. Biol. Med. 2007, 232, 1121–1129. [Google Scholar] [CrossRef] [PubMed]
- Isabella, A.J.; Horne-Badovinac, S. Building from the Ground Up; Elsevier Ltd: Amsterdam, The Netherlands, 2015; Volume 76. [Google Scholar]
- Yurchenco, P.D. Basement Membranes: Cell Scaffoldings and Signaling Platforms. Cold Spring Harb. Perspect. Biol. 2011, 3, a004911. [Google Scholar] [CrossRef] [PubMed]
- Haag, T.A.; Haag, N.P.; Lekven, A.C.; Hartenstein, V. The role of cell adhesion molecules in Drosophila heart morphogenesis: Faint sausage, shotgun/DE-cadherin, and laminin A are required for discrete stages in heart development. Dev. Biol. 1999, 208, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Harpaz, N.; Ordan, E.; Ocorr, K.; Bodmer, R.; Volk, T. Multiplexin Promotes Heart but Not Aorta Morphogenesis by Polarized Enhancement of Slit/Robo Activity at the Heart Lumen. PLoS Genet. 2013, 9, e1003597. [Google Scholar] [CrossRef] [PubMed]
- Hollfelder, D.; Frasch, M.; Reim, I. Distinct functions of the laminin β LN domain and collagen IV during cardiac extracellular matrix formation and stabilization of alary muscle attachments revealed by EMS mutagenesis in Drosophila. BMC Dev. Biol. 2014, 14, 26. [Google Scholar] [CrossRef] [PubMed]
- Medioni, C.; Noselli, S. Dynamics of the basement membrane in invasive epithelial clusters in Drosophila. Development 2005, 132, 3069–3077. [Google Scholar] [CrossRef] [PubMed]
- Sessions, A.O.; Kaushik, G.; Parker, S.; Raedschelders, K.; Bodmer, R.; Van Eyk, J.E.; Engler, A.J. Extracellular matrix downregulation in the Drosophila heart preserves contractile function and improves lifespan. Matrix Biol. 2016, in press. [Google Scholar]
- Morishita, N.; Kusachi, S.; Yamasaki, S.; Kondo, J.; Tsuji, T. Sequential Changes in Laminin and Type IV Collagen in the Infarct Zone. Jpn. Circ. J. 1996, 60, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Kusachi, S.; Yamanishi, A.; Kumashiro, H.; Nunoyama, H.; Sano, I.; Nakahama, M.; Murakami, T.; Naito, I.; Ninomiya, Y.; et al. Localization of type IV collagen alpha chain in the myocardium of dilated and hypertrophic cardiomyopathy. Jpn. Heart J. 1998, 39, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Yamanishi, A.; Kusachi, S.; Nakahama, M.; Ninomiya, Y.; Watanabe, T.; Kumashiro, H.; Nunoyama, H.; Kondo, J.; Naito, I.; Tsuji, T. Sequential changes in the localization of the type IV collagen alpha chain in the infarct zone: Immunohistochemical study of experimental myocardial infarction in the rat. Pathol. Res. Pr. 1998, 194, 413–422. [Google Scholar] [CrossRef]
- Rasi, K.; Piuhola, J.; Czabanka, M.; Sormunen, R.; Ilves, M.; Leskinen, H.; Rysä, J.; Kerkelä, R.; Janmey, P.; Heljasvaara, R.; et al. Collagen XV is necessary for modeling of the extracellular matrix and its deficiency predisposes to cardiomyopathy. Circ. Res. 2010, 107, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Utriainen, A.; Sormunen, R.; Kettunen, M.; Carvalhaes, L.S.; Sajanti, E.; Eklund, L.; Kauppinen, R.; Kitten, G.T.; Pihlajaniemi, T. Structurally altered basement membranes and hydrocephalus in a type XVIII collagen deficient mouse line. Hum. Mol. Genet. 2004, 13, 2089–2099. [Google Scholar] [CrossRef] [PubMed]
- Isobe, K.; Kuba, K.; Maejima, Y.; Suzuki, J.; Kubota, S.; Isobe, M. Inhibition of endostatin/collagen XVIII deteriorates left ventricular remodeling and heart failure in rat myocardial infarction model. Circ. J. 2010, 74, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Urbano, J.M.; Torgler, C.N.; Molnar, C.; Tepass, U.; López-varea, A.; Brown, N.H.; De Celis, J.F.; Martín-bermudo, M.D. Drosophila laminins act as key regulators of basement membrane assembly and morphogenesis. Development 2009, 136, 4165–4176. [Google Scholar] [CrossRef] [PubMed]
- Wolfstetter, G.; Holz, A. The role of LamininB2 (LanB2) during mesoderm differentiation in Drosophila. Cell. Mol. Life Sci. 2012, 69, 267–282. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.H.; Galchev, V.I.; Kim, J.Y.; Misek, S.A.; Stevenson, T.K.; Campbell, M.D.; Pagani, F.D.; Day, S.M.; Johnson, T.C.; Washburn, J.G.; et al. Differential protein expression and basal lamina remodeling in human heart failure. Proteom. Clin. Appl. 2016, 10, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hoshijima, M.; Lam, J.; Zhou, Z.; Jokiel, A.; Dalton, N.D.; Hultenby, K.; Ruiz-Lozano, P.; Ross, J.; Tryggvason, K.; et al. Cardiomyopathy associated with microcirculation dysfunction in laminin a4 chain-deficient mice. J. Biol. Chem. 2006, 281, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Martinek, N.; Shahab, J.; Saathoff, M.; Ringuette, M. Haemocyte-derived SPARC is required for collagen-IV-dependent stability of basal laminae in Drosophila embryos. J. Cell Sci. 2008, 121, 1671–1680. [Google Scholar] [CrossRef] [PubMed]
- Hartley, P.S.; Motamedchaboki, K.; Bodmer, R.; Ocorr, K. SPARC-dependent cardiomyopathy in drosophila. Circ. Cardiovasc. Genet. 2016, 9, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, A.D. The role of secreted protein acidic and rich in cysteine (SPARC) in cardiac repair and fibrosis: Does expression of SPARC by macrophages influence outcomes? J. Mol. Cell. Cardiol. 2016, 93, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Chanana, B.; Graf, R.; Koledachkina, T.; Pflanz, R.; Vorbrüggen, G. αPS2 integrin-mediated muscle attachment in Drosophila requires the ECM protein Thrombospondin. Mech. Dev. 2007, 124, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Bunch, T.; Wayburn, B.; Volk, T. Thrombospondin-mediated adhesion is essential for the formation of the myotendinous junction in. Development 2007, 1278, 1269–1278. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G.; Ren, G.; Dewald, O.; Zymek, P.; Haudek, S.; Koerting, A.; Winkelmann, K.; Michael, L.H.; Lawler, J.; Entman, M.L. Critical role of endogenous thrombospondin-1 in preventing expansion of healing myocardial infarcts. Circulation 2005, 111, 2935–2942. [Google Scholar] [CrossRef] [PubMed]
- Mustonen, E.; Aro, J.; Puhakka, J.; Ilves, M.; Soini, Y.; Leskinen, H.; Ruskoaho, H.; Rysä, J. Thrombospondin-4 expression is rapidly upregulated by cardiac overload. Biochem. Biophys. Res. Commun. 2008, 373, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Frolova, E.G.; Sopko, N.; Blech, L.; Popovic, Z.B.; Li, J.; Vasanji, A.; Drumm, C.; Krukovets, I.; Jain, M.K.; Penn, M.S.; et al. Thrombospondin-4 regulates fibrosis and remodeling of the myocardium in response to pressure overload. FASEB J. 2012, 26, 2363–2373. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, K.; Manabe, R.I.; Yamada, T.; Nakano, I.; Oguri, Y.; Keene, D.R.; Sengle, G.; Sakai, L.Y.; Sekiguchi, K. ADAMTSL-6 is a novel extracellular matrix protein that binds to fibrillin-1 and promotes fibrillin-1 fibril formation. J. Biol. Chem. 2010, 285, 4870–4882. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, M.; Kumsta, C.; Kaushik, G.; Diop, S.B.; Ding, Y.; Bisharat-Kernizan, J.; Catan, H.; Cammarato, A.; Ross, R.S.; Engler, A.J.; et al. A dual role for integrin-linked kinase and β1-integrin in modulating cardiac aging. Aging Cell 2014, 13, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Zhu, Y.; Sun, Z.; Trzeciakowski, J.P.; Gansner, M.; Depre, C.; Resuello, R.R.G.; Natividad, F.F.; Hunter, W.C.; Genin, G.M.; et al. Vascular smooth muscle cell stiffness as a mechanism for increased aortic stiffness with aging. Circ. Res. 2010, 107, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Shai, S.Y.; Harpf, A.E.; Babbitt, C.J.; Jordan, M.C.; Fishbein, M.C.; Chen, J.; Omura, M.; Leil, T.A.; Becker, K.D.; Jiang, M.; et al. Cardiac myocyte-specific excision of the β1 integrin gene results in myocardial fibrosis and cardiac failure. Circ. Res. 2002, 90, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Shcherbata, H.R.; Yatsenko, A.S.; Patterson, L.; Sood, V.D.; Nudel, U.; Yaffe, D.; Baker, D.; Ruohola-Baker, H. Dissecting muscle and neuronal disorders in a Drosophila model of muscular dystrophy. EMBO J. 2007, 26, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Lefeber, D.J.; de Brouwer, A.P.M.; Morava, E.; Riemersma, M.; Schuurs-Hoeijmakers, J.H.M.; Absmanner, B.; Verrijp, K.; van den Akker, W.M.R.; Huijben, K.; Steenbergen, G.; et al. Autosomal recessive dilated cardiomyopathy due to DOLK mutations results from abnormal dystroglycan O-mannosylation. PLoS Genet. 2011, 7, e1002427. [Google Scholar] [CrossRef] [PubMed]
- Grossman, T.R.; Gamliel, A.; Wessells, R.J.; Taghli-Lamallem, O.; Jepsen, K.; Ocorr, K.; Korenberg, J.R.; Peterson, K.L.; Rosenfeld, M.G.; Bodmer, R.; et al. Over-expression of DSCAM and COL6A2 cooperatively generates congenital heart defects. PLoS Genet. 2011, 7, e1002344. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.L.; Clemens, J.C.; Neves, G.; Hattori, D.; Flanagan, J.J.; Hummel, T.; Vasconcelos, M.L.; Chess, A.; Zipursky, S.L. Analysis of Dscam diversity in regulating axon guidance in Drosophila mushroom bodies. Neuron 2004, 43, 673–686. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; You, X.; Gogol-Dö ring, A.; He, H.; Kise, Y.; Sohn, M.; Chen, T.; Klebes, A.; Schmucker, D.; Chen, W. Ultra-deep profiling of alternatively spliced Drosophila Dscam isoforms by circularization- assisted multi-segment sequencing. EMBO J. 2013, 32, 2029–2038. [Google Scholar] [CrossRef] [PubMed]
- Barlow, G.M.; Chen, X.N.; Shi, Z.Y.; Lyons, G.E.; Kurnit, D.M.; Celle, L.; Spinner, N.B.; Zackai, E.; Pettenati, M.J.; Van Riper, A.J.; et al. Down syndrome congenital heart disease: A narrowed region and a candidate gene. Genet. Med. 2001, 3, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Knox, J.; Moyer, K.; Yacoub, N.; Soldaat, C.; Komosa, M.; Vassilieva, K.; Wilk, R.; Hu, J.; de Vazquez Paz, L.L.; Syed, Q.; et al. Syndecan contributes to heart cell specification and lumen formation during Drosophila cardiogenesis. Dev. Biol. 2011, 356, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Lunde, I.G.; Herum, K.M.; Carlson, C.C.; Christensen, G. Syndecans in heart fibrosis. Cell Tissue Res. 2016, 365, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Liu, J.; Bodmer, R. Slit and Robo control cardiac cell polarity and morphogenesis. Curr. Biol. 2005, 15, 2271–2278. [Google Scholar] [CrossRef] [PubMed]
- Medioni, C.; Astier, M.; Zmojdzian, M.; Jagla, K.; Sémériva, M. Genetic control of cell morphogenesis during Drosophila melanogaster cardiac tube formation. J. Cell Biol. 2008, 182, 249–261. [Google Scholar] [CrossRef] [PubMed]
- MacMullin, A.; Jacobs, J.R. Slit coordinates cardiac morphogenesis in Drosophila. Dev. Biol. 2006, 293, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Mommersteeg, M.T.M.; Andrews, W.D.; Ypsilanti, A.R.; Zelina, P.; Yeh, M.L.; Norden, J.; Kispert, A.; Chédotal, A.; Christoffels, V.M.; Parnavelas, J.G. Slit-roundabout signaling regulates the development of the cardiac systemic venous return and pericardium. Circ. Res. 2013, 112, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Mommersteeg, M.T.M.; Yeh, M.L.; Parnavelas, J.G.; Andrews, W.D. Disrupted Slit-Robo signalling results in membranous ventricular septum defects and bicuspid aortic valves. Cardiovasc. Res. 2015, 106, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Macabenta, F.D.; Jensen, A.G.; Cheng, Y.S.; Kramer, J.J.; Kramer, S.G. Frazzled/DCC facilitates cardiac cell outgrowth and attachment during Drosophila dorsal vessel formation. Dev. Biol. 2013, 380, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, P.; Ye, K.; Cai, H. Central role of SIAH inhibition in DCC-dependent cardioprotection provoked by netrin-1/NO. Proc. Natl. Acad. Sci. USA 2015, 112, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Raza, Q.S.; Vanderploeg, J.L.; Jacobs, J.R. Matrix Metalloproteinases are required for membrane motility and lumenogenesis during Drosophila heart development. PLoS ONE 2017, 12, e0171905. [Google Scholar] [CrossRef] [PubMed]
- Raza, Q.; Vanderploeg, J.; Jacobs, J.R. Transmembrane and Secreted MMPs are Required for Heart Morphogenesis. In Proceedings of the Drosophila Research Conference, Chicago, IN, USA, 4–8 March 2015. [Google Scholar]
- Fonfara, S.; Hetzel, U.; Tew, S.R.; Cripps, P.; Dukes-McEwan, J.; Clegg, P.D. Expression of matrix metalloproteinases, their inhibitors, and lysyl oxidase in myocardial samples from dogs with end-stage systemic and cardiac diseases. Am. J. Vet. Res. 2013, 74, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Ducharme, A.; Frantz, S.; Aikawa, M.; Rabkin, E.; Lindsey, M.; Rohde, L.E.; Schoen, F.J.; Kelly, R.A.; Werb, Z.; Libby, P.; et al. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J. Clin. Investig. 2000, 106, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Hayashidani, S.; Tsutsui, H.; Ikeuchi, M.; Shiomi, T.; Matsusaka, H.; Kubota, T.; Imanaka-Yoshida, K.; Itoh, T.; Takeshita, A. Targeted deletion of MMP-2 attenuates early LV rupture and late remodeling after experimental myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1229–H1235. [Google Scholar] [CrossRef] [PubMed]
- Etoh, T.; Joffs, C.; Deschamps, A.M.; Davis, J.; Dowdy, K.; Hendrick, J.; Baicu, S.; Mukherjee, R.; Manhaini, M.; Spinale, F.G. Myocardial and interstitial matrix metalloproteinase activity after acute myocardial infarction in pigs. Am. J. Physiol. Circ. Physiol. 2001, 281, H987–H994. [Google Scholar]
- Tian, H.; Cimini, M.; Fedak, P.W.M.; Altamentova, S.; Fazel, S.; Huang, M.L.; Weisel, R.D.; Li, R.K. TIMP-3 deficiency accelerates cardiac remodeling after myocardial infarction. J. Mol. Cell. Cardiol. 2007, 43, 733–743. [Google Scholar] [CrossRef] [PubMed]
- Kandalam, V.; Basu, R.; Abraham, T.; Wang, X.; Soloway, P.D.; Jaworski, D.M.; Oudit, G.Y.; Kassiri, Z. TIMP2 deficiency accelerates adverse post-myocardial infarction remodeling because of enhanced MT1-MMP activity despite lack of MMP2 activation. Circ. Res. 2010, 106, 796–808. [Google Scholar] [CrossRef] [PubMed]
- Creemers, E.E.J.M.; Davis, J.N.; Parkhurst, A.M.; Leenders, P.; Dowdy, K.B.; Hapke, E.; Hauet, A.M.; Escobar, P.G.; Cleutjens, J.P.M.; Smits, J.F.M.; et al. Deficiency of TIMP-1 exacerbates LV remodeling after myocardial infarction in mice. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H364–H371. [Google Scholar] [CrossRef] [PubMed]
- Fedak, P.W.M.; Smookler, D.S.; Kassiri, Z.; Ohno, N.; Leco, K.J.; Verma, S.; Mickle, D.A.G.; Watson, K.L.; Hojilla, C.V.; Cruz, W.; et al. TIMP-3 deficiency leads to dilated cardiomyopathy. Circulation 2004, 110, 2401–2409. [Google Scholar] [CrossRef] [PubMed]
- Spinale, F.G. Myocardial Matrix Remodeling and the Matrix Metalloproteinases: Influence on Cardiac Form and Function. Physiol. Rev. 2007, 87, 1285–1342. [Google Scholar] [CrossRef] [PubMed]
- Myllyharju, J.; Kivirikko, K.I. Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet. 2004, 20, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Volk, T.; Wang, S.; Rotstein, B.; Paululat, A. Matricellular proteins in development: Perspectives from the Drosophila heart. Matrix Biol. 2014, 37, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Meyer, F.; Moussian, B. Drosophila multiplexin (Dmp) modulates motor axon pathfinding accuracy. Dev. Growth Differ. 2009, 51, 483–498. [Google Scholar] [CrossRef] [PubMed]
- Price, R.L.; Nakagawa, M.; Terracio, L.; Borg, T.K. Ultrastructural localization of laminin on in vivo embryonic, neonatal, and adult rat cardiac myocytes and in early rat embryos raised in whole-embryo culture. J. Histochem. Cytochem. 1992, 40, 1373–1381. [Google Scholar] [CrossRef] [PubMed]
- Davis, L.A.; Ogle, R.C.; Little, C.D. Embryonic heart mesenchymal cell migration in laminin. Dev. Biol. 1989, 133, 37–43. [Google Scholar] [CrossRef]
- Colognato, H.; Yurchenco, P.D. Form and function: The laminin family of heterotrimers. Dev. Dyn. 2000, 218, 213–234. [Google Scholar] [CrossRef]
- Adams, J.C. Thrombospondins: Multifunctional Regulators of Cell Interactions. Annu. Rev. Cell Dev. Biol. 2001, 17, 25–51. [Google Scholar] [CrossRef] [PubMed]
- Manso, A.M.; Elsherif, L.; Kang, S.M.; Ross, R.S. Integrins, membrane-type matrix metalloproteinases and ADAMs: Potential implications for cardiac remodeling. Cardiovasc. Res. 2006, 69, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Lapidos, K.A.; Kakkar, R.; McNally, E.M. The Dystrophin Glycoprotein Complex: Signaling Strength and Integrity for the Sarcolemma. Circ. Res. 2004, 94, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Kreipke, R.E.; Kwon, Y.V.; Shcherbata, H.R.; Ruohola-Baker, H. Drosophila melanogaster as a Model of Muscle Degeneration Disorders. Curr. Top. Dev. Biol. 2017, 121, 83–109. [Google Scholar] [PubMed]
- Allikian, M.J.; Bhabha, G.; Dospoy, P.; Heydemann, A.; Ryder, P.; Earley, J.U.; Wolf, M.J.; Rockman, H.A.; McNally, E.M. Reduced life span with heart and muscle dysfunction in Drosophila sarcoglycan mutants. Hum. Mol. Genet. 2007, 16, 2933–2943. [Google Scholar] [CrossRef] [PubMed]
- Durbeej, M.; Larsson, E.; Ibraghimov-Beskrovnaya, O.; Roberds, S.L.; Campbell, K.P.; Ekblom, P. Non-muscle α-dystroglycan is involved in epithelial development. J. Cell Biol. 1995, 130, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Ibraghimov-Beskrovnaya, O.; Ervasti, J.M.; Leveille, C.J.; Slaughter, C.A.; Sernett, S.W.; Campbell, K.P. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature 1992, 355, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Tögel, M.; Meyer, H.; Lehmacher, C.; Heinisch, J.J.; Pass, G.; Paululat, A. The bHLH transcription factor hand is required for proper wing heart formation in Drosophila. Dev. Biol. 2013, 381, 446–459. [Google Scholar] [CrossRef] [PubMed]
- Visse, R.; Nagase, H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: Structure, function, and biochemistry. Circ. Res. 2003, 92, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Spinale, F.G.; Coker, M.L.; Thomas, C.V.; Walker, J.D.; Mukherjee, R.; Hebbar, L. Time-dependent changes in matrix metalloproteinase activity and expression during the progression of congestive heart failure: Relation to ventricular and myocyte function. Circ. Res. 1998, 82, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Coker, M.L.; Thomas, C.V.; Clair, M.J.; Hendrick, J.W.; Krombach, R.S.; Galis, Z.S.; Spinale, F.G. Myocardial matrix metalloproteinase activity and abundance with congestive heart failure. Am. J. Physiol. 1998, 274, H1516–H1523. [Google Scholar] [PubMed]
- Reinhardt, D.; Sigusch, H.H.; Henβe, J.; Tyagi, S.C.; Körfer, R.; Figulla, H.R. Cardiac remodelling in end stage heart failure: Upregulation of matrix metalloproteinase (MMP) irrespective of the underlying disease, and evidence for a direct inhibitory effect of ACE inhibitors on MMP. Heart 2002, 88, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.; Fan, D.; Basu, R.; Kandalam, V.; Kassiri, Z. Tissue inhibitor of metalloproteinases (TIMPs) in heart failure. Heart Fail. Rev. 2012, 17, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Fondard, O.; Detaint, D.; Iung, B.; Choqueux, C.; Adle-biassette, H.; Jarraya, M.; Hvass, U.; Couetil, J.; Henin, D.; Michel, J.; et al. Extracellular matrix remodelling in human aortic valve disease: The role of matrix metalloproteinases and their tissue inhibitors. Eur. Heart J. 2005, 26, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Sakata, Y.; Yamamoto, K.; Mano, T.; Nishikawa, N.; Yoshida, J.; Hori, M.; Miwa, T.; Masuyama, T. Activation of Matrix Metalloproteinases Precedes Left Ventricular Remodeling in Hypertensive Heart Failure Rats: Its Inhibition as a Primary Effect of Angiotensin-Converting Enzyme Inhibitor. Circulation 2004, 109, 2143–2149. [Google Scholar] [CrossRef] [PubMed]
- Van Wart, H.E.; Birkedal-Hansen, H. The cysteine switch: A principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. Proc. Natl. Acad. Sci. USA 1990, 87, 5578–5582. [Google Scholar] [CrossRef] [PubMed]
- Arpino, V.; Brock, M.; Gill, S.E. The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix Biol. 2015, 44-46, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Lemaître, V.; D’Armiento, J. Matrix metalloproteinases in development and disease. Birth Defects Res. 2006, 78, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Knorr, E.; Schmidtberg, H.; Vilcinskas, A.; Altincicek, B. MMPs Regulate both Development and Immunity in the Tribolium Model Insect. PLoS ONE 2009, 4, e4751. [Google Scholar] [CrossRef] [PubMed]
- Ra, H.J.; Parks, W.C. Control of matrix metalloproteinase catalytic activity. Matrix Biol. 2007, 26, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Parks, W.C.; Wilson, C.L.; Lopez-Boado, Y.S. Matrix metalloproteinases as modulators of inflammation. Nat. Rev. Immunol. 2004, 4, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Spinale, F.G. Matrix metalloproteinases: Regulation and dysregulation in the failing heart. Circ. Res. 2002, 90, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; McTiernan, C.F.; Feldman, A.M. Proinflammatory cytokines regulate tissue inhibitors of metalloproteinases and disintegrin metalloproteinase in cardiac cells. Cardiovasc. Res. 1999, 42, 162–172. [Google Scholar] [CrossRef]
- Koskivirta, I.; Kassiri, Z.; Rahkonen, O.; Kiviranta, R.; Oudit, G.Y.; McKee, T.D.; Kytö, V.; Saraste, A.; Jokinen, E.; Liu, P.P.; et al. Mice with tissue inhibitor of metalloproteinases 4 (Timp4) deletion succumb to induced myocardial infarction but not to cardiac pressure overload. J. Biol. Chem. 2010, 285, 24487–24493. [Google Scholar] [CrossRef] [PubMed]
- Moe, G.W.; Laurent, G.; Doumanovskaia, L.; Konig, A.; Hu, X.; Dorian, P. Matrix Metalloproteinase Inhibition Attenuates Atrial Remodeling and Vulnerability to Atrial Fibrillation in a Canine Model of Heart Failure. J. Card. Fail. 2008, 14, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Linke, A.; Müller, P.; Nurzynska, D.; Casarsa, C.; Torella, D.; Nascimbene, A.; Castaldo, C.; Cascapera, S.; Böhm, M.; Quaini, F.; et al. Stem cells in the dog heart are self-renewing, clonogenic, and multipotent and regenerate infarcted myocardium, improving cardiac function. Proc. Natl. Acad. Sci. USA 2005, 102, 8966–8971. [Google Scholar] [CrossRef] [PubMed]
- Nagase, H.; Visse, R.; Murphy, G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 2006, 69, 562–573. [Google Scholar] [CrossRef] [PubMed]
- Llano, E.; Pendas, A.M.; Aza-Blanc, P.; Kornberg, T.B.; Lopez-Otin, C. Dm1-MMP, a matrix metalloproteinase from Drosophila with a potential role in extracellular matrix remodeling during neural development. J. Biol. Chem. 2000, 275, 35978–35985. [Google Scholar] [CrossRef] [PubMed]
- Llano, E.; Adam, G.; Pendás, A.M.; Quesada, V.; Sánchez, L.M.; Santamaría, I.; Noselli, S.; López-Otín, C.; De Oviedo, U. Structural and enzymatic characterization of Drosophila Dm2-MMP, a membrane-bound matrix metalloproteinase with tissue-specific expression. J. Biol. Chem. 2002, 277, 23321–23329. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.M.; Page-McCaw, A.; Broihier, H.T. Matrix metalloproteinases promote motor axon fasciculation in the Drosophila embryo. Development 2008, 135, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Horn, M.A. Cardiac Physiology of Aging: Extracellular Considerations. Compr. Physiol. 2015, 5, 1069–1121. [Google Scholar] [PubMed]
- Brancaccio, M.; Hirsch, E.; Notte, A.; Selvetella, G.; Lembo, G.; Tarone, G. Integrin signalling: The tug-of-war in heart hypertrophy. Cardiovasc. Res. 2006, 70, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Sessions, A.O.; Engler, A.J. Mechanical Regulation of Cardiac Aging in Model Systems. Circ. Res. 2016, 118, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Eghbali, M.; Czaja, M.J.; Zeydel, M.; Weiner, F.R.; Zern, M.A.; Seifter, S.; Blumenfeld, O.O. Collagen chain mRNAs in isolated heart cells from young and adult rats. J. Mol. Cell. Cardiol. 1988, 20, 267–276. [Google Scholar] [CrossRef]
- Li, D.; Fareh, S.; Leung, T.K.; Nattel, S. Promotion of atrial fibrillation by heart failure in dogs: Atrial remodeling of a different sort. Circulation 1999, 100, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.A.; Copeland, C.R.; Reich, D.H.; Tung, L. Mechanical Coupling Between Myofibroblasts and Cardiomyocytes Slows Electrical Conduction in Fibrotic Cell Monolayers. Circulation 2012, 123, 2083–2093. [Google Scholar] [CrossRef] [PubMed]
- Burstein, B.; Nattel, S. Atrial Fibrosis: Mechanisms and Clinical Relevance in Atrial Fibrillation. J. Am. Coll. Cardiol. 2008, 51, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Morita, N.; Mandel, W.J.; Kobayashi, Y.; Karagueuzian, H.S. Cardiac fibrosis as a determinant of ventricular tachyarrhythmias. J. Arrhythm. 2014, 30, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Conrad, C.H.; Brooks, W.W.; Hayes, J.A.; Sen, S.; Robinson, K.G.; Bing, O.H.L. Myocardial Fibrosis and Stiffness With Hypertrophy and Heart Failure in the Spontaneously Hypertensive Rat. Circulation 1995, 91, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Segura, A.M.; Frazier, O.H.; Buja, L.M. Fibrosis and heart failure. Heart Fail. Rev. 2014, 19, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Anversa, P.; Hiller, B.; Ricci, R.; Guideri, G.; Olivetti, G. Myocyte cell loss and myocyte hypertrophy in the aging rat heart. J. Am. Coll. Cardiol. 1986, 8, 1441–1448. [Google Scholar] [CrossRef]
- Kapelko, V.I. Extracellular matrix alterations in cardiomyopathy: The possible crucial role in the dilative form. Exp. Clin. Cardiol. 2001, 6, 41–49. [Google Scholar] [PubMed]
- Brooks, A.; Schinde, V.; Bateman, A.C.; Gallagher, P.J. Interstitial fibrosis in the dilated non-ischaemic myocardium. Heart 2003, 89, 1255–1256. [Google Scholar] [CrossRef] [PubMed]
- Biernacka, A.; Frangogiannis, N.G. Aging and Cardiac Fibrosis. Aging Dis. 2011, 2, 158–173. [Google Scholar] [PubMed]
- Burgess, M.L.; Mccrea, J.C.; Hedrick, H.L. Age-associated changes in cardiac matrix and integrins. Mech. Ageing Dev. 2001, 122, 1739–1756. [Google Scholar]
- de Castro Brás, L.E.; Toba, H.; Baicu, C.F.; Zile, M.R.; Weintraub, S.T.; Lindsey, M.L.; Bradshaw, A.D. Age and SPARC change the extracellular matrix composition of the left ventricle. Biomed Res. Int. 2014, 2014, 810562. [Google Scholar] [CrossRef] [PubMed]
- Horn, M.A.; Graham, H.K.; Richards, M.A.; Clarke, J.D.; Greensmith, D.J.; Briston, S.J.; Hall, M.C.S.; Dibb, K.M.; Trafford, A.W. Age-related divergent remodeling of the cardiac extracellular matrix in heart failure: Collagen accumulation in the young and loss in the aged. J. Mol. Cell. Cardiol. 2012, 53, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Na, J.; Musselman, L.P.; Pendse, J.; Baranski, T.J.; Bodmer, R.; Ocorr, K.; Cagan, R. A Drosophila Model of High Sugar Diet-Induced Cardiomyopathy. PLoS Genet. 2013, 9, e1003175. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, Z.; Feng, Z.; Wang, J.; Ouyang, C.; Liu, W.; Fu, B.; Cai, G.; Wu, C.; Wei, R.; et al. Integrin-linked kinase induces both senescence-associated alterations and extracellular fibronectin assembly in aging cardiac fibroblasts. J. Gerontol. Biol. Sci. 2006, 61A, 1232–1245. [Google Scholar] [CrossRef]
- White, D.E.; Coutu, P.; Shi, Y.F.; Tardif, J.C.; Nattel, S.; St. Arnaud, R.; Dedhar, S.; Muller, W.J. Targeted ablation of ILK from the murine heart results in dilated cardiomyopathy and spontaneous heart failure. Genes Dev. 2006, 20, 2355–2360. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, G.; Zambon, A.C.; Fuhrmann, A.; Bernstein, S.I.; Bodmer, R.; Engler, A.J.; Cammarato, A. Measuring passive myocardial stiffness in Drosophila melanogaster to investigate diastolic dysfunction. J. Cell. Mol. Med. 2012, 16, 1656–1662. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, G.; Spenlehauer, A.; Sessions, A.O.; Trujillo, A.S.; Fuhrmann, A.; Fu, Z.; Venkatraman, V.; Pohl, D.; Tuler, J.; Wang, M.; et al. Vinculin network-mediated cytoskeletal remodeling regulates contractile function in the aging heart. Sci. Transl. Med. 2015, 7, 292ra99. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J.; Stöllberger, C. The heart in human dystrophinopathies. Cardiology 2003, 99, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Monkley, S.J.; Pritchard, C.A.; Critchley, D.R. Analysis of the Mammalian Talin2 Gene TLN2. Biochem. Biophys. Res. Commun. 2001, 286, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Bonnema, D.D.; Webb, C.S.; Pennington, W.R.; Stroud, R.E.; Leonardi, A.E.; Clark, L.L.; Mcclure, C.D.; Spinale, F.G.; Zile, M.R. Effects of age on plasma matrix metalloproteinases (MMPs) and tissue inhibitor of metalloproteinases (TIMPs). J. Card. Fail. 2007, 13, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Apple, K.A.; Yarbrough, W.M.; Mukherjee, R.; Deschamps, A.M.; Escobar, P.G.; Mingoia, J.T.; Sample, J.A.; Hendrick, J.W.; Dowdy, K.B.; McLean, J.E.; et al. Selective targeting of matrix metalloproteinase inhibition in post-infarction myocardial remodeling. J. Cardiovasc. Pharmacol. 2006, 47, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Spinale, F.G.; Coker, M.L.; Krombach, S.R.; Mukherjee, R.; Hallak, H.; Houck, W.V.; Clair, M.J.; Kribbs, S.B.; Johnson, L.L.; Peterson, J.T.; et al. Matrix metalloproteinase inhibition during the development of congestive heart failure: Effects on left ventricular dimensions and function. Circ. Res. 1999, 85, 364–376. [Google Scholar] [CrossRef] [PubMed]
- LaFever, K. S.; Wang, X.; Page-McCaw, P.; Bhave, G.; Page-McCaw, A. Both Drosophila matrix metalloproteinases have released and membrane-tethered forms but have different substrates. Sci. Rep. 2017, 7, 44560. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).